History
Patient presents with an acquired skull deformity, as a result of being placed in a fixed supine position for sleep.
Exam
Unilateral features noted on physical examination include occipital flattening; anterior displacement of the ipsilateral ear, forehead, and zygoma; and widening of the ipsilateral palpebral fissure, causing a parallelogram-shaped cranium.
Treatment
If diagnosed early, treatment that involves repeatedly repositioning the child out of the flat spot will be sufficient. Cranial-molding helmets are used for more severe cases or for those who are diagnosed late.
1.1.2 Diagnosis: Anterior Plagiocephaly (Unilateral Coronal Synostosis (UCS))
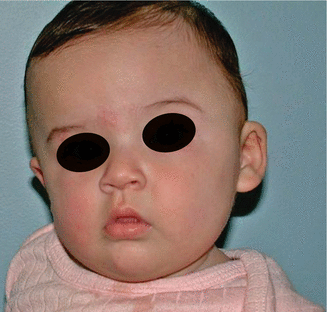
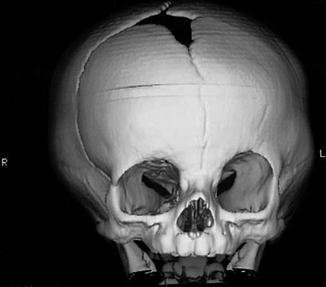
History
Patients present with progressive facial deformities that are not seen with other nonsyndromic craniosynostosis.
Exam
Anterior plagiocephaly is the result of unilateral coronal synostosis, characterized by superior and posterior displacement of the supraorbital rim and eyebrow on the ipsilateral side, widening of the ipsilateral palpebral fissure, frontal bossing of the contralateral side, deviation of the nasal root toward the affected side, occipital protrusion of the ipsilateral side, and flattening of the contralateral occiput.
Treatment
Surgical intervention entails release of the synostosed suture with fronto-orbital advancement, usually between 3 and 6 months of age.
1.1.3 Diagnosis: Scaphocephaly (Sagittal Synostosis)
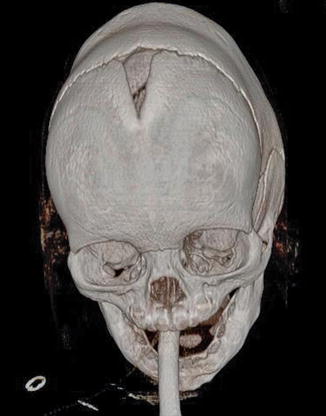
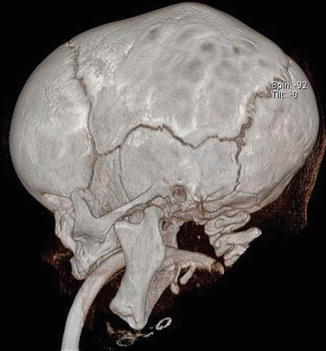
History
Patients present with boat-shaped skull deformity, hence the term scaphocephaly. It is the most frequent nonsyndromic synostoses with a male predominance of 4:1.
Exam
The fusion of the sagittal suture impairs expansion of the skull width. The cranium is therefore long, narrow, and keel-shaped. This is accompanied by frontal and occipital bossing.
Treatment
Cranial vault remodeling with barrel-staving technique and subtotal cranial vault remodeling are the main surgical options. Endoscopic correction has been described in patients between 2 and 4 months of age.
1.1.4 Diagnosis: Trigonocephaly (Metopic Synostosis)
History
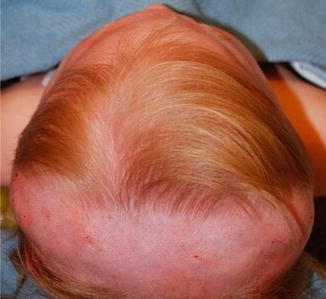
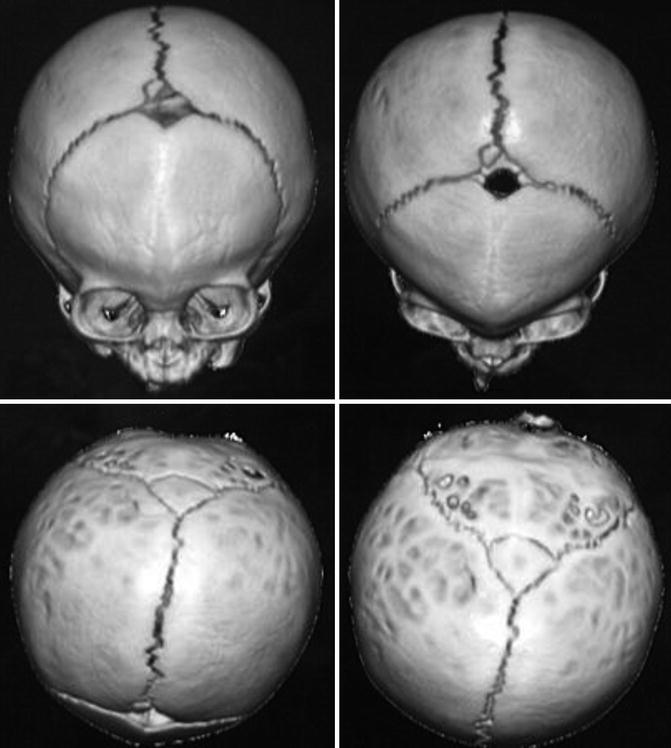
Patients present with triangular-shaped skull deformity when viewed from above, hence the term trigonocephaly. Metopic synostosis represents the third most common nonsyndromic synostosis with male predominance.
Exam
Physical examination shows a midfrontal keel with bitemporal narrowing and orbital hypotelorism, epicanthal folds, and low nasal dorsum.
Treatment
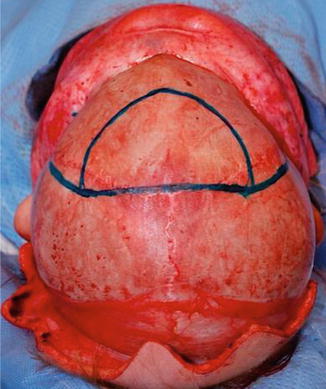
The risk for congenital and behavioral impairment is greater in metopic synostosis than in other nonsyndromic synostosis. Surgical treatment includes bifrontal craniotomy, frontal reshaping and radial osteotomies, and fronto-orbital advancement.
1.1.5 Diagnosis: Kleeblattschadel Skull Deformity
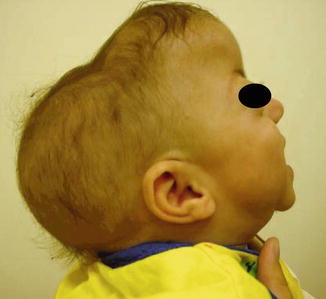
History
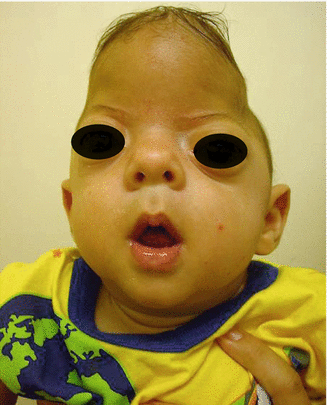
Patient presents with a cloverleaf-shaped skull deformity (Kleeblattschadel).
