Introduction888
INTRODUCTION
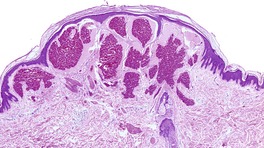
Fig. 38.1
HAMARTOMAS AND MALFORMATIONS
ECCRINE ANGIOMATOUS HAMARTOMA
PHAKOMATOSIS PIGMENTOVASCULARIS
CAPILLARY MALFORMATIONS (NEVUS FLAMMEUS)
Sturge–Weber syndrome
Klippel–Trenaunay syndrome
Cobb’s syndrome
Capillary malformation – AVM syndrome
Histopathology of capillary malformations
VENOUS MALFORMATIONS
‘Blue rubber bleb’ nevus syndrome
Maffucci’s syndrome
Venous malformations with cutaneous and mucosal involvement
Histopathology of venous malformations
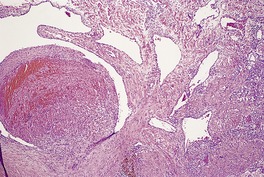
Fig. 38.2
Electron microscopy
CUTIS MARMORATA TELANGIECTATICA CONGENITA
Histopathology
GLOMULOVENOUS MALFORMATION
Histopathology
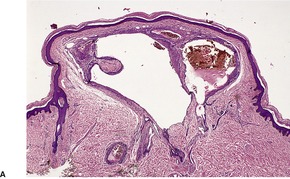
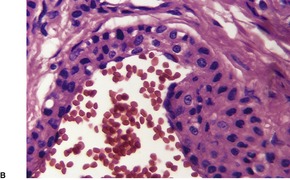
Fig. 38.3
LYMPHANGIOMA (CYSTIC LYMPHATIC MALFORMATION)
Superficial lymphangioma
Histopathology
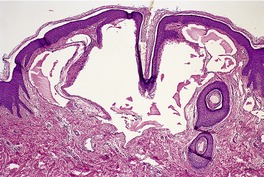
Fig. 38.4
Deep lymphangioma
Histopathology
Lymphangiomatosis
Histopathology
VERRUCOUS HEMANGIOMA
Histopathology
CALIBER-PERSISTENT ARTERY
VASCULAR DILATATIONS (TELANGIECTASES)
HEREDITARY HEMORRHAGIC TELANGIECTASIA
GENERALIZED ESSENTIAL TELANGIECTASIA
HEREDITARY BENIGN TELANGIECTASIA
UNILATERAL NEVOID TELANGIECTASIA
ATAXIA–TELANGIECTASIA
‘SPIDER’ ANGIOMA
Histopathology
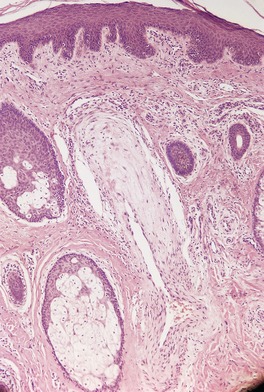
Fig. 38.6
VENOUS LAKE
Histopathology
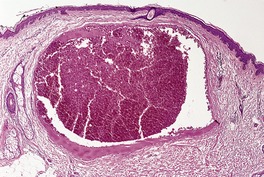
Fig. 38.7
ANGIOKERATOMA
Histopathology
Fig. 38.8
Electron microscopy
MISCELLANEOUS TELANGIECTASES
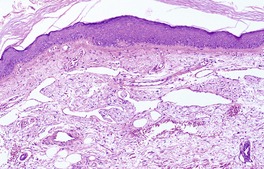
Fig. 38.9
DRUG-INDUCED TELANGIECTASIA
RETICULATE TELANGIECTATIC ERYTHEMA (STERNAL ERYTHEMA)
Histopathology
COSTAL FRINGE
LYMPHANGIECTASES
VASCULAR PROLIFERATIONS (HYPERPLASIAS AND BENIGN NEOPLASMS)
INFANTILE HEMANGIOMA
Diffuse neonatal hemangiomatosis
Histopathology of infantile hemangioma
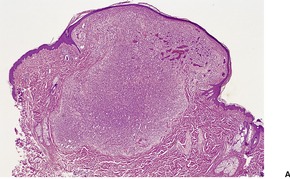
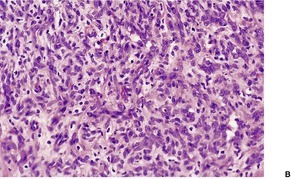
Fig. 38.10
Electron microscopy
SINUSOIDAL HEMANGIOMA
Histopathology433
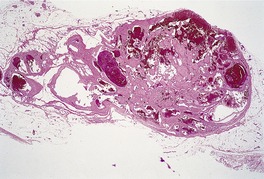
Fig. 38.11
HEMANGIOBLASTOMA
Histopathology
‘CHERRY’ ANGIOMA
Histopathology
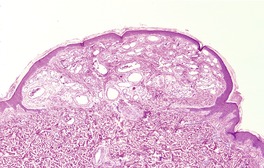
Fig. 38.12
GLOMERULOID HEMANGIOMA
Histopathology443
PAPILLARY HEMANGIOMA
Histopathology
ARTERIOVENOUS HEMANGIOMA
Histopathology466. and 467.
