Syndactyly (MIM) [64]
Sub-groups
Gene
Loci
Phenotype
SD1/Zygodactyly
–
2q34-q36
Syndactyly of the third + fourth finger web space and/or the web between the second and third toes
(MIM 185900)
Zygodactyly 1
–
3p21.31
Foot zygodactyly without hand or bony involvement
Zygodactyly 2
–
–
Bilateral cutaneous and/or bony hand and foot involvement
Zygodactyly 3
–
–
Specific bilateral webbing, cutaneous or bony, of the third + fourth finger
Zygodactyly 4
–
–
Bilateral cutaneous webbing of the fourth + fifth toe
SD2/synpolydactyly (MIM 185900)
SPD 1
Homeobox D 13
Syndactyly of the third + fourth fingers associated with polydactyly of all components or of part of the fourth finger in the web. Foot polydactyly of the fifth toe included in a web of syndactyly of the fourth + fifth toes
SPD 2
Fibulin 1
Syndactyly of the third/fourth finger web space and synostosis of the metacarpal and metatarsal bones
SPD3
14q11.2-q12
Third and fourth finger syndactyly with varying degrees of polydactyly of the fourth finger web space. There is also polydactyly of the fifth toe commonly
SD3 (of the ODDD spectrum) (MIM 186100)
Gap Junction Protein Alpha 1
Complete/bilateral, generally soft tissue syndactyly between the fourth and fifth fingers. The fifth finger is short with absent or rudimentary middle phalanx
SD4/Haas type (MIM 186200)
LMBR1
Complete syndactyly, bilateral with polydactyly, generally six metacarpals and six digits
SD5 (MIM 186300)
Homeobox D 13
Soft tissue syndactyly usually affects the third and fourth fingers and second and third toes with associated metatarsal and metacarpal fusion (fourth and fifth or the third and fourth)
SD6/Mitten Hand (MIM n/a)
–
–
Unilateral syndactyly of digits 2–5
SD7/Cenani–Lenz (MIM 212780)
LRP4
Severe shortening of the ulna and radius with fusion, fusion of the metacarpals and “disorganization” of phalangeal development including syndactyly
SD8 (MIM n/a)
MF4
? Xq26
Fusion of the fourth and fifth metacarpals
SD9/Mesoaxial Synostotic (MIM 609432)
–
17p13.3
Complete syndactyly and synostosis of the third and fourth fingers with severe bone reduction in the proximal phalanges, hypoplasia of the thumbs and halluces, aplasia/hypoplasia of the middle phalanges of the second and fifth fingers, and complete or partial soft tissue syndactyly of the toes
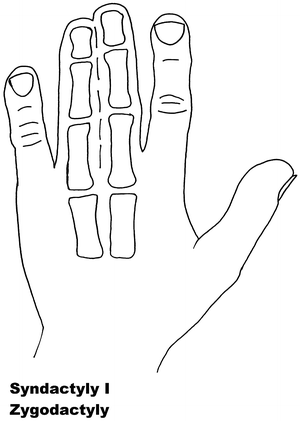
Fig. 13.1
Syndactyly 1. Reprinted from Jordan D, Hindocha S, Dhital M, Saleh M, Khan W. The epidemiology, genetics and future management of syndactyly. Open Orthop J. 2012;6:14–27. Copyright © Jordan et al.; Licensee Bentham Open
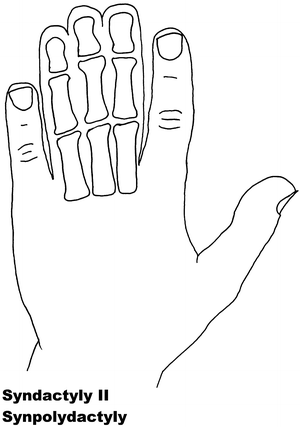