Exam
Kleeblattschadel skull deformity is a result of multiple-suture craniosynostoses characterized by frontal bossing, bitemporal bulging, orbital exposure, and downward displacement of the ears. Increase in intracranial pressure results in visual compromise, venous hypertension, and hindbrain herniation.
Treatment
Staged calvarial decompression and remodeling are the treatment of choice.
1.1.6 Diagnosis: Turricephaly (or Oxycephaly)
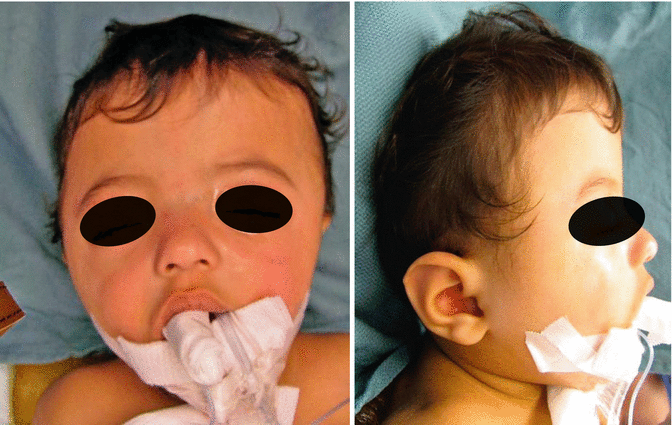
History
Patients present with a tall, tower-shaped skull.
Exam
The vertically tall head shape, accompanied by short anterior-posterior dimension, is a result of bicoronal syndromic craniosynostoses such as Apert, Crouzon, or Pfeiffer syndromes. Other clinical findings include elevated intracranial pressure, hydrocephalus, Chiari malformations, and ocular exposure.
Treatment
Treatment is typically addressed before 12 months of age with resection of the suture, fronto-orbital advancement, and cranial vault remodeling, though other techniques have been described. Staged procedures may be required.
1.1.7 Diagnosis: Apert Syndrome
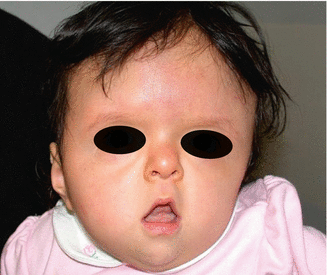
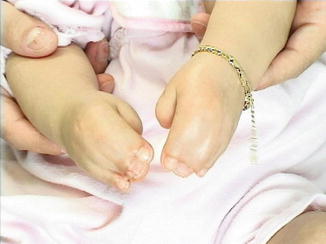
History
It is an autosomal dominant syndrome with incidence of 1:100,000 caused by mutations in fibroblast growth factor gene 2 (FGFR2).
Exam
Physical findings show significant turribrachycephaly, proptosis, hypertelorism, and down slanting of palpebral fissures. The midface hypoplasia is accompanied by depressed nasal dorsum and septal deviation and acne. Patients also present with complex syndactyly of the hands and feet. Mental impairment is common with a high likelihood of increased ICP and cerebral palsy.
Treatment
Treatment is typically addressed before 12 months of age with resection of the suture, fronto-orbital advancement, and cranial vault remodeling. Airway and visual compromise may prompt for emergent treatment. Le Fort III advancement of the midface is often performed at 6–8 years of age.
The craniofacial dysostosis syndromes have several common features: brachycephaly (premature fusion of the coronal sutures), variable severity midface hypoplasia, variable involvement of other cranial sutures, and identification of the responsible genetic mutations. Some have characteristic extremity deformities.
1.1.8 Diagnosis: Crouzon Syndrome
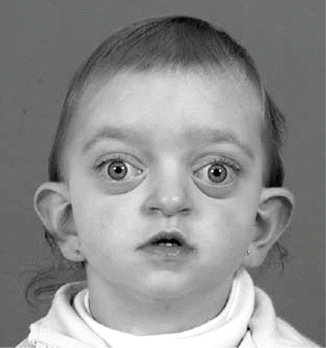
History
It is an autosomal dominant syndrome with an incidence of 1:25,000, as a result of multiple mutations in the fibroblast growth factor receptor 2 (FGFR-2) gene.
Exam
Characterized by a brachycephaly due to premature fusion of both coronal sutures, Crouzon syndrome is a frequent form of craniofacial dysostosis. Other cranial sutures may also be involved. There is also midface hypoplasia, exorbitism, and proptosis. The extremities are normal. These patients may also exhibit mild mental status impairment, hydrocephalus, and elevated intracranial pressure in almost half of the cases.
Treatment
Surgical treatment is typically addressed before 12 months of age with resection of the suture, fronto-orbital advancement, and cranial vault remodeling. Airway and visual compromise may dictate emergent treatment. Le Fort III advancement of the midface is often performed at 6–8 years of age.
1.1.9 Diagnosis: Pfeiffer Syndrome
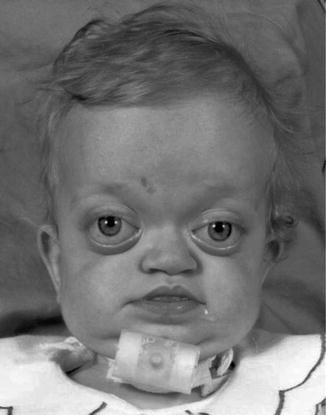
History
An autosomal dominant craniosynostosis syndrome. Patients present with normal mental status in the majority of the cases.
Exam
Clinical findings include turribrachycephaly with coronal or sagittal synostosis. Orbital and midface features are similar to Apert syndrome with an additional distinct findings of broad thumbs and halluces, mild simple syndactyly, and tracheal cartilage anomalies.
Treatment
Cranial vault remodeling is usually performed before 18 months of age. Le Fort III advancement is indicated for those with worsening nasopharyngeal airway obstruction.
1.1.10 Diagnosis: Carpenter Syndrome
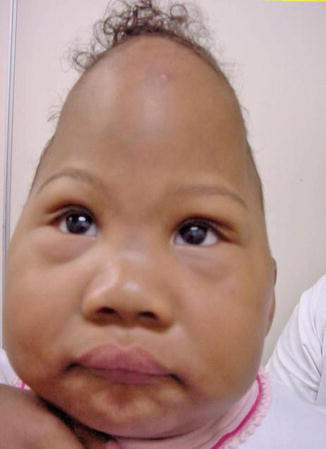
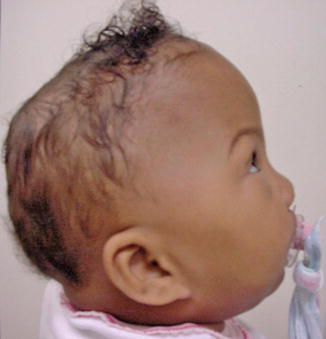

History
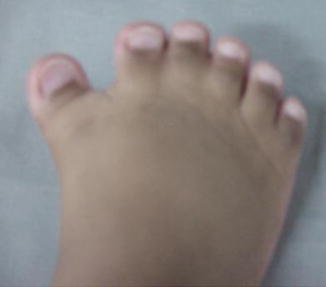
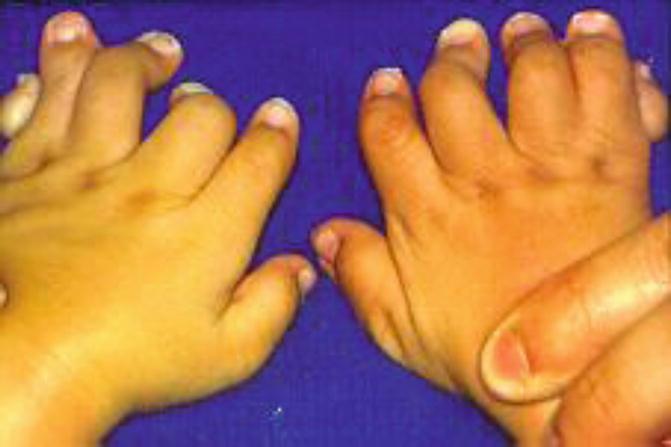
Carpenter syndrome, also known as acrocephalopolysyndactyly type II, is a rare congenital disorder characterized by craniosynostosis and musculoskeletal abnormalities. Unlike other acrocephalosyndactylies, it is autosomal recessive and has recently been linked to mutations in RAB23 and the hedgehog signaling pathway.