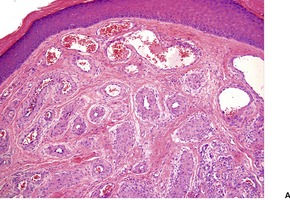
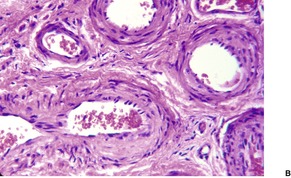
Fig. 38.13
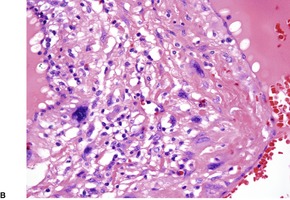
Fig. 38.14
MICROVENULAR HEMANGIOMA
Histopathology479
TARGETOID HEMOSIDEROTIC (HOBNAIL) ‘HEMANGIOMA’
Histopathology492
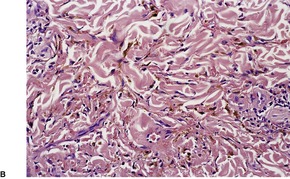
Fig. 38.15
SPINDLE-CELL HEMANGIOENDOTHELIOMA (HEMANGIOMA)
Histopathology501. and 502.
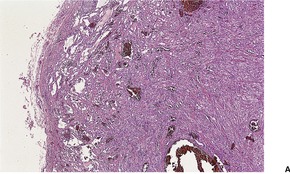
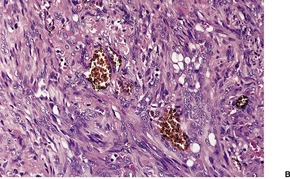
Fig. 38.16
Electron microscopy
ACQUIRED ELASTOTIC HEMANGIOMA
Histopathology
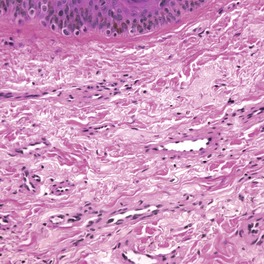
Fig. 38.17
ANGIOMA SERPIGINOSUM
Histopathology
GIANT CELL ANGIOBLASTOMA
Histopathology526
EPITHELIOID ANGIOMATOUS NODULE
Histopathology527. and 529.
ANGIOLYMPHOID HYPERPLASIA WITH EOSINOPHILIA
Histopathology
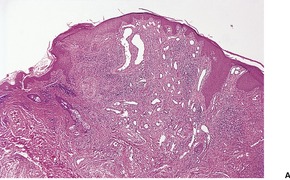
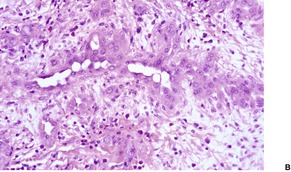
Fig. 38.18
LOBULAR CAPILLARY HEMANGIOMAS
Pyogenic granuloma and variants
Histopathology
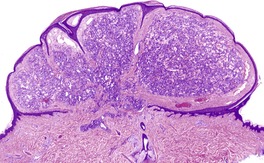
Fig. 38.19
Acquired tufted angioma (angioblastoma)
Histopathology
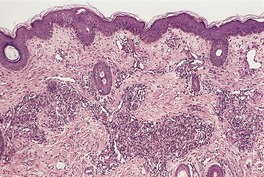
Fig. 38.20
GLOMUS TUMOR
Histopathology
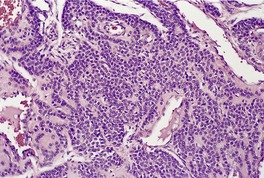
Fig. 38.21
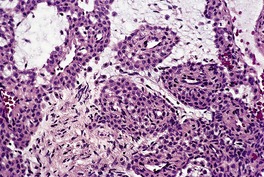
Fig. 38.22
Electron microscopy
Glomus coccygeum
Histopathology
ERUPTIVE PSEUDOANGIOMATOSIS
Histopathology
PAPULAR ANGIOPLASIA
MULTINUCLEATE CELL ANGIOHISTIOCYTOMA
Histopathology764
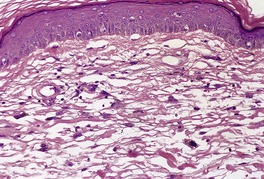
Fig. 38.23
REACTIVE ANGIOENDOTHELIOMATOSIS
Histopathology
Diffuse dermal angiomatosis
Histopathology
ACROANGIODERMATITIS
Histopathology821. and 822.