Fig. 13.2
Syndactyly 2. Reprinted from Jordan D, Hindocha S, Dhital M, Saleh M, Khan W. The epidemiology, genetics and future management of syndactyly. Open Orthop J. 2012;6:14–27. Copyright © Jordan et al.; Licensee Bentham Open
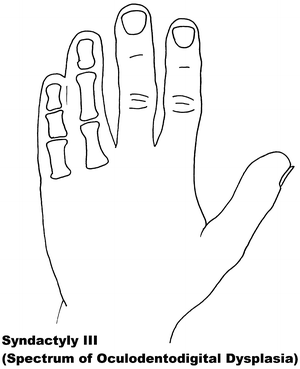
Fig. 13.3
Syndactyly 3. Reprinted from Jordan D, Hindocha S, Dhital M, Saleh M, Khan W. The epidemiology, genetics and future management of syndactyly. Open Orthop J. 2012;6:14–27. Copyright © Jordan et al.; Licensee Bentham Open
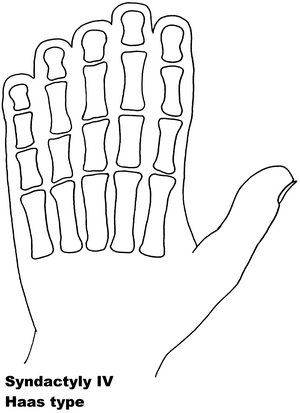
Fig. 13.4
Syndactyly 4. Reprinted from Jordan D, Hindocha S, Dhital M, Saleh M, Khan W. The epidemiology, genetics and future management of syndactyly. Open Orthop J. 2012;6:14–27. Copyright © Jordan et al.; Licensee Bentham Open
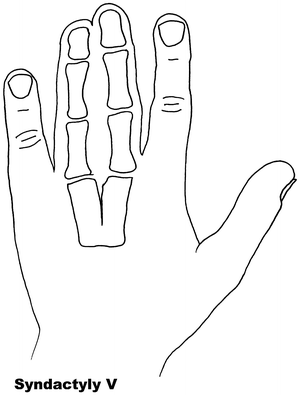
Fig. 13.5
Syndactyly 5. Reprinted from Jordan D, Hindocha S, Dhital M, Saleh M, Khan W. The epidemiology, genetics and future management of syndactyly. Open Orthop J. 2012;6:14–27. Copyright © Jordan et al.; Licensee Bentham Open
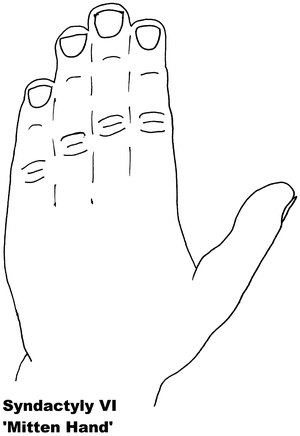
Fig. 13.6
Syndactyly 6. Reprinted from Jordan D, Hindocha S, Dhital M, Saleh M, Khan W. The epidemiology, genetics and future management of syndactyly. Open Orthop J. 2012;6:14–27. Copyright © Jordan et al.; Licensee Bentham Open
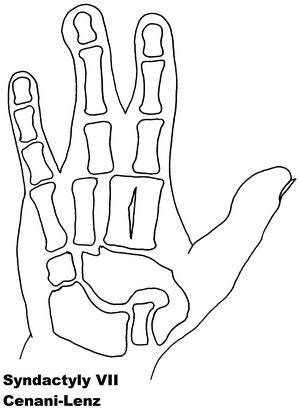
Fig. 13.7
Syndactyly 7. Reprinted from Jordan D, Hindocha S, Dhital M, Saleh M, Khan W. The epidemiology, genetics and future management of syndactyly. Open Orthop J. 2012;6:14–27. Copyright © Jordan et al.; Licensee Bentham Open
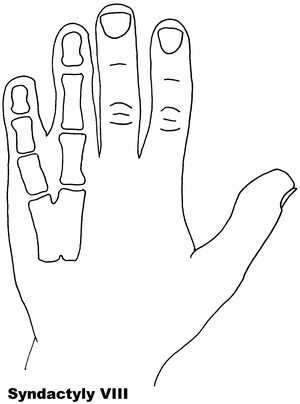
Fig. 13.8
Syndactyly 8. Reprinted from Jordan D, Hindocha S, Dhital M, Saleh M, Khan W. The epidemiology, genetics and future management of syndactyly. Open Orthop J. 2012;6:14–27. Copyright © Jordan et al.; Licensee Bentham Open
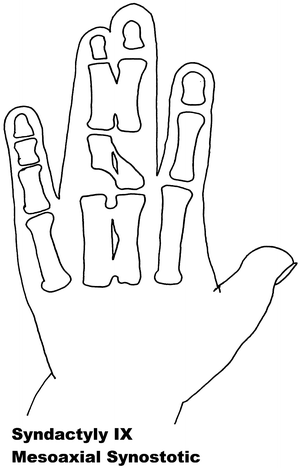
Fig. 13.9
Syndactyly 9. Reprinted from Jordan D, Hindocha S, Dhital M, Saleh M, Khan W. The epidemiology, genetics and future management of syndactyly. Open Orthop J. 2012;6:14–27. Copyright © Jordan et al.; Licensee Bentham Open
Syndromic syndactyly describes syndactyly discovered alongside additional malformations of the body. This list is extensive and continues to expand as syndactyly is discovered alongside other abnormalities, the majority of which appear to develop at a time during the fetal development alongside the digit anomaly formation. In this chapter, we will note some of the more well-known syndromes and review their currently known associated traits and the genes suggested as being causative.
