Keratosis Pilaris and Other Inflammatory Follicular Keratotic Syndromes: Introduction
|
Follicular keratosis refers to orthokeratosis involving the follicular ostium and infundibulum. Horny plugs protrude from the orifices, producing a rough sensation on palpation of the skin. It may be isolated [keratosis pilaris (KP)] or associated with other pathologic processes, including follicular inflammation, atrophy, scarring, and alopecia [keratosis pilaris atrophicans (KPA)]. These are reaction patterns that occur alone or as part of a wide variety of syndromes.
Keratosis Pilaris
Typical KP is a common condition of keratotic follicular plugging with varying degrees of surrounding erythema. Sometimes, the erythema is so striking that it is the main complaint. The clinical expression of KP varies from subtle to conspicuous, which, along with possible racial differences, may explain the great range of reported prevalences, from 1% to 42%.1–4 It involves most commonly the extensor aspects of the upper arms (Fig. 87-1) and thighs as well as the face but may rarely be more extensive, extending to the distal limbs and the trunk. There seem to be two patterns. In early childhood, the face and arms are mainly affected and gradual improvement is seen in most cases by later childhood or adolescence. In the other pattern, the onset is in teenage years, and the extensor arms and legs are predominantly involved. It usually improves by the mid-20s. However, in both patterns, the condition may be persistent into later adult life.5 The histopathologic pattern in skin biopsy specimens is nonspecific, simply showing the follicular orifice distended by a keratin plug.

Treatment is usually with various keratolytics, from simple urea, lactic acid, or salicylic acid preparations to topical retinoids and tazarotene.6,7 These preparations may aggravate associated erythema, limiting their value.
Clinical ichthyosis vulgaris (see Chapter 49) is associated with KP8; in one series KP was found in 74.3% of patients (Fig. 87-1).2 Other conditions in which KP is more prevalent or more prominent are atopic disorders, hypothyroidism, Cushing syndrome, insulin dependent diabetes, obesity or high body mass index, and Down syndrome.9 Follicular keratosis, which may simulate KP, can occur in several nutritional deficiencies, although vitamin A deficiency is most commonly cited (see Chapter 130).10
Keratosis Pilaris Variants
Erythromelanosis follicularis faciei et colli (EEFC) is a condition probably related to KP.11 The suffix “colli” refers to the neck. EEFC It is seen primarily in adolescents and young adults, most commonly in males. Well-demarcated erythema, hyperpigmentation, and follicular papules involve the preauricular and maxillary areas, usually in a symmetric distribution, with spread in some cases to the temples and sides of the neck and trunk. Atrophy is not a feature. Histopathology is nonspecific, demonstrating a variable degree of follicular hyperkeratosis, dilation of upper dermal vessels, some perivascular inflammatory infiltrate, and hyperpigmentation of the basal layer. There are few reports of EEFC in the literature, however it may be underreported.
Although erythema is often present in typical KP, it is usually mild and limited to the perifollicular skin. When perifollicular erythema is more noticeable, the disorder has been called keratosis pilaris rubra (KPR) or less commonly keratosis follicularis rubra.12 These findings are usually limited to the cheeks, forehead, and neck (Fig. 87-2). Features that differentiate EFFC from KPR are a lack of reported involvement on the torso and the presence of hyperpigmentation. However the hyperpigmentation noted in EFFC may, at least in part, be related to skin pigmentation type, with darker skin types showing more evidence of hyperpigmentation. Thus it is likely that KPR and EFFC are variants of the same clinical spectrum.