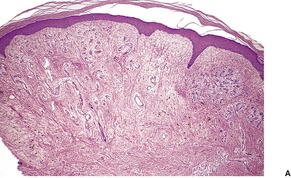
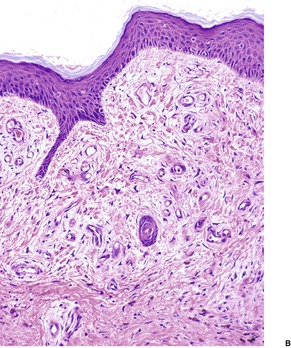
Fig. 38.24
MISCELLANEOUS LESIONS
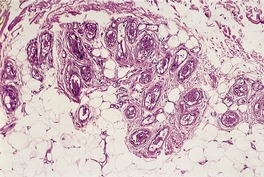
Fig. 38.25
PENILE MYOINTIMOMA
INTRAVASCULAR PAPILLARY ENDOTHELIAL HYPERPLASIA
Histopathology
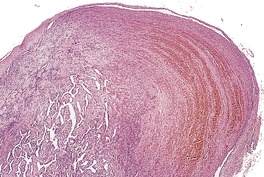
Fig. 38.26
Electron microscopy
BACILLARY ANGIOMATOSIS
Histopathology850. and 863.
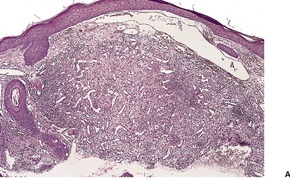
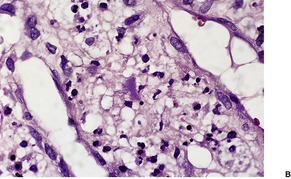
Fig. 38.27
Differential diagnosis
VERRUGA PERUANA
Histopathology
LYMPHANGIOENDOTHELIOMA (ACQUIRED PROGRESSIVE LYMPHANGIOMA)
Histopathology841
TUMORS WITH VARIABLE OR UNCERTAIN BEHAVIOR
KAPOSI’S SARCOMA
Classic type
African (endemic) type
Kaposi’s sarcoma associated with immunosuppressive therapy
Epidemic (HIV-associated) type
Etiology and pathogenesis
Treatment of Kaposi’s sarcoma
Histopathology
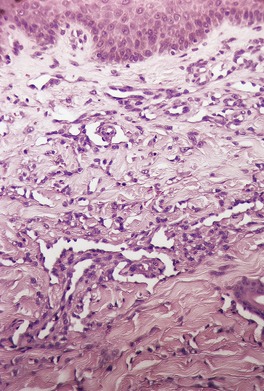
Fig. 38.28
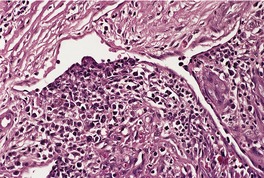
Fig. 38.29
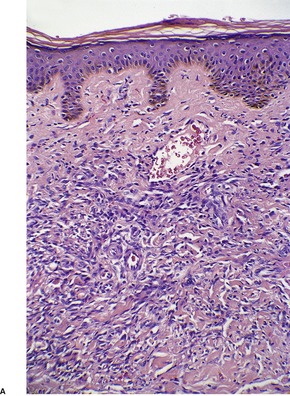
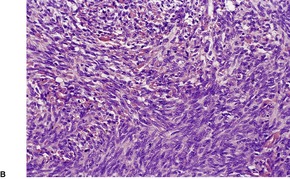
Fig. 38.30
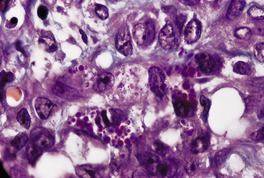
Fig. 38.31
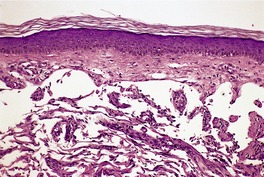
Fig. 38.32
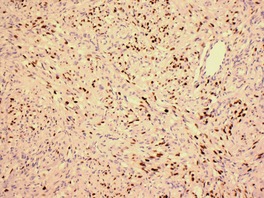
Fig. 38.33
Differential diagnosis
HEMANGIOPERICYTOMA
Histopathology1112
Electron microscopy
KAPOSIFORM HEMANGIOENDOTHELIOMA
Histopathology1145
ENDOVASCULAR PAPILLARY ANGIOENDOTHELIOMA OF CHILDHOOD
Histopathology
MALIGNANT TUMORS
ANGIOSARCOMA AND LYMPHANGIOSARCOMA
Idiopathic cutaneous angiosarcoma of the head and neck1162.1163.1164. and 1165.