Syndactyly: Non-syndromic Forms [68]
Syndactyly, in this and syndromic form, is seen to have an autosomal dominant transmission with variable expression and penetrance [1–4]. This is best represented with the increased prevalence in male offspring, possibly due to reduced penetrance in females. Occasionally skipping generations, it can present in a reduced form indicating variable phenotype.
The non-syndromic forms of syndactyly which are genetically distinct have been expanded from five to nine since the first discussion of syndactyly phenotypes by Temtamy and McKusick [65] and are summarized individually below.
Syndactyly Type I (SD1)
SD1 is characterized by involvement of the third and fourth finger web space and/or the web between the second and third toes. The most common non-syndromic presentation of syndactyly, it has been described with involvement of other digits and the underlying bones [69]. It is also known under the name zygodactyly.
The phenotype of zygodactyly has been seen to vary. It has been seen to affect the upper or lower limb, both simultaneously and independently. SD1 appears to be inherited only as an autosomal dominant trait. Initial genetic studies localized the 2q34-q36 region of the second chromosome, mapped during studies involving both a large German and a non-related Iranian family [70, 71]. This locus has also been linked to a Philadelphia type of craniosynostosis with associated syndactyly [72, 73].
Mouse studies have shown a chemically induced mutation on the chromosome 6 causes syndactyly of digits 2 and 3 of the hind legs (Sndy Jrt/Sndy +). This varies from simple complete to incomplete phenotype, and although sparing the front limbs appears to correlate well with the characteristics of SD1. The homologous region of this chromosomal mutation in humans would be found on 3p25.1 [74].
Malik et al. [75] postulated that SD1 can be further divided into four subtypes:
Subtype 1: Foot zygodactyly without hand or bony involvement
Subtype 2: Bilateral cutaneous and/or bony hand and foot involvement
Subtype 3: Specific bilateral webbing, cutaneous or bony, of the third and fourth finger
Subtype 4: Bilateral cutaneous webbing of the fourth and fifth toe
They designated the 3p21.31 locus to be specific for this first subtype and named it zygodactyly 1 (ZD1). This appeared to be a new locus for the same phenotype previously described in the German family by Bosse et al. [70].
Syndactyly Type II (SD2)
Synpolydactyly (SPD) is, in terms of both genetic and clinical terms, one of the most heterogeneous malformations of the non-syndromic syndactyly types. It appears to lack penetrance within SPD-affected families, with its typical signs including third and fourth finger syndactyly associated with varying degrees of polydactyly of the fourth finger web space. Polydactyly of the fifth toe is often seen.
SPD has been categorized several times in the literature. There is agreement that SPD is inherited in an autosomal dominant manner. Subtypes have been constructed as new genetic sources have been found.
The Homeobox family of genes were the first group to be acknowledged in relation to Synpolydactyly. Located on the 5′ region of the A- and D-clusters of human chromosomes 7 and 2, respectively, several distinct genes have been recognized [39]. These genes appear to influence limb patterning and of particular interest is the Homeobox D gene (HOXD), and precisely that related to the loci at 2q31 [76].
Following this theory, research into the HOXD13 gene found in one family a relation to polyalanine expansion [77–81]. Specifically, the N terminal region of the protein, involved in binding to DNA, is disturbed. With this there appears to be a correlation between expansion size and the appearance and severity of the SPD phenotype in affected patients, with a greater number of limb involvement seen with increasing expansion size [82]. It has correspondingly been found that minimal duplication does not seem to cause the phenotypical deformity [83]. Since its finding, HOXD13 has been linked with multiple limb deformities including SD5, brachydactyly, and several syndromic forms of syndactyly [84, 85].
The HOXD13 gene link has been supplemented by the discovery that a translocation between Chromosomes 12 and 22 resulting in a defect in the Fibulin gene, normally located on the latter, was found to cause SPD [86, 87]. Debeer and Schoenmakers team published further papers examining this translocation within the FBLN1 gene and localized specific involvement of an area represented by EST R72964, as well as ruling out several previously characterized genes [88].