Exam
Craniosynostosis patterns in Carpenter syndrome are variable but often involve lambdoid, sagittal, and metopic sutures, the latter resulting in hypotelorism. There is underdevelopment of the anterior cranial fossa and expansion of the middle cranial fossa giving the classic “diamond-shaped” face originally described by Dr. Carpenter in 1909. Common limb anomalies include syndactyly and polydactyly of the hands and feet but can also include clinodactyly and brachydactyly.
Treatment
Treatment of Carpenter syndrome is similar to other craniosynostotic syndromes and includes cranial vault remodeling usually between 3 and 9 months of age. Limb anomalies should also be treated as necessary as the child develops.
-
Perlyn, C.A., Marsh, J.L.. Craniofacial Dysmorphology of Carpenter Syndrome: Lessons from Three Affected Siblings. Plast Reconsr Surg. March 2008; 121: 971.
-
Cottrill C., et al. Carpenter Syndrome. The Journal of Craniofacial Surgery. January 2009 20(1): 254–256
-
Carpenter, G. Case of acrocephaly with other congenital malformations. Proc R Soc Med Sect Study Dis Child. 1909; 2:45–53
1.1.11 Diagnosis: Stickler Syndrome
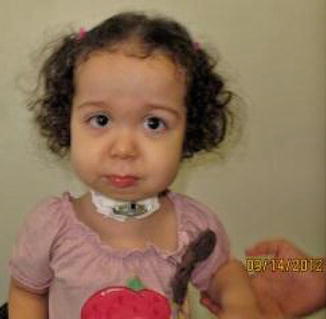
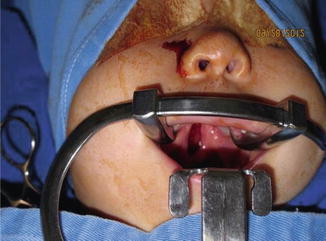
History
Progressive autosomal dominant connective tissue disorder (collagen) with variable penetrance. It is the most common genetic syndrome associated with Pierre Robin sequence ~1–3/10,000 births.
Exam
Can exhibit hypotonia, micrognathia, flat facies with mild to moderate midface hypoplasia, shallow orbits with proptosis, cleft of secondary palate, flat nasal bridge with epicanthal folds, hypermobility of joints with enlarged wrist/knee/ankle joints, scoliosis, mitral valve prolapse, hearing loss (conductive/sensorineural/mixed), retinal detachment, glaucoma, cataracts, and strabismus.
Treatment
Airway maintenance for micrognathia, vigilant ophthalmic and otologic examinations for treatment of above, cleft palate repair, maxillofacial surgery, and orthodontics as needed.
1.1.12 Diagnosis: Van der Woude Syndrome
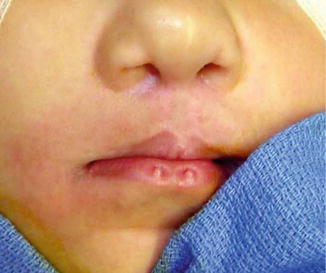
History
Autosomal dominant with variable penetrance that has been linked to a mutation of interferon regulatory factor 6 (IRF6) gene on chromosome 1.
Exam
Usually symmetrical bilateral paramedian sinuses or depressions of the vermilion lower lip (lip pits), possible tongue tie (short lingual frenulum), cleft lip, palate, or submucous cleft palate.
Treatment
Direct excision of sinus pits and treatment of associated cleft.
1.1.13 Diagnosis: Binder Syndrome (Maxillonasal Dysplasia)
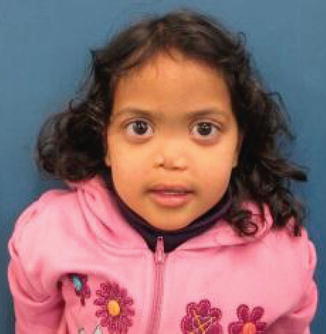
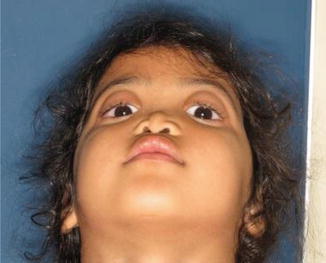
History
It is generally a sporadically occurring syndrome characterized by varying degrees of nasomaxillary hypoplasia, absent anterior nasal spine, and small frontal sinuses.
Exam
Midface hypoplasia; obtuse frontonasal angle; a short, vertical nose with acute nasolabial angle; and perialar and nasal tip flattening. Patient may also have class III occlusion with an anterior open bite.
Treatment
Staged reconstruction during adolescence, including orthodontic treatment, possibly orthognathic surgery (Le Fort I), and eventual nasal reconstruction.
1.1.14 Diagnosis: Saethre-Chotzen (Acrocephalosyndactyly Type III)
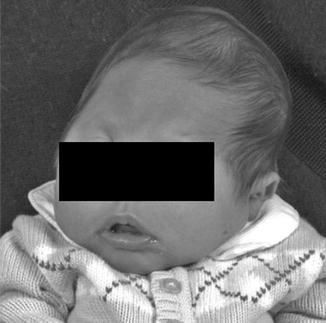
History
An autosomal dominant syndrome with an incidence of 1:25,000–50,000. Patients usually present with normal mental status.
Exam
Asymmetric brachycephaly, ptosis of the eyelids, antimongoloid slanting of palpebral fissures, midface hypoplasia, narrow palate, low-set hairline, and partial syndactyly.
Treatment
Cranial vault remodeling with fronto-orbital advancement is the mainstay of treatment. Patients with syndactyly will also require syndactyly release.