Lymphangiosarcoma arising in chronic lymphedematous limbs
Postirradiation angiosarcoma
Miscellaneous angiosarcoma
Histopathology
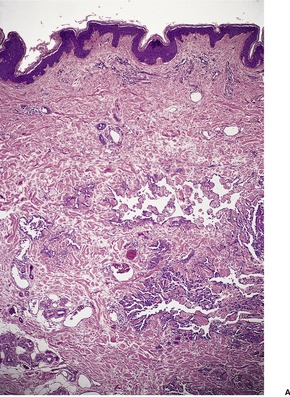
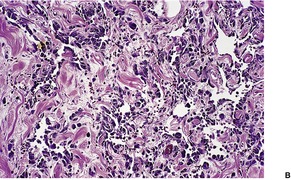
Fig. 38.34
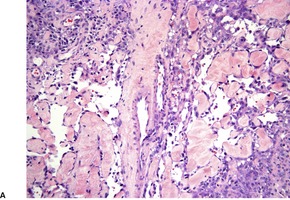
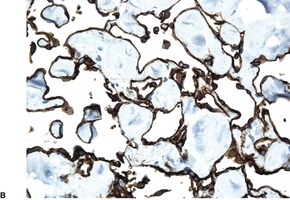
Fig. 38.35
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



Vascular tumors
Vascular dilatations (telangiectases)894
Hereditary hemorrhagic telangiectasia894
Generalized essential telangiectasia894
Hereditary benign telangiectasia894
Unilateral nevoid telangiectasia894
Ataxia–telangiectasia894
‘Spider’ angioma895
Venous lake895
Angiokeratoma896
Miscellaneous telangiectases897
Drug-induced telangiectasia897
Reticulate telangiectatic erythema (sternal erythema)897
Costal fringe897
Lymphangiectases897
Vascular proliferations (hyperplasias and benign neoplasms)897
Sinusoidal hemangioma899
Hemangioblastoma900
‘Cherry’ angioma900
Glomeruloid hemangioma900
Papillary hemangioma901
Arteriovenous hemangioma901
Microvenular hemangioma902
Targetoid hemosiderotic (hobnail) ‘hemangioma’902
Spindle-cell hemangioendothelioma (hemangioma)902
Acquired elastotic hemangioma904
Angioma serpiginosum904
Giant cell angioblastoma904
Epithelioid angiomatous nodule904
Angiolymphoid hyperplasia with eosinophilia904
Eruptive pseudoangiomatosis908
Papular angioplasia909
Multinucleate cell angiohistiocytoma909
Acroangiodermatitis910
Miscellaneous lesions911
Penile myointimoma911
Intravascular papillary endothelial hyperplasia911
Bacillary angiomatosis911
Verruga peruana (bartonellosis)912
Lymphangioendothelioma (acquired progressive lymphangioma)913
A renewed interest in vascular tumors was provoked by the emergence of AIDS-related Kaposi’s sarcoma in the 1980s, and since then a number of new entities have been described. 1 Although many of these are rare, they may produce diagnostic dilemmas, several having many features in common with Kaposi’s sarcoma.
The classification of the vascular tumors and ectasias is far from straightforward. First, there is difficulty in separating true neoplasms from reactive proliferations or developmental abnormalities. Secondly, some vascular lesions represent a dilatation of pre-existing vessels rather than a proliferation of new vessels. Finally, it may be difficult to distinguish between a lesion showing blood vessel differentiation and one with lymphatic features as, for example, in the case of Kaposi’s sarcoma. Immunohistochemical, ultrastructural, and morphometric studies are helping to resolve some of these difficulties. An excellent, but selective review of vascular tumors was published by Hunt and Santa Cruz in 2004. 2
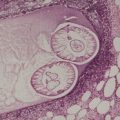
Vascular abnormalities of the skin are common. Approximately 50% of all neonates have some type of congenital vascular lesion. 3 The classification adopted here is based on clinical classifications that were delineated comparatively recently. 4 The distinction between malformations and vascular proliferations (tumors) has important clinical consequences. The distinction is more clear cut clinically than it is on a shave or punch biopsy of skin. This is a dilemma that remains to be resolved (Fig. 38.1). Furthermore, some overlap does occur between vascular tumors and malformations.5. and 6. The following categories will be considered:
• hamartomas and malformations
• vascular dilatations (telangiectases)
• vascular proliferations
• tumors with variable or uncertain behavior
• malignant tumors
• tumors with a significant vascular component.