This finding initially complicated the SPD phenotype, and resulted in the commonly recognized classification SPD 1–3. With this description, SPD 3 correlates to the more classical presentation of SPD and has been linked to the 14q11.2-q12 loci [89].
Likewise, the grouping of phenotype to gene of SPD 2 to the Fibulin 1 gene on Chromosome 12 (MIM 608180) and SPD 1 with Homeobox D13 (MIM 186000) is now widely accepted. SPD2 is generally thought to include synostosis of the metacarpal and metatarsal bones.
A more recent paper [90] has stated that SPD should be sub-classed more specifically relating to phenotype, stating genotype–phenotype correlation is weak when looking only at the HOXD13 mutation. They propose the phenotypic variant being classed as (1) typical SPD features, (2) minor variants, and (3) unusual phenotypes.
A further SPD subtype is described by one paper [91] where a new distinct clinical form involving a complicated and distinctive hypoplastic synpolydactyly was found. This currently does not appear to have been investigated on a genetic basis, and further research into this will help define this new phenotype as a new or mixed entity.
Continuing research has led to other genes being suggested as causative in SPD, although all of these have been found to involve the Shh pathway on one level or another [92].
Syndactyly Type III (SD3)
In syndactyly type III, the typical and first described phenotype involves complete and bilateral syndactyly between the fourth and fifth fingers. This is a soft tissue syndactyly but has been seen with the distal phalanges fused. An absent or rudimentary middle phalanx results in an often seen shorter fifth digit. The feet are generally not affected. Johnston and Kirby [93] presented a family which was one of the largest fully described pedigrees, involving seven affected males and seven affected females over five generations in a pattern compatible with an autosomal dominant inheritance [65].
Other papers to describe SD3 as a single entity, as opposed to as being part of a syndrome include De Smet et al. [94].
Isolated SD3 appears to be in a disease spectrum that includes oculodentodigital dysplasia (ODDD; MIM 164200), which commonly involves digit as well as craniofacial dysmorphia and neurological degeneration [95]. ODDD has complete penetrance but a varying phenotype. Gene research has led to the locus 6q21-q23 being associated with SD3, with significant crossover of the locus 6q22-q24, linked to ODDD, and in particular the Connexin 43 (Cx43) gene and its involvement with the gap junction protein, alpha 1 (GJA1) [96–98].
With six types, it has been found the Connexin family are key in forming gap junctions allowing small molecule and ion passage, with Cx43 being expressed in the developing limb bud and in particular relating to digit and cartilage condensation [99]. Further studies into both the phenotype and genetic regions above have found localized missense mutations causative for ODDD, of which over eight have been described, as well as tested in animal studies [100–104].
Dobrowolski et al. [105] have described ODDD phenotype in specific mutations (131M and G138R) whilst mutations at other points appear to result in no syndactyly (H194P) or solely facial abnormality (G143S). This has led to a belief that increased hemi-channel activity may strengthen ODDD phenotype in Cx43 gap junction deficient patients. Other studies have also confirmed a highly variable phenotype of Cx43 mutations which includes ODDD [106–108].
Syndactyly Type IV (SD4)
Haas [109] was first to describe this phenotype, referred to as Haas type polysyndactyly, with the syndactyly described as complete, affecting the fingers of both hands, with associated polydactyly, generally involving six metacarpals and six digits. Flexion of the fingers results in the hands forming a cup shape. In contradistinction to the type of syndactyly in Apert syndrome, there is no bone fusion. In the reports, there is no mention of SD4 affecting the feet, with descriptions noting there were no associated malformations.
Syndactyly Type V (SD5)
Another rare form of syndactyly, SD5 is as a rule characterized by the presence of an associated metacarpal and metatarsal fusion. The fourth and fifth, or third and fourth, metacarpals and metatarsals are most commonly fused, with soft tissue syndactyly usually affecting the third and fourth fingers and the second and third toes. In this form, the syndactyly tends to be more extensive and complete. In 1932, Kemp and Ravn [116] described this anomaly in five generations of a family from the island of Seeland. Other descriptions without metatarsal fusion have been documented, but these are usually seen with other foot abnormalities [117].
Syndactyly type V has an autosomal dominant trait but has been described as X-linked recessive. Research has linked SD5 to the locus at 2q31-q32 as well as mutations in the HOXD13 gene, including the pathogenicity of a c.950A→G (p.Q317R) mutation [84]. In this paper, the authors called for a genotype classification of HOXD13 limb morphologies, again confusing the genotype-phenotype boundaries of the syndactylies.