1.1.15 Diagnosis: Nager Syndrome
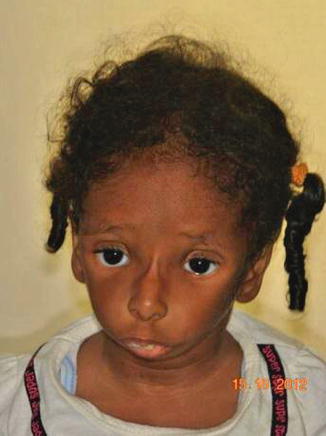
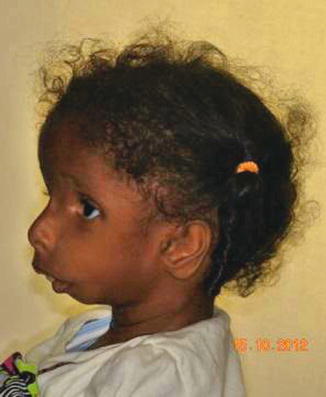
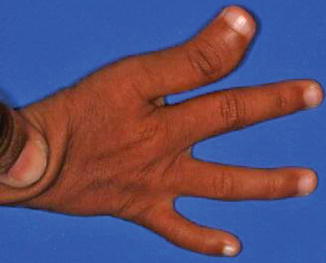
History
A rare and sporadic congenital condition that involves preaxial upper limb abnormalities and mandibulofacial dysostosis.
Exam
Similar craniofacial findings to Treacher Collins syndrome: mandibular, malar, and maxillary hypoplasia, colobomas, downslanting palpebral fissures, absent eyelashes medially, low-set malformed ears, and palatal defects. Airway compromise due to micrognathia and glossoptosis may occur, as well as velopharyngeal incompetence and conductive hearing loss. Although preaxial abnormalities (hypoplastic thumb/radius) are described classically, a variety to hand anomalies may occur, as well as urogenital anomalies. Patients usually have normal intelligence.
Treatment
Initial management includes assessment and management of the airway (positioning, lip-tongue adhesion, tracheostomy). Management of the craniofacial anomalies may include repair of the palate, mandibular distraction osteogenesis, midface reconstruction with bone grafts or custom implants, and ear reconstruction. Directed surgical correction of the upper limb and urogenital anomalies is also done.
1.1.16 Diagnosis: Möbius Syndrome
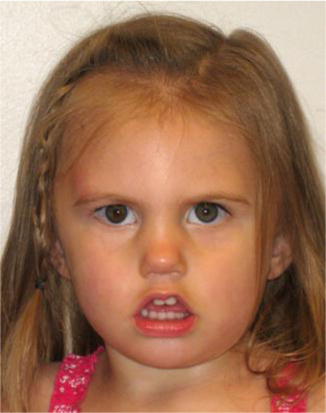
History
Möbius syndrome is a rare congenital disorder characterized by a variety of cranial nerve (CN) defects, usually the facial and abducens cranial nerve paralysis. The syndrome is usually bilateral. However, unilateral cases have been described.
Exam
Diagnosis is based on physical examination alone and depends on the cranial nerve involved. As CN VI and VII are the most involved, the majority of these patients display the absence of facial expression with an inability to smile and lack of eye movement. A masked appearance, especially while crying, is typical of this syndrome.
Treatment
Traditionally, local muscle transfers involving the masseter, temporalis, and the platysma have been used to restore functions lost due to facial paralysis. More recently, transfer of free neurovascularized muscle such as the free gracilis muscle transfer has been used, and it has become first line of treatment to restore partial function of the facial nerve allowing for the ability to smile, adequate excursion of the commissure, oral competence, and improved speech.
1.1.17 Diagnosis: Frontonasal Dysplasia/Median Cleft Face Syndrome
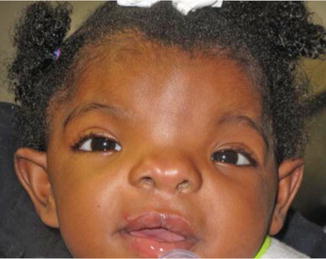
History
Congenital malformation with variable degrees of severity of Tessier 0/14 median facial cleft; may be associated with other syndromes.
Exam
Orbital hypertelorism, broad nasal bridge, cleft lip/palate/maxilla, bifid nose, +/− other associated anomalies (encephalocele, absent corpus callosum, ocular abnormalities, hearing loss).
Treatment
Surgical treatment of hypertelorism, nasal abnormalities, and cleft lip/palate as appropriate.
1.1.18 Fibrous Dysplasia
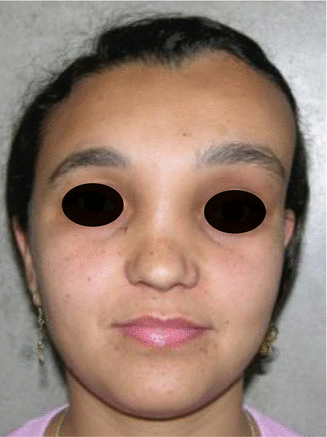
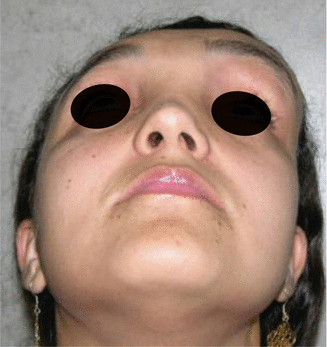
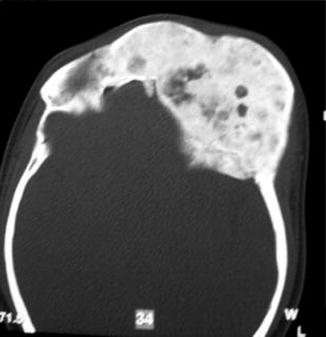
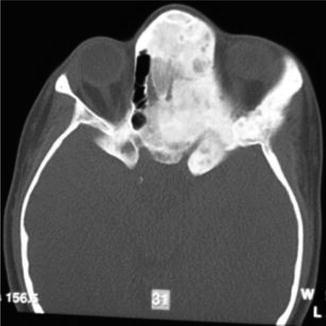
History
Patient presents with craniomaxillofacial or axial bony abnormalities.
Exam
It is a progressive benign bone disorder that occurs sporadically, often presenting in late childhood or adolescence. It may be classified as monostotic, polyostotic, or a part of McCune-Albright syndrome (polyostotic fibrous dysplasia, precocious puberty, skin pigmentation). Histologically, normal bone is replaced with an unorganized mixture of fibrous connective tissue and structurally weak trabecular bone. For craniomaxillofacial involvement, thorough head and neck, ocular, and otologic examinations are performed. CT face will demonstrate a ground glass appearance of woven bone.
Treatment
As this is a benign and unpredictable process, surgical management must consider the functional and aesthetic concerns of the area involved, skeletal maturity of the patient, and type of disease process (mono- vs. polyostotic). Complete resection is not always mandated, particularly if vital structures are not compromised. If subtotal resection is done, long-term monitoring is warranted, especially if the orbit is involved. Threatened optic nerves require orbital decompression. Reconstruction is tailored toward the nature of the defect and includes free tissue transfer, autologous bone grafts, as well as alloplastic materials in select cases.
1.1.19 Diagnosis: Goldenhar Syndrome
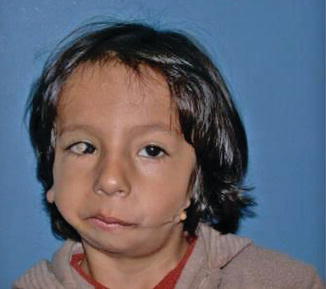
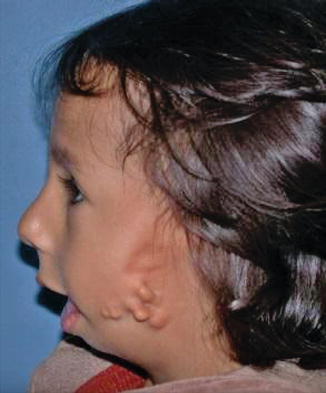
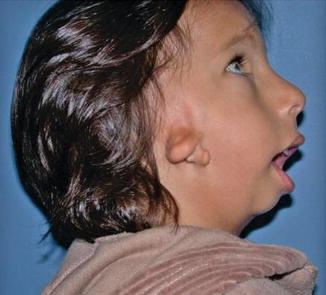
History
Patients present with asymmetry of the bony and soft tissues of the face.