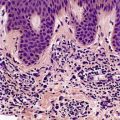
This lesion illustrates some of the problems with nomenclature. It would have been called cavernous hemangioma in the past. It does not fit neatly into the venous malformations or other entities. (H & E)
Before doing so, a brief comment will be made on tumor markers for vascular neoplasms. Although retained in this book, the first-generation vascular markers such as von Willebrand factor (factor VIII-related antigen), and the lectin Ulex europaeus are rarely used these days because of their non-specificity or unreliable staining properties. They have largely been replaced by CD31 (platelet endothelial cell adhesion molecule-1) and CD34. CD31 does not usually stain glomus tumors. It is negative also in some lymphatic lesions. On the other hand, CD34 also stains a number of soft tissue neoplasms. It also gives variable staining of lymphatic endothelium, 7 but this has become less significant with the commercial availability of D2-40, an antibody to a sialoglycoprotein that reacts with an epitope on lymphatic endothelium. 8 Vascular endothelial growth factor receptor type 3 was an initial marker, thought to be specific for lymphatic endothelium, 9 that did not live up to its expectations. Anti-thrombomodulin antibody is another. It detects thrombomodulin, a cell-surface glycoprotein that is present mainly on the luminal surface of endothelial cells of arteries, veins, capillaries, and lymphatics. 7 A recent trial of the antibody revealed moderately intense staining in 89% of benign and malignant vascular tumors tested, while CD34 stained 98% of the same tumors. 7 CD99 which has been said to be positive in ‘epithelioid vascular neoplasms’ has no place in the diagnosis of vascular neoplasms. 10
Hamartomas result from an error in embryological development and are present at birth. In the case of vascular hamartomas and malformations, they may become more obvious clinically some time after birth as a consequence of progressive ectasia. The constituent vessels may be capillaries, veins, arteries, lymphatics, or a combination of these vessels. Vascular malformations are associated with a range of dysmorphic syndromes such as the Sturge–Weber syndrome and ‘blue rubber bleb’ nevus syndrome (see below).
Eccrine angiomatous hamartoma is a rare malformation characterized by an increased number of small blood vessels, admixed with or adjacent to an increased number of eccrine glands (see p. 794). Rarely it may have a segmental distribution and present as a capillary malformation (nevus flammeus). 11 There may also be an increase in mucin, fat, or nerve fibers.
The term ‘phakomatosis pigmentovascularis’ refers to the coexistence of a vascular hamartoma, in the form of a capillary malformation (nevus flammeus), with a melanocytic lesion – usually a Mongolian spot or nevus spilus. Nevus anemicus, a functional abnormality of blood vessels (see p. 290), is often present. Five types of phakomatosis pigmentovascularis have been recognized based on the nature of the constituent abnormalities. 12 A new classification has recently been proposed (see p. 731). The condition is thought to result from an abnormality of neural crest development. The capillary malformation (nevus flammeus) is similar to the port wine stain (see below) although there may also be an increase in perivascular nerve fibers. 12 In type V, the only variant without a port wine stain, there are Mongolian spots associated with cutis marmorata telangiectatica congenita. 13
Capillary malformation (nevus flammeus) is a generic term for a group of congenital vascular malformations that commonly involve the forehead, face, and neck of newborns. The term includes the ‘port wine’ stain, the ‘salmon’ patch, and the ‘stork bite’, the latter representing the combination of glabellar lesions with lesions on the nape of the neck. The lesions generally grow proportionately with the child’s development and there is usually no tendency for spontaneous involution, although the small ‘salmon’ patch usually fades early in life (see below). 14 Recently, Happle has questioned the use of the term capillary malformation for a specific cutaneous entity. 15 He believes it should be used as an ‘umbrella term’ for at least nine different conditions. 15
The term nevus flammeus has largely disappeared; it has been replaced by the terms capillary malformation or port wine stain. 16 Capillary malformation is gaining a wider meaning and now includes the telangiectases in some reports. 17 Capillary malformations are believed to represent an error in vascular development occurring during embryogenesis.