Interestingly, as in SPD, evidence of HOXD13 polyalanine expansion has been found in the Seeland family [118].
Syndactyly Type VI (SD6)
Also known as mitten hand syndactyly, this form consists of unilateral syndactyly of digits 2–5 [65]. One family has been described with this anomaly, where an autosomal dominant inheritance, but with variable expression and incomplete penetrance, is likely. Tentamy and McKusick included this phenotype in their initial classification but, even since their description, due to its rarity it remains the least researched non-syndromic syndactyly.
Syndactyly Type VII (SD7)
In 1967 two brothers with an Apert syndrome-like form of syndactyly were described by Cenani and Lenz [119]. They noted however, that additional features including severe shortening of the ulna and radius with fusion, as well as fusion of the metacarpals and “disorganization” of phalangeal development were present. The feet of both brothers were less severely affected. They identified similar cases reported by Liebenam [120], Borsky [121], and Yelton [122].
Cenani–Lenz syndrome, named after the pair’s description, is a very rare phenotype and has been reported to show an autosomal recessive inheritance. There have been accounts of varying phenotypes, including a description of a patient with features consistent with Cenani–Lenz type but also displaying a severe form of SPD1 [69].
The LRP4 gene has been linked to syndactyly in cattle [123, 124], and it is reported with multiple mutations on Chromosome 11p12-p11.2 to be the causative factor in SD7 [125]. In the study group, two families did not exhibit LRP4 mutations, suggesting further gene involvement. Bachelli et al. found that this is unlikely to be related to the pathways involving Formin or Gremlin expression [126]. A more recent paper suggests a mutation involving the loci of these bmp antagonists can result in a phenotype similar to Cenani–Lenz syndrome [127].
Within the Cenani–Lenz syndactyly group, there appears to be two grossly variant phenotypes: one involving a spoon hand type, and the other an oligodactyly type [128].
Syndactyly Type VIII (SD8)
Fusion of the fourth and fifth metacarpals is an uncommon presentation of syndactyly. First described by Orel in 1928 [129], it was thought to have an X-linked recessive trait, which has been supported by later papers [130, 131].
An autosomal dominant inheritance has been suggested by Lerch [132] after he found a family with male–male transmission as well as female member being affected.
Xq26 has been suggested as a starting point for analysis, a known mapped area for split-hand/foot malformation (SHFM2), with the gene allocated as MF4 (MIM 309630), although there is general consensus that this syndactyly needs further research before its relationship is fully understood [133].
Syndactyly Type IX (SD9)
Type IX, mesoaxial synostotic syndactyly (MSSD) has been described only in two families. The characteristic features consist of complete syndactyly and synostosis of the third and fourth fingers with severe bone reduction in the proximal phalanges, hypoplasia of the thumbs and halluces, aplasia/hypoplasia of the middle phalanges of the second and fifth fingers, and complete or partial soft tissue syndactyly of the toes. Percin initially believed, with family members known to have SD1 trait, this to be a severe form of SD1 having a possible homozygous origin [134].
Malik et al. [135] found similar findings in another family, with an autosomal recessive trait, and ruled out genome candidates at 2q34-q36, 2q31, and 6q22-q23. The previous family had also had HOXD13 and the genome associated with 2q31 disproved as causative by Percin et al. Merging the two families into one study has revealed a likelihood of a causal gene being mapped to chromosome 17p13.3 [67].
Syndactyly: The Syndromic Forms
Syndactyly often presents as part of a syndrome, usually with other congenital abnormalities. Some of the more common syndromes are reviewed as follows.
Acrosyndactyly describes syndactyly associated with congenital constriction bands. It appears to lack a genetic basis, with Tentamy and McKusick [65] being first to find little or no evidence of a clear or simple genetic link. The formation of syndactyly in this syndrome is thought to be as a result of inflammatory changes resulting in scar formation fusing the digits [136–138]. This is reinforced by the appearance of dorsal to palmar epithelium lined sinuses lying proximal to the scar fusion site in these patients.
The ischemic insult after initial digit formation causes digit deformity although Patterson [137] has also noted the high incidence of deformity in other anatomical regions and raises the possibility of a molecular tissue defect. However, it is noted that any deformity is not usually seen to be symmetrical in the opposite limb pointing away from a genetic source.