Exam
Also known as oculo-auriculo-vertebral (OAV) syndrome, this rare congenital abnormality is a subset of hemifacial microsomia and is a result of anomalous development of the first and second branchial arch. It is characterized by epibulbar dermoid/lipodermoid/colobomas, preauricular skin tags, unilateral facial hypoplasia, unilateral macrostomia, vertebral anomalies, mandibular/maxillary hypoplasia, and/or abnormalities of internal organs.
Treatment
Patients with Goldenhar syndrome should be treated in a multistage, multidisciplinary fashion with a long-term plan. Although there is no consensus on the methods of surgical treatment, priority should be given to deficits causing functional problems such as airway compromise in maxillary/mandibular hypoplasia. Commonly employed surgical options include costochondral bone grafts or classic osteotomy.
1.1.20 Diagnosis: Progressive Hemifacial Atrophy/Romberg Disease/Parry-Romberg Syndrome

History
Patient presents with progressive hemifacial atrophy in the first or early second decade of life.
Exam
It is a rare neurocutaneous syndrome characterized by progressive shrinkage and degeneration of progressive atrophy of skin and subcutaneous tissue within the dermatome of one of the branches of the trigeminal nerve on unilateral face (95 %) but can occasionally extend to other parts of the body. Findings include a circumscribed patch of frontal scleroderma with hair loss and a depressed linear scar extending down through the midface on the affected side, hence the term “coup de sabre.”
Treatment
There is no adequate medical treatment for Parry-Romberg syndrome. After it is allowed to run its course, reconstruction options include fat injection, soft tissue augmentation with free tissue transfer, bone grafting, and orthognathic surgery as indicated.
1.1.21 Diagnosis: Unilateral Cleft Lip
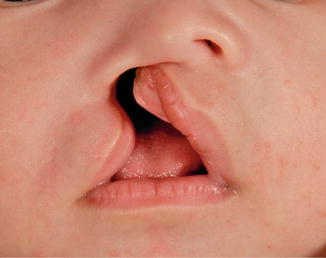
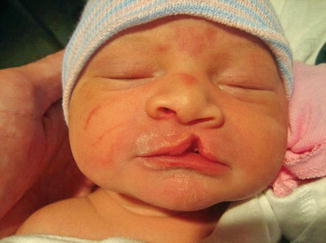
History
A cleft lip represents a deformity affecting the projection and outward rotation of the premaxilla with abnormal insertion of the orbicularis muscle. The incidence of cleft lip, with or without a cleft palate, is higher among Asians, males, and is most frequently found on the left side. Cleft lips are associated with a syndrome in only 15 % of the cases.
Exam
A unilateral complete cleft lip is characterized by a disruption of the lip, nostril sill, and alveolus. A unilateral incomplete cleft lip does not extend through the entire lip to the nasal floor (Simonart band). An abnormal attachment of the orbicularis oris muscles is present on both sides of the cleft. The vertical height of the lip is decreased. On the non-cleft side, the philtral column, philtral dimple, and two-thirds of Cupid’s bow are preserved.
Treatment
A multidisciplinary approach is needed for the care of the patient with cleft lip. Children with this condition should be assessed shortly after birth, and parents should be counseled before birth if cleft lip is suspected. Special feeding bottles and nipples are needed by the patient to ensure adequate growth and weight gain. Presurgical orthopedics such as a Latham device or nasoalvelolar molding (NAM) can be used to narrow and align cleft segments. Primary cleft repair is often performed at the age of 3 months. For unilateral cleft lips, the four main techniques for repair include straight line repair (Rose-Thompson), triangular flap repair (Randall-Tennison), the rotation-advancement repair (Millard) and the anatomical subunit approximation technique (Fisher). It is common for these patients to require secondary procedures. The nasal deformity is addressed at the same time. Some recommend primary alveolar cleft repair before 2 years of age. Secondary bone grafting is recommended during age of transitional dentition (between ages 8–12). Definitive rhinoplasty is performed at adolescence.
1.1.22 Diagnosis: Bilateral Cleft Lip
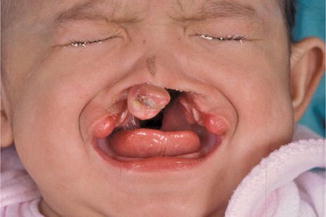
History
Bilateral lip deformity
Exam
Bilateral cleft lip results from failure of fusion of the maxillary prominences with the medial nasal prominence. The deformity is characterized by a disruption of the lip, nostril sill, and alveolus. The unattached premaxillary segment results in varying degrees of protrusion and angulation.
Treatment
As in unilateral cleft lip, treatment requires a multidisciplinary approach addressing the associated anomalies, genetic assessment, feeding techniques, and presurgical orthopedics. The timing of the repair is ideally around 3 months of age. Different techniques of repair have been described, but common principles of repair include bilateral cleft repair, reduction of the prolabium, formation of Cupid’s bow and tubercle from lateral lip elements, repair of orbicularis muscle, deepening of the central gingivolabial sulcus using prolabial mucosa, and primary rhinoplasty.
1.1.23 Diagnosis: Primary Cleft Palate
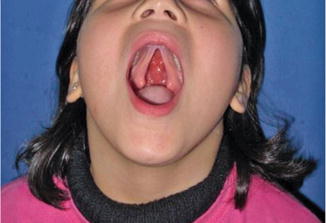
History
A cleft palate deformity is caused by a disruption in the migration and fusion of the medial and lateral nasal prominences of the frontonasal process with the maxillary prominence. The incidence of isolated cleft palate with associated anomalies is approximately 70 %. Patients with an untreated cleft palate will present with feeding difficulties, impaired facial growth, and speech abnormalities.
Exam
The lip, alveolus, and hard palate anterior to the incisive foramen are affected.
Treatment
Surgical management of a primary cleft palate requires a repair of the anterior hard palate. A two-flap palatoplasty is preferred with bilateral mucoperiosteal flaps based on the greater palatine arteries. The flaps will extend to the cleft margin and provide adequate tissue coverage with tension-free closure.
1.1.24 Diagnosis: Submucous Cleft
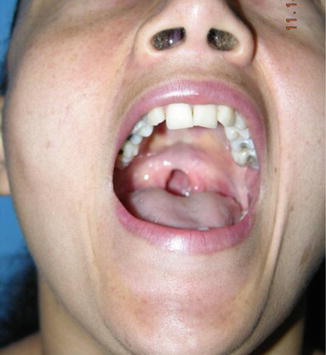
History
Submucous cleft palate occurs in 1:10,000–1:20,000 births. Patients often present because of velopharyngeal insufficiency and other speech/resonance disorders. Initial evaluation focuses on extent of nasal air flow escape and speech difficulties. Results of these studies are used to guide treatment.