The port wine stain occurs in 0.3–0.5% of newborns.18.19. and 20. There is no sex predilection. Familial occurrence is rare. 21 An acquired variant, which follows antecedent trauma, is said to occur very rarely.22.23. and 24. It may also follow oral isotretinoin. 25 Although any area may be affected, the lesion occurs most frequently on the face and neck. Single or multiple lesions may be present, and they are often sharply unilateral or segmental. Small or extensive areas of skin may be involved. At birth the lesions are flat and light pink in color. Lesions may become darker and thickened with time with undulation or cobblestoning. 26 This change is most common in facial lesions in the area of the second and third branches of the facial nerve. 27 An eczematous dermatitis develops within a port wine stain in a small number of cases. 28
The pulsed dye laser remains the standard of care for patients with port wine stains.20.29. and 30. Complete resolution is rarely seen, 31 and up to 20% hardly lighten at all with laser therapy. 30 Videomicroscopy shows when no further response can be expected. Other lasers have been tried,28.31. and 32. but the Nd:YAG laser, which is sometimes used for cases that do not respond, will result in scarring if fluences higher than the minimum purpura dose (MPD) are used. 33 The further management of non-responding cases is beyond the scope of this book. This topic was discussed in two excellent reviews in 200730 and 2008. 20
The ‘salmon’ patch (medial telangiectatic nevus) is a pink macular area present at birth in approximately 40% of the population. The nape of the neck, the eyelids, or the skin over the glabella may be involved. 34 In the majority of cases the lesions fade in the first year of life. 34 There may be associated congenital abnormalities.35. and 36. In a small number of cases the condition persists for life, particularly a lesion in the nuchal region.
Clinical syndromes associated with capillary malformations include:
• Sturge–Weber syndrome
• Klippel–Trenaunay syndrome
• Cobb’s syndrome
• Capillary malformation – AVM syndrome.
The essential components of Sturge–Weber syndrome, also known as encephalotrigeminal angiomatosis, are:
• a unilateral facial port wine stain which includes that area of skin supplied by the ophthalmic branch of the trigeminal nerve (forehead and upper eyelid)
• an ipsilateral vascular abnormality of the leptomeninges
• an ipsilateral vascular abnormality of the choroid of the eye. 37
The port wine stain may be associated with only one or other of the other components of the syndrome. It is present at birth. The syndrome is often associated with port wine stains on other areas of the body. 38 Sometimes facial lesions are bilateral. Such cases are associated with a higher incidence of eye and brain abnormalities. 16 Glaucoma is the most common ophthalmologic abnormality found in this syndrome. Pyogenic granulomas and an arteriovenous malformation may develop in the lesions. 39
The pulsed dye laser is the treatment of choice for cutaneous lesions. The Sturge–Weber Foundation is an active support group for patients and families (website: www.sturge-weber.org).
The literature on Klippel–Trenaunay syndrome (OMIM 149000), also known as angio-osteohypertrophy, is somewhat confusing. However, the major elements are a port wine stain, usually on a limb, associated with varicose veins (present in 76–100% of cases), and limb overgrowth due to hypertrophy of bone and the associated soft tissues. 40 The hypertrophy is usually localized, rather than generalized hemihypertrophy, a feature associated with Wilms’ tumor. Only one case of Wilms’ tumor has been seen in this syndrome. 41
Since the original description, a number of other abnormalities have been described in conjunction with this syndrome. It should be distinguished from Parkes Weber syndrome (OMIM 608355), in which there is significant arteriovenous (high flow) shunting in the limb.38.42. and 43. Parkes Weber syndrome is due to a mutation in the RASA1 gene on chromosome 5q13.3.
The majority of cases of the Klippel–Trenaunay syndrome are sporadic, but familial cases have also been described.44. and 45. Capillary hemangiomas and venous malformations may be present. 46 The cutaneous capillary malformations may have a dermatomal distribution. 43 Lymphatic malformations are commonly present in this syndrome. They are usually present in patients with a ‘geographic’ cutaneous stain, rather than a blotchy pattern. 47 Various techniques such as MRI scanning and computed tomography can be used to assess the extent of the disease. 48 There are many incomplete forms of this syndrome, and in one large series a port wine stain was present in only 32% of cases. 49 Thrombosis may occur in the abnormal vascular channels, resulting in some cases in pulmonary emboli. 43
The genetic defect is currently unknown. A supernumerary ring chromosome 18 has been reported in this syndrome. 38
The capillary malformation – arteriovenous malformation syndrome (OMIM 608354) was described in 2003. 52 The capillary malformations are small pink-red macules distributed widely over the skin, and the high-flow malformations may be in the skin or deeper bone or muscle. It shares phenotypic features with Parkes Weber syndrome, which is not surprising, as it is allelic to it, with mutations in the RASA1 gene at 5q13.3. 53 It encodes the protein RAS p21. This genetic abnormality has also been found recently in capillary malformations without AVMs. 53
High-flow arteriovenous malformations have been found in over half the patients with the PTEN hamartoma–tumor syndrome, the term now used for the allelic disorders Bannayan–Riley–Ruvalcaba syndrome (OMIM 153480) and Cowden’s disease (OMIM 158350). 54 Intracranial venous anomalies are often present. 54 Excessive ectopic fat is often present in the cutaneous lesions. 54
Initially, there is a barely detectable dilatation of the thin-walled vessels of the superficial vascular plexus. Progressive ectasia occurs and there is obvious erythrocyte stasis. 55 There is no significant increase in thickness or number of vessels with age in typical superficial lesions. In some cases there is an underlying cavernous hemangioma, which may blend with the superficial lesion. Localized exaggeration of the vascular ectasia produces the roughened surface of the older lesions.27. and 56. In one case these late changes were associated with epithelial and mesenchymal hamartomatous changes. 57 Secondary angiomatous lesions and pyogenic granulomas may occur within the main lesion.58. and 59.