Keratosis Pilaris Atrophicans
|
The general term keratosis pilaris atrophicans is a group of rare genodermatoses in which the clinical hallmarks is follicular keratosis with variable degrees of inflammation, and secondary atrophic scarring and/or alopecia.13–15 Three distinct clinical entities that fall under the umbrella of keratosis pilaris atrophicans include ulerythema ophryogenes [UO or keratosis pilaris atrophicans faciei (KPAF)], atrophoderma vermiculatum, and keratosis follicularis spinulosa decalvans (KFSD).
The prefix ‘ophryo-’ refers to the eyebrow. UO is a form of KPA affecting particularly and initially the eyebrow areas, in some cases extending later to the cheeks and forehead.13 Scalp and eyelash hair is normal. The onset is within months of birth, with erythema and small keratotic follicular papules involving the lateral one-third of the eyebrows. It may slowly progress through childhood to involve more of the eyebrows and sometimes beyond, leading to alopecia (Fig. 87-3). Progression usually ceases after puberty but the sequelae are permanent. The condition is frequently associated with standard KP that may involve the arms and legs or may be more generalized. The term keratosis pilaris rubra atrophicans faciei is used interchangeably with UO, however some prefer this diagnosis for patients in whom the initial erythema starts on the cheeks as opposed to the eyebrows.
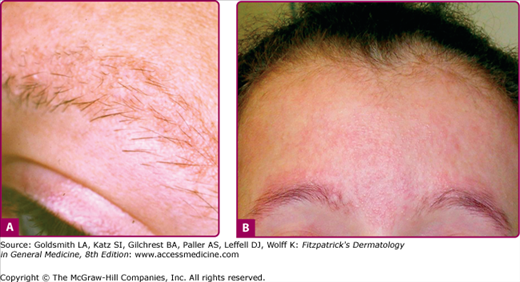
Most cases have followed an autosomal dominant inheritance pattern, with incomplete penetrance.13 Response to topical corticosteroids, topical retinoids, and keratolytics has been poor. There is one report of a good response to several months’ therapy with oral isotretinoin, with lessening of the horny plugs and the erythema.16 The improvement was maintained for some months after therapy had ceased. Some improvement in erythema was reported with the pulsed tunable dye laser at 585 nm, but the follicular plugging was unchanged.17
UO has been reported in association with isolated woolly hair,18,19 Noonan syndrome,19,20 cardiofaciocutaneous (CFC) syndrome,21,22 Rubenstein–Taybi syndrome,23 Cornelia de Lange syndrome,24 and 18p deletion.25,26 CFC and Noonan syndrome have overlapping clinical features, and both have mutations in genes of the Ras pathways.27,28 In CFC, mutations have been described in BRAF, KRAS, MEK1, and MEK2 genes.27 Whereas in Noonan syndrome, there is primarily mutations of PTPN11, but also KRAS and SOS1 genes.29,30
Follicular keratosis in Noonan syndrome, if present, is usually limited to the face (Fig. 87-3B) and resembles UO with loss of eyebrow hairs.19,20 In CFC syndrome, follicular keratosis is often much more extensive, and there may be striking alopecia involving the scalp.22 The remaining scalp hair in CFC syndrome is brittle and curly.
AV is a form of KPA in which the cheeks are predominantly involved, with follicular plugs and pit-like depressions up to 1.5 mm in diameter and a background of erythema. Alopecia and follicular papules are not features of the condition,22,31 but sparse open and closed comedones and milia may be found.32 The depressions merge into each other, producing a “worm-eaten,” “honeycombed,” or reticular appearance (Fig. 87-4). Involvement of the forehead, arm, and leg, as well as the typical cheek area, has been described.14 However, in general, it is rare for the condition to involve the limbs, and eyebrow involvement does not occur. Standard KP of the extensor aspects of arms and legs is a common associated finding.31 Unlike the other KPA disorders in which the onset is in infancy, AV usually starts in childhood, between 5 and 12 years old, although later onset has been described14; the course is usually one of inexorable worsening. The condition is often sporadic but, in some cases, appears to be inherited as an autosomal dominant trait.33 Several more or less linear unilateral cases have been described,34 suggesting a possible mosaic form of the condition.35 However, it is possible that at least some of these cases may represent examples of nevus comedonicus.35

Histopathologic evaluation demonstrates atrophic pilosebaceous units with mild perifollicular and perivascular inflammation. The follicular orifices are dilated and filled with keratin plugs. Numerous dermal horn cysts may be found. There is perifollicular fibrosis and a decrease in elastic fibers in the dermis.31,36
Therapy is uniformly unsuccessful and has included topical steroids, topical retinoids, cryotherapy, and derm-abrasion. Favorable results have also been described with carbon dioxide laser in a case in which atrophy was prominent, and pulsed dye laser in a case with a marked erythematous component.32 Oral isotretinoin suppressed the inflammation in one patient.37
Follicular atrophoderma occurs as a feature of several rare syndromes38–43 and in nevus comedonicus (Table 87-1).44 As such, these conditions may come into the differential diagnosis of atrophoderma vermiculatum.
Condition | Inheritance | Main Features | Comments |
|---|---|---|---|
Bazex syndrome (Bazex–Dupre—Christol syndrome)41 | X-linked dominant | Hypotrichosis, pili torti, hypohidrosis, milia, development of basal cell carcinomas in adolescence. | — |
Rombo syndrome38 | Autosomal dominant | Hypotrichosis, especially of eyelashes, peripheral cyanosis, basal cell carcinomas, and trichoepitheliomas. | — |
X-linked dominant chondrodysplasia punctata (Conradi–Hünermann syndrome)39 | X-linked dominant, lethal in males | Congenital Blaschko-distributed red scaly lesions that resolve in months, replaced by follicular atrophoderma. Cataracts, shortening of limbs, frontal bossing, saddle nose, low-set ears. | Mutation in emopamil binding protein gene, relevant in cholesterol biosynthesis |
Congenital ichthyosis, follicular atrophoderma, hypotrichosis, and hypohidrosis42,43 | Probably autosomal recessive | Congenital, diffuse ichthyosis, hypohidrosis, hypotrichosis. Woolly hair in one pedigree. | Only described in two families so far |
Hereditary perioral pigmented follicular atrophoderma associated with milia and epidermoid cysts40 | Autosomal dominant | Epidermoid cysts and milia. | Follicular atrophoderma limited to face, especially periorally |
Nevus comedonicus44 | Somatic mutation | Blaschko-distributed comedo-like plugs in dilated follicular orifices. Cribriform atrophy follows extrusion of comedones. | Due to somatic mutation of fibroblast growth factor receptor 2, identical to that, which, if present in the germline, causes Apert syndrome |
The term KFSD was first used by Siemens in 1926 who described a scarring follicular condition in 20 members of a large family.45 The condition begins in infancy with noninflammatory, flesh-colored, spiny lesions affecting hair-bearing areas, especially the scalp (Fig. 87-5) and later eyebrows, eyelashes, and the dorsal of the hands and fingers. Sometimes, more proximal limbs and even the trunk become involved.45–47 An associated plantar keratoderma may occur, especially over the heels.15 By puberty, many of the follicular spines have disappeared and are replaced by atrophy. Scarring alopecia of scalp (see Fig. 87-5), eyebrows, and eyelashes becomes apparent in childhood and progresses till puberty. It is often patchy and is rarely total. Facial lanugo hair is absent, and axillary and pubic hair is often thinned.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