Exam
The classical form of submucous cleft is defined by the presence of the following anatomical features: (1) a bifid or distorted uvula, (2) a bony notch at the back of the hard palate, and (3) a translucent zone in the midline of the soft palate due to separation of normally fused muscles. Occult forms may exist that lack these clinical findings, but the patient presents with velopharyngeal insufficiency.
Treatment
The goal of treatment centers on establishment of velopharyngeal competence. Surgical treatment includes palatal-lengthening procedures, repositioning of the levator muscles of the velum and sphincter, or pharyngoplasty. Techniques that have been described include Furlow Z-plasty, Wardill technique, minimal incision palatopharyngoplasty, and variations of the above.
1.1.25 Diagnosis: Alveolar Cleft
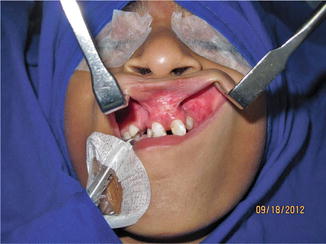
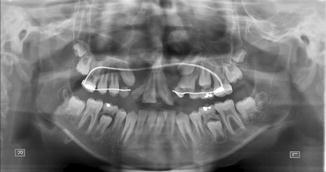
History
Patient presents with a gap in the alveolus.
Exam
An alveolar cleft or gap is the result of abnormal primary palate formation during weeks 4–12 of gestation. Alveolar gap can be unilateral/ bilateral, and it may or may not be associated with a palatal defect. This defect is also associated with mostly anterior nasal septal deviation, which is more extreme and posterior as well when associated with a palate defect. Exam should also include a hearing exam.
Treatment
The rationale for closure of alveolar clefts includes stabilizing the maxillary arch, permitting support for tooth eruption, eliminating oronasal fistulae, and providing improved aesthetic results. Surgical treatment includes (1) appropriate flap design, (2) wide exposure, (3) nasal floor reconstruction, (4) closure of oronasal fistula, (5) packing bony defect with cancellous bone, and (6) coverage of bone graft with gingival mucoperiosteal flaps. Orthodontic preparation may be required for certain alveolar clefts. Surgery should be performed prior to permanent teeth eruption.
1.1.26 Diagnosis: Pierre Robin Sequence
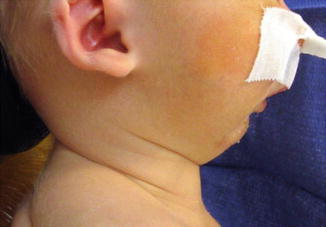
History
A sequence of events that occur secondary to a small mandible and includes: glossoptosis, cleft palate, and possible airway obstruction.
Exam
Micrognathia, retrodisplaced tongue, possible cleft palate (often U shaped), and possible difficulty breathing. Must rule out other associated syndromes: Stickler, velocardiofacial, Treacher Collins, hemifacial microsomia, trisomy 18, and Nager.
Treatment
Successful management of airway includes prone or semi-prone positioning (70 %), nasopharyngeal airway, palatal plates, tongue-lip adhesion, or a short course of intubation. Tracheostomy may be required in severe cases (~10 %), and mandibular distraction is reserved for refractory ventilator-dependent patients. Cleft palate repair as per protocol.
1.1.27 Diagnosis: Craniofacial Clefts
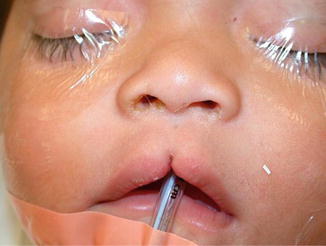
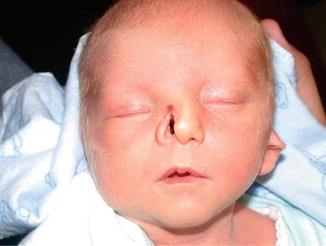
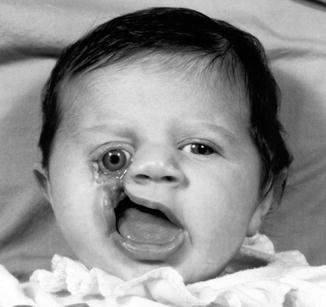
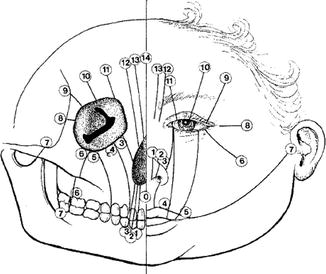
History
Patient presents with anatomic distortions of the face and cranium with deficiencies in a linear pattern.
Exam
Craniofacial clefts exist in a multitude of locations with varying degrees of severity. In the Tessier classification, clefts are numbered from 0 to 14. The orbits separate the cranial clefts (8–14) from the facial clefts (0–7). The clefts are numbered so that the facial and the cranial component of the cleft always add up to 14. Tessier cleft number 7 is the lateral-most craniofacial cleft. CT scan is obtained to evaluate underlying skeletal deformities.
Treatment
Treatment of craniofacial clefts requires a multidisciplinary approach. Timing of surgery depends on severity of the problem. Functional problems like ocular exposure are treated early. Cranial defects and soft tissue clefts are usually corrected during infancy. Midface reconstruction and bone grafting are performed around 6–9 years of age. Orthognathic procedures are deferred until skeletal maturity.
Photos showing examples of facial clefts.
1.1.28 Diagnosis: Treacher Collins Syndrome
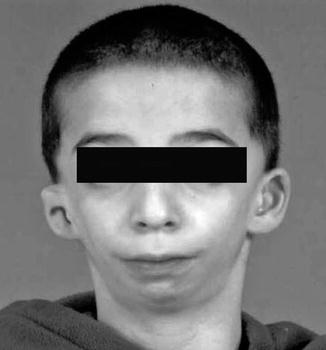
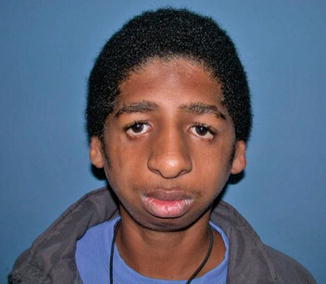
History
A common autosomal dominant bilateral craniofacial cleft syndrome with an incidence of 1:10,000. Initially described by Treacher Collins in 1900, it has later been recognized as a combination of Tessier cleft numbers 6, 7, and 8.
Exam
Multiple facial findings of malar and mandibular bone hypoplasia, colobomas, and lower lid retraction with an antimongoloid slant as the lateral canthus is displaced inferiorly. Loss of facial width with the absence of the zygomatic arch and hypoplasia of the temporalis muscle and ear abnormalities.
Treatment
Given the narrow pharyngeal size and mandibular hypoplasia, airway management is imperative. Facial reconstruction may occur which includes eyelid reconstruction for colobomas, distraction osteogenesis for mandibular hypoplasia, skeletal augmentation, and orthognathic surgery.
1.1.29 Diagnosis: Prominent Ear
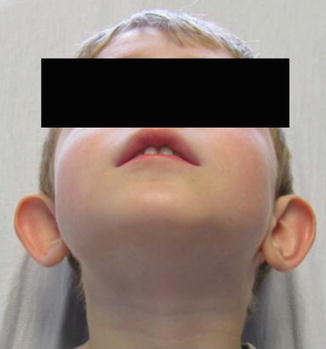
History
A congenital ear deformity that results from embryonic arrest during the final convolutions of the ear, with failure of folding of the antihelix.