One study has shown that the lesions are produced by dilatations of postcapillary venules of the superficial horizontal plexus, with no evidence of new vessel formation, although most authorities now believe that there is an increase in the number of vessels. 17 The walls of the venules are thickened by basement membrane-like material and reticulin fibers. 60 Progressive dilatation does not appear to be related to decreased fibronectin or type IV collagen in the vessel walls. 61 A decreased nerve density has been demonstrated within affected areas, and it has been proposed that abnormal neural control of blood flow may be important in the pathogenesis of this lesion. 62 One immunohistochemical study showed that vessels were typical of capillaries, postcapillary venules, and small veins. 63 They are GLUT-1 negative.
Venous malformations are slow-flow vascular malformations that are present at birth. They are non-proliferating vascular birthmarks composed of anomalous ectatic venous channels. 17 It has been known in the past as cavernous hemangioma. Superficial venous malformations are blue or purple papules or nodules, which may have a grouped configuration. Deeper lesions may impart little color to the skin. 64 Multiple lesions are present in the ‘blue rubber bleb’ nevus syndrome, Maffucci’s syndrome, and the condition known as venous malformations with cutaneous and mucosal involvement (see below). Occasionally venous malformations have a unilateral dermatomal (zosteriform) distribution. 65 This may represent a form of mosaicism of a more generalized process. 66 Most lesions arise sporadically but familial cases with autosomal dominant inheritance occur. Unlike (capillary) hemangiomas, venous malformations lack a proliferative phase with thymidine incorporation and have little tendency to regress with time. Venous malformations are generally at a deeper level in the skin than hemangiomas. They may often involve muscle and be associated with painful thromboses. Coagulation disorders are often present in these patients as well;67.68. and 69. they may also complicate extensive venous/lymphatic malformations. 70
The distinction between venous malformations and hemangiomas is not quite as clear cut as the classification system of Mulliken et al would suggest.14. and 71. Dysmorphic syndromes occur with hemangiomas as well as with venous malformations and in some, such as Maffucci’s syndrome, overlap lesions may occur. Arteriovenous malformations may also be present, leading to fast-flow lesions. 72 Vascular malformations that may also involve lymphatic channels are seen in Proteus syndrome (OMIM 176920), a rare, sporadic overgrowth disorder that is probably caused by a somatic mosaicism lethal in the non-mosaic state.73. and 74. The gene has been mapped to 10q23.31. In addition to the vascular/lymphatic malformations, there are soft tissue swellings (lipomas), macrodactyly, macrocephaly, and epidermal nevi. 74 The diagnostic criteria for this syndrome have been listed in a recent review. 38
Cutaneous vascular malformations are rare in Gorham–Stout syndrome (OMIM 123880) in which diffuse skeletal muscular (venous, capillary, and lymphatic) abnormalities lead to osteolysis (disappearing bone disease).38.75. and 76. A recent study has suggested that the cutaneous lesions are lymphatic malformations. 77
Diffuse phlebectasia or Bockenheimer syndrome is characterized by a blue network of dilated veins. 78 This venous malformation involves more commonly upper, but also lower, extremities. It may involve muscles. 78 It is present at birth. 38
Cutaneous capillary-venous malformations which are hyperkeratotic are seen in a small percentage of persons with inherited cerebral capillary malformations (OMIM 116860). 79 The condition is due to mutations in the CCM-1 gene that encodes for the KRIT1 protein. It is involved in the Ras signaling pathway and is important in cerebral and cutaneous vascular development. 38
There are several, somewhat overlapping syndromes in which multiple venous malformations form the major abnormality. They include:
• the ‘blue rubber bleb’ nevus syndrome
• Maffucci’s syndrome
• venous malformations with cutaneous and mucosal involvement.