Dependant on the degree of bone involvement, acrosyndactyly can be described as mild, moderate, or severe [139–141]. Mild deformity involves normal metacarpal structure with three well-formed digits, meaning three phalanges and two joints, whereas there is loss of a phalangeal bone resulting in one joint in the moderate form. The severe form relates to little or no digit presence with only small phalanges present, and occasionally metacarpal involvement. The variance in acrosyndactyly, as opposed to the other forms of syndactyly tends to involve no extra-skeletal parts and the fusion involving a scar lying either side-to-side or an on-top position.
Poland’s syndrome (MIM 173800) presents with unilateral hypoplasia or absence of pectoralis muscle with ipsilateral hand and digit anomalies. The syndrome is named after Alfred Poland, who reported on George Elt’s absent pectoralis major [142]. Patrick Clarkson later described the syndrome, including its hand anomalies [143]. As of yet no gene or loci has been implicated in its origin. It is believed that there may be a causative source in a disruption sequence related to the brachiocephalic arterial system [144–146].
The radial fingers are more typically involved in Poland’s syndrome and hypoplasia of the digits is frequent. Breast hypoplasia, in varying degrees, is often a common presentation as well as involvement of the latissimus dorsi, deltoid and/or serratus anterior muscles [147]. Poland syndrome has also been reported with evidence of dextrocardia and sternal deformity. Karnak et al. described a bilateral Poland syndrome [148]; however, most presently agree that Poland syndrome is solely a unilateral disease.
Acrocephalosyndactyly (ACS), a condition involving syndactyly and craniosynostosis, where there is a premature fusion of one or more of the fibrous suture lines of the skull. Five types have been described, each having variances on the hand and skull deformity. There is confusion where the distinction between the ACS group and the syndromes involving craniosynostosis, syndactyly, and polydactyly (ACPS), which incorporates a different four syndromes into a further five types, ends. The main ACS/ACPS syndromes are commented on below.
Apert syndrome (MIM 101200) ACS type I is synonymous with the term acrocephalosyndactyly. Associated with the FGFR2 gene, and the loci 10q26, includes midface hypoplasia, foot and hand syndactyly with a trend for distal bony fusion [149].
A subgroup of the Crouzon syndrome linked to FGFR2 is termed ACS type 2. Although Crouzon syndrome usually involves only a craniofacial dysostosis, Crouzon type 2 also involves mild soft tissue syndactyly.
Saethre–Chotzen syndrome, ACS type III (MIM 101400), involves syndactyly of the second and third fingers, as well as the third and fourth toes, as well as eyelid anomalies and cranial abnormalities. It has been linked to the loci 7p21.2 and 10q26 involving the TWIST 1 and FGFR2 genes, respectively [150, 151].
ACS type V, also known as Pfeiffer syndrome (MIM 101600) has been linked to the FGFR 1 and 2 genes [152, 153]. This is likewise classified as ACPS 5, and has since had Noack syndrome, previously ACPS 1, grouped with it.
ACPS 2, Carpenter syndrome (MIM 201000), has been linked to RAB23 gene originating from 6p11, with malformations including foot and hand syndactyly/brachydactyly and acrocephaly [154]. ACPS 4 was known as Goodman syndrome (MIM 201020) but is thought now to be a variant of type II [155].
Other syndromes and chromosomal location include Acropectorevertebral dysplasia (MIM 102510) and 2q36 and Fraser syndrome (MIM 219000) associated with both the sites 4q21 and 13q13, involving the FRAS1 and FREM2 basement membrane genes, respectively [156, 157], which have also been shown to be linked to fin deformity in zebrafish [158].
Greig cephalopolysyndactyly (MIM 175700) is an autosomal dominant disorder associated with haplo-insufficiency of GLI3. This appears to be caused by deletions, truncations, or point mutations of the associated Gli3 gene. Similarly the zinc finger domain of Gli3 has been found to be causative in Pallister Hall syndrome whose phenotype includes central nervous system and craniofacial deformities, as well as anal defects [52].
Research into the individual phenotypes appears to complicating phenotypical classification as new genes are found both linked, and not linked, to each malformation.
Environmental Influence on Limb Formation
It should be noted that sporadic distinct syndactyly with no familial history has been documented. In utero environmental factors that predispose the fetus to syndactyly and other congenital hand abnormalities have been evaluated. Man conducted a study that reports a probable association with these conditions and maternal smoking [161], and there are suggestions that syndactyly occurrence is associated with lower nutritional and economic status, including increased meat and egg intake whilst pregnant, although more research is required before suggesting that these are causative factors [162].