Exam
The most common feature of a prominent ear is the effacement of the antihelical fold with a conchoscaphal angle >90°. Other features include conchal hypertrophy defined as a deep conchal bowl with excessive height of the conchal wall >1.5 cm.
Treatment
Many authors advocate combining techniques to minimize relapse. Commonly used techniques for the antihelical fold deformity are the conchoscaphal suture technique (Mustarde), antihelical scoring (Stenstrom), or antihelical excision (Luckett). The conchal bowl hypertrophy is managed with conchomastoid suture technique (Furnas) or conchal bawl excision (Davis).
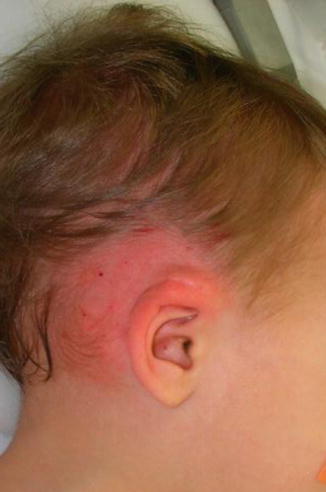
1.1.30 Cryptotia
History
An abnormal adherence of the superior ear to the temporal skin with varying degrees of severity. Cryptotia is commonly seen in Asians with an incidence of 1:400. The deformity is bilateral in 40 % of the cases with the right ear being more affected than the left.
Exam
Cartilaginous malformation in the scaphal-antihelix complex with loss of the post-auricular sulcus definition.
Treatment
Splinting in the neonatal period can be effective. Surgical intervention requires division of the abnormally positioned transverse and oblique ear muscles. The surgically created retroauricular sulcus is then covered with a skin graft.
1.1.31 Microtia
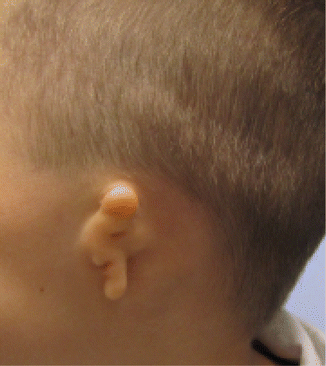
History
It is the most common major congenital anomaly of the external ear. The etiology is likely a result of in utero tissue ischemia secondary to obliteration of the stapedial artery or actual hemorrhage into the local tissues. Microtia is also associated with several craniofacial syndromes. The classification system is based on a gradient from mild ear deformity to total absence of the external ear (anotia).
Exam
-
Grade I: The pinna is mildly deformed and smaller than normal.
-
Grade II: The pinna is smaller than in grade II. The helix may not be fully developed. The triangular fossa, scaphae, and antihelix have much less definition.
-
Grade III: The pinna is essentially absent, except for a vertical sausage-shaped skin remnant. The superior aspect of this sausage-shaped skin remnant consists of underlying unorganized cartilage, and the inferior aspect of this remnant consists of a relatively well-formed lobule.
-
Grade IV: Total absence of the pinna.
Treatment
Staged reconstruction of the external ear begins with excision of the remnant cartilaginous component and placement of a cartilagenous framwork made of rib grafts. In subsequent procedures, the lobule and tragus are reconstructed. Creation of a post-auricular sulcus then follows with placement of a skin graft. Surgical management of the middle ear should be delayed until after the auricle is reconstructed.
1.1.32 Stahl’s Ear
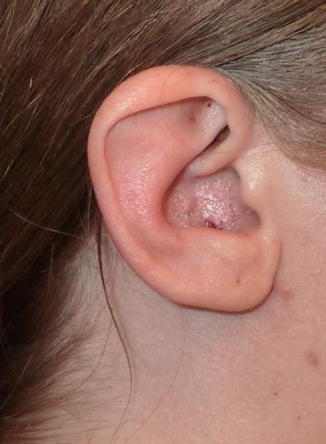
History
A congenital hereditary auricular deformity of the helical rim commonly found in patients of Asian descent. Stahl’s ear deformity, first described in the nineteenth century, is also known as “Spock’s ear.”
Exam
The deformed ear is recognized by the presence of an abnormal cartilaginous pleat, known as the third crus, which extends from the antihelix to the edge of the helix. In addition, a flat helix and an anomalous scaphoid are present.
Treatment
In the neonatal age, it can be treated with splinting given the pliability of the cartilage found in the early months of life. Surgical management is indicated for children and young adults. Operative techniques include wedge excision of the third crus, third crus helical advancement, Z-plasty, cartilage reversal, or periosteal tethering.
1.1.33 Cup/Constricted Ear
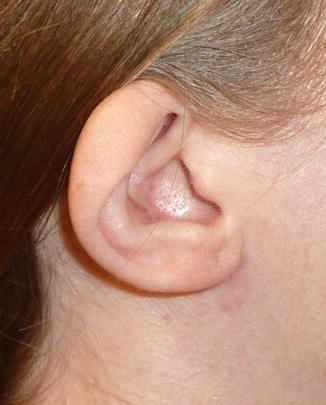
History
Constricted ear anomalies can be categorized as either lop or cup ear deformities. The lop ear is a malformed auricle with an acute downward folding of the superior ear. A cup ear is a malformed ear that combines the features of a lop ear and a protruding ear.
Anatomy/Exam
In a lop ear, the folding or deficiency is in the helix and scapha at the level of Darwin’s tubercle. Malformed superior crus of the antihelix may also be present. The features of a cup ear are overdeveloped, cup-shaped concha, a deficient superior aspect of the helical margin and antihelical crus, and a small vertical height.
Treatment
In cases of helical involvement only, the folded helix is detached and reattached in an upright position. Scaphal involvement is treated with the use of local tissues such as banner flaps of cartilage and skin flaps from the medial surface of the ear. In severe cases involving the antihelix and conchal wall, Kislov’s technique is utilized with unfurling of the superior pole, the use of remnant tissue for the middle portion of the ear, and contralateral conchal cartilage for missing tissue.
1.2 Craniofacial, Trauma
1.2.1 Frontal Sinus Fracture
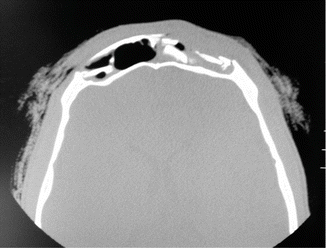
History
Severe trauma to the head and/or face.
Exam
Lacerations, palpable step-offs, or contour irregularities of the forehead should prompt for a craniomaxillofacial CT scan (1.5 mm cuts) with axial and sagittal views. Key features of the fracture are the degree of comminution, displacement of the anterior and/or posterior tables, and involvement of the frontal sinus outflow tract (FSOT or nasofrontal duct). Nasal discharge may be present, and CSF leak should be ruled out with beta-transferrin.
Treatment
Depends on the characteristics of the fracture with the goal of minimizing contour deformities and late complications (mucocele, osteomyelitis, epidural abscess, CSF leak, etc.). Observation is acceptable for minimally displaced anterior or posterior wall fractures without FSOT involvement or CSF leak. Open reduction and fixation of anterior table fractures with microplates are done for anterior wall irregularities. For fractures that involve the anterior wall and FSOT, obliteration of the sinus and outflow tract (with bone, fat, pericranial flap), exenteration of all sinus mucosa, and plating of anterior wall are done. CSF leak and severe bone loss require cranialization of the frontal sinus where the posterior table is removed, in addition to obliteration of the sinus.