Anatomy and Management
The normal position of the web commissure lies at the midpoint of the proximal phalanx if looked at from a lateral view. From a distal view the space appears to be shaped like an hourglass with a larger area within the second and fourth web space when compared to the third. The web space appears to normally slope, towards this distal view, from the dorsal aspect of the hand at an angle of 45° (Figs. 13.10 and 13.11).
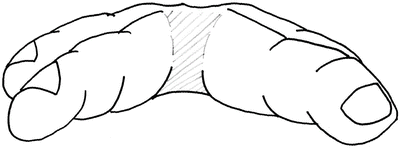
Fig. 13.10
Showing lateral view of interdigital web space. Note 45° dorsal to palmar fall finishing at midpoint of the proximal phalanx
Fig. 13.11
Hourglass shape of interdigital space
The mainstay of treatment for syndactyly remains surgical. Indications for operative intervention run along the same principles as that of all hand anomalies:
Function—to allow hand function and the development of normal grip
Cosmesis—to improve the aesthetic appearance of the hand to minimize the psychological and social effects of the deformity
The timing of the surgical intervention needs to be optimized in order to reduce long-term complications and improve outcome. Many centers begin corrective surgery by 12–18 months of age and aim to complete reconstruction by the time the child reaches school, helping social and functional tasks at this time. It is thought that there is less risk of scar contracture in comparison to younger age groups. However, it is imperative that patients be assessed on an individual basis and reconstruction tailored as to the complexity of the syndactyly. Some forms of syndactyly are operated on at 6 months of age [163] or earlier [164].
Involvement of border digits, complex syndactyly, and flexion contractures are all indications for early repair. The aim of early intervention is to reduce the loss of function associated with the deformity and provide normal grip development.
On the contrary, Kettlekamp and Flatt [165] found that surgery performed at less than 18 months of age was associated with a higher complication rate and poorer aesthetic outcome, particularly in relation to the web commissure. Timing of surgery, therefore, is often down to individual surgeon preference.
Multiple-digit syndactyly should be corrected as part of a multistage procedure. Release should be performed on only one side of a digit at a time so as not to risk necrosis, particularly in those supplied by only one artery [166]. As a rule, border digits should be released first followed by a second procedure performed at least 4 months later [167].
Surgical Technique
Surgical correction of syndactyly requires the separation of digits and the creation of a new web space. The main concern with syndactyly is the greater surface area encountered on separation of the digits, with a circumference of approximately 1.4 times the preoperative state. Technique for repair therefore, must provide a means for adequate resurfacing.
Over the last two centuries, techniques for syndactyly repair have evolved significantly [163]. Many successful methods are described in the literature. Most employ a variant of the procedure described below (Figs. 13.12, 13.13, 13.14, 13.15, 13.16, 13.17, 13.18, 13.19, and 13.20):
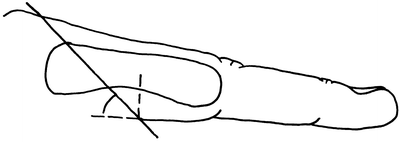
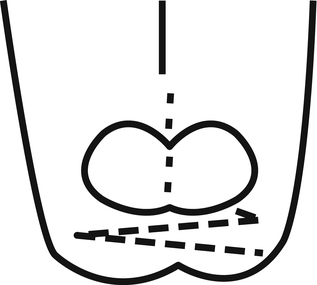
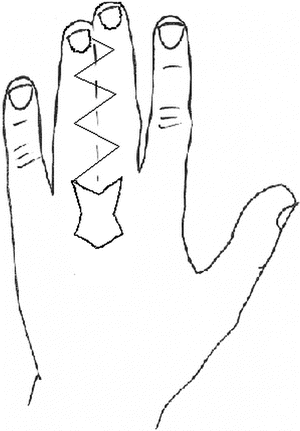
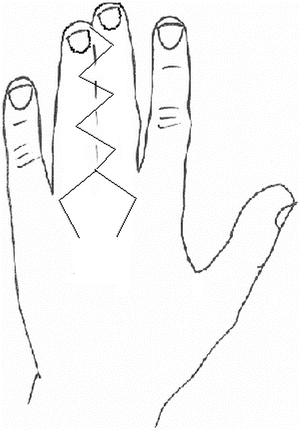
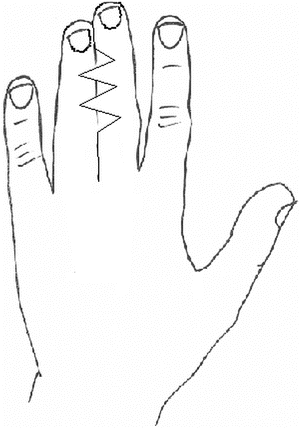
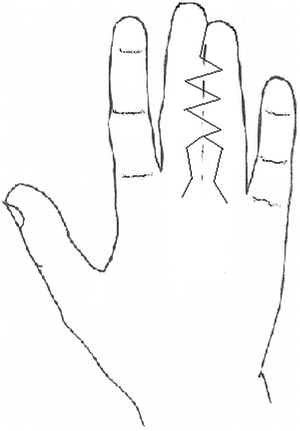
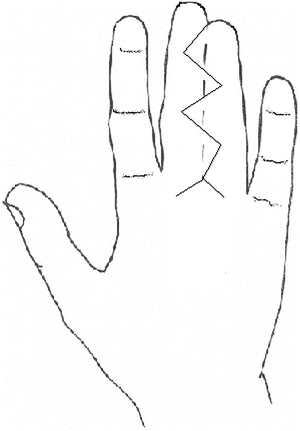
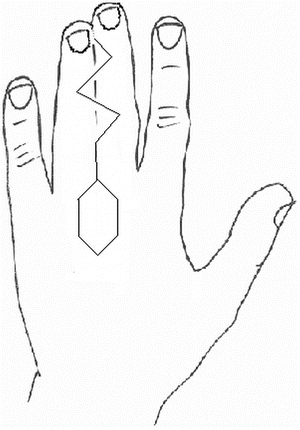
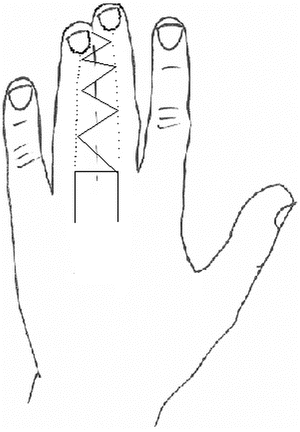
Fig. 13.12
Showing lateral view of interdigital web space. Note 45° dorsal to palmar fall finishing at midpoint of the proximal phalanx
Fig. 13.13
Buck Gramcko nail fold creation
Fig. 13.14
Dorsal Island flap
Fig. 13.15
Dorsal pentagonal flap
Fig. 13.16
Dorsal view of Jose et al. flap
Fig. 13.17
Palmar view of palmar-shaped flap
Fig. 13.18
Volar zigzag approach to release
Fig. 13.19
Diagram of V-Y advancement flap for release
Fig. 13.20
Zigzag dorsal flap
1.
A zigzag incision for the separation of digits
2.
A dorsal flap for the creation of a web commissure
3.
A skin graft to resurface raw areas
Skin grafts are associated with various complications: graft loss, hair growth from donor sites, scar contracture, web creep as well as general surgical risks including donor site infection. In general, full thickness grafts are used for resurfacing. Split thickness grafts have been shown to have higher complications from scar contracture [168] and have therefore fallen out of favor. Full thickness grafts may be taken from the dorsum, hypothenar region, antecubital fossa, and the groin. Although widespread use of the groin as a donor site, it has been recommended that more medial areas are avoided so as to avoid excessive hair growth on the hand [169].
Another consideration with the use of skin grafts is the problems associated with graft management in patients of a young age, mainly due to difficulties with immobilization. Recently, Kamath et al. [170] describe the use of a mini external fixator to facilitate the maintenance of the neo-web space by allowing accurate positioning of the graft and make dressing changes easier and pain free.
Related posts:
 Anesthesia Concerns in Congenital Anomalies of the Upper Extremity
General Skeletal Disorders
Anesthesia Concerns in Congenital Anomalies of the Upper Extremity
General Skeletal Disorders
 Therapy Management of Children with Congenital Anomalies of the Upper Extremity
Therapy Management of Children with Congenital Anomalies of the Upper Extremity
 Physical Medicine and Rehabilitation Management of Children with Congenital Anomalies of the Upper Extremity
Physical Medicine and Rehabilitation Management of Children with Congenital Anomalies of the Upper Extremity
 Ulnar Polydactyly and Ulnar Dimelia
Ulnar Polydactyly and Ulnar Dimelia
 Macrodactyly
Macrodactyly
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