1.2.2 Orbital Floor: Blow-Out Fracture
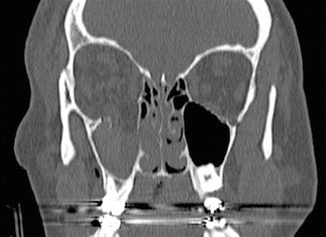
History
Periorbital trauma and possible diplopia.
Physical Exam
Periorbital edema/ecchymosis. Swelling may mask enophthalmos, which may be severe enough to result in proptosis and possible diplopia, Forced duction test will differentiate swelling from entrapment of extraocular muscles. Blow-out fractures involve isolated fractures of the orbital walls—typically the floor and medial wall. CT face will show displacement of the orbital floor into maxillary sinus with herniation of orbital contents.
Treatment
Preoperative ophthalmic exam is warranted to rule out globe injury. Thin cut CT with axial, coronal, and sagittal views define extent of injury and need for repair. Operative repair is performed to restore orbital anatomy and avoid enophthalmos.
1.2.3 NOE Fracture
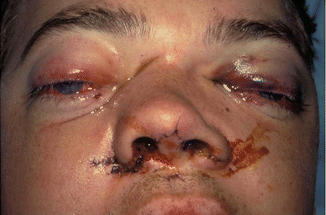
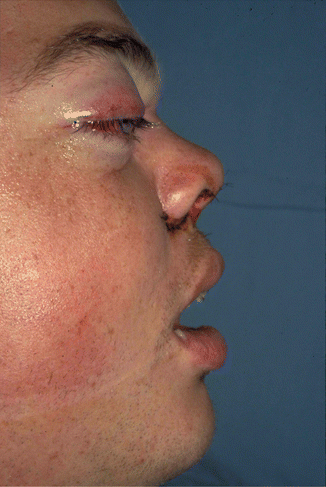
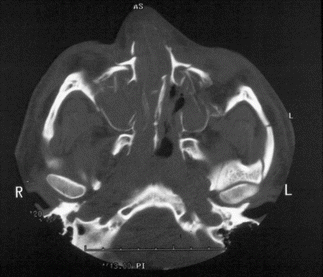
History
Central midface impact.
Physical Exam
Periorbital edema/ecchymosis, subconjunctival hematoma, telecanthus, shortened palpebral fissures, epiphora, and displaced nasal bridge (saddle nose deformity).
X-Ray Exam
Displacement of canthal bearing bone segments.
Treatment
Surgical goals include restoration of the intercanthal distance, palpebral shape, nasal projection/width/height/length, and ORIF of fractured segment(s) to adjacent stable bone. Intercanthal wires must be placed superior and posterior to the lacrimal fossa.
1.2.4 Nasal Fractures
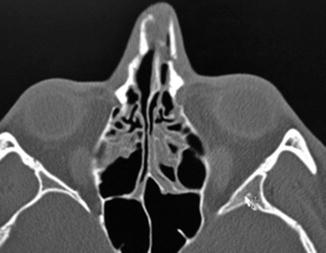
History
Trauma to the nose; determine age of injury and prior nasal surgery.
Exam
Epistaxis, bony crepitus, periorbital ecchymosis, asymmetries or step-offs, and septal deviation. Must rule out a septal hematoma, and evaluate bony and cartilaginous nasal septum and vault.
Treatment
Septal hematomas are drained immediately to avoid long-term sequelae, especially necrosis. Closed nasal reduction is successful in most isolated nasal bone fractures and is best done within 7–10 days post-injury when most edema has resolved. Obtain pre-injury photos if possible.
1.2.5 Maxillary Fractures
1.2.5.1 Le Fort 1
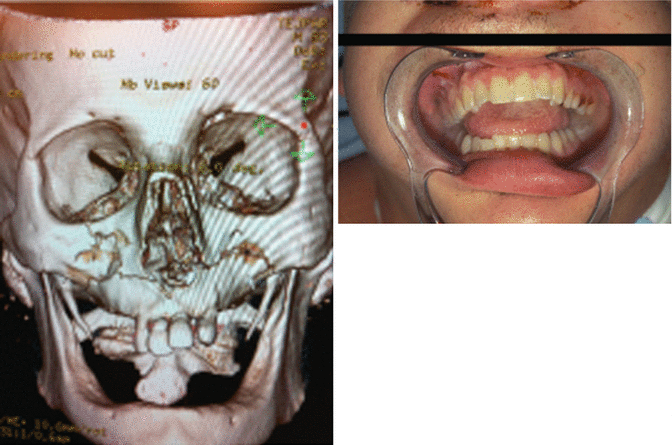
1.2.5.2 Le Fort 2
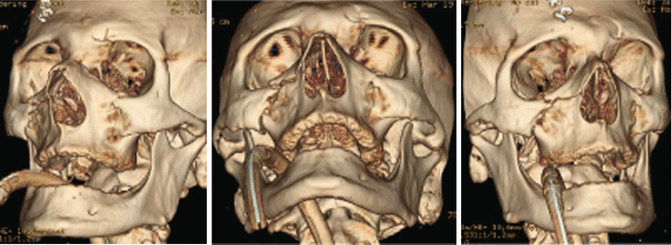
History
Facial trauma.
Exam
Midface instability, malocclusion, trismus, enophthalmos, restricted ocular movement, V2 paresthesia, loss of facial projection/height, increased facial width, and CSF leak. Maxillofacial CT with three views (axial, sagittal, coronal) and 3D reconstruction will show type of Le Fort fracture on each side, as bilateral Le Fort fractures at the same level are less common.
Treatment
Initial control of airway and hemorrhage is paramount, followed by a coordinated operative plan with concomitant injures. Operative goals are restoration of dental occlusion, facial height/width/projection, and orbital anatomy. Nasotracheal intubation, dental splints, and multiple approaches to the facial skeleton may be necessary.
1.2.6 Mandibular Fractures: Parasymphyseal
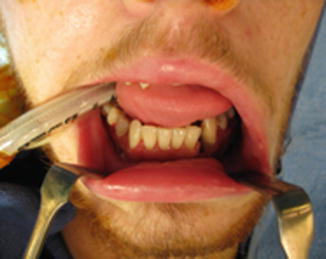
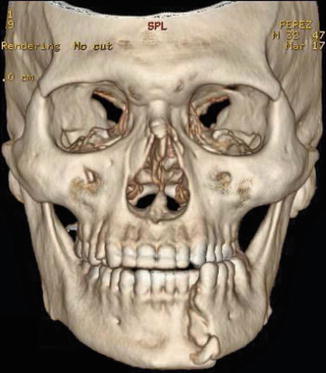
History
Trauma to the lower face and malocclusion.
Physical Exam
Intraoral lacerations, palpable bony step-offs or mobility, malocclusion, diminished range of motion, crossbite, anterior or posterior open bite, and V3 paresthesia.
X-Ray Exam
Imaging should include maxillofacial CT (with axial, coronal, and 3D reconstruction views) and/or plain X-rays (panoramic and posteroanterior views).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