Before they are discussed, mention will be made of the classification used for GLUT-1-negative congenital vascular malformations of skin and soft tissue in a recent paper from Amsterdam. 80 It forms the likely basis for future classifications of these lesions:
• venous vascular malformation (veins of variable size, often with thick walls)
• lymphatic malformations (mixed lesions with a substantial component of dilated thin-walled lymphatic channels and likely to increase in number with the advent of lymphatic markers) 81
• deep arteriovenous malformations (tortuous arteries and veins often with fibrointimal thickening)
• superficial arteriovenous malformation/acral arteriovenous tumor (localized nodular tumor of thick-walled arteries and veins of the skin and subcutis).
This latter category is discussed in this chapter as arteriovenous hemangioma (see p. 901).
The ‘blue rubber bleb’ nevus syndrome (OMIM 112200) was first named by Bean. 82 It is a rare disorder, characterized by multiple compressible blue rubbery venous malformations of the skin and of the gastrointestinal tract, and occasionally other organs.83.84.85.86.87. and 88. Cortical blindness was present in one case. 89 Sinus pericranii is a rare association. 90 The skin lesions may be present at birth or develop in childhood. They do not regress. Some, but not all, are characteristically painful or tender on palpation. There may be associated hyperhidrosis in the region of the tumors. 91 Iron-deficiency anemia sometimes results from gastrointestinal hemorrhage. 92 Most cases are sporadic, but there is also evidence for an autosomal dominant mode of inheritance. 92 In one family males only were affected. 93
The cutaneous lesions are composed of irregular cavernous channels in the deep dermis and subcutis. There is smooth muscle in the vessel walls. In some cases vessels may be intimately related to dermal sweat glands. 91 They represent true venous malformations and not ‘cavernous hemangiomas’ as labeled in the past.
Destructive modalities, including carbon dioxide laser, sclerotherapy, and surgical excision, have been used to treat cutaneous lesions. 38
Maffucci’s syndrome is characterized by multiple vascular tumors of the skin and subcutis associated with multiple enchondromas of bone (OMIM 166000), particularly the long bones. The vascular tumors are usually venous malformations, but capillary hemangiomas, phlebectasias (dilated venules and veins), and lymphangiomas also occur. Spindle-cell hemangioendothelioma is also associated with this syndrome. Phlebolith-like bodies may develop in vascular channels. In two cases, only lymphangiomas were present. 94 The vascular tumors may be present at birth but most appear during early childhood; they do not regress.95. and 96. Mucous membrane and visceral hemangiomas have also been reported. 96
Enchondromatosis results in variable shortening and deformity of the extremities. Skeletal lesions are predominantly unilateral in approximately half the reported cases. 96 There is no anatomical relationship between the osseous and vascular components.
There is probably an equal sex incidence and no familial grouping of cases. 96 Chondrosarcomas develop in approximately 15–30% or more of cases, and other cancers have also been reported.96. and 97. These associated tumors were reviewed in 2005. 98 The condition appears to be a generalized disorder of mesenchymal tissues. 99
This disorder (VMCM – OMIM 600195) is a rare condition that was described by Boon et al in 1994. 100 It is characterized by ‘slow-flow’ venous malformations of the skin and oral mucosa. 101 In the original family, a few lesions were present at birth, but most appeared by puberty. It was not associated with gastrointestinal bleeding. It is caused by an activating mutation in the receptor tyrosine kinase (TIE2/TEK) gene on chromosome 9p21. 100
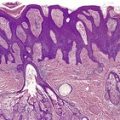
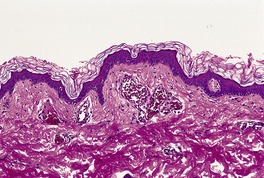
The malformations may be found at any level of the skin, but there is a tendency for them to occur in the deep dermis and subcutis. They consist of large dilated vascular channels lined by flat endothelium (Fig. 38.2). The walls of the vessels vary in thickness, but they are generally thin and fibrous. Some vessels may have smooth muscle in their walls and resemble dilated veins. Thrombosis may complicate these lesions. Calcification of the walls and phlebolith-like calcific bodies in the lumina may be found.
Venous malformation. There are dilated vascular channels in the deep dermis and subcutis. One of the channels contains a thrombus. (H & E)
The endothelium is flattened and the basal lamina duplicated, with interspersed collagen fibrils. 102
Cutis marmorata telangiectatica congenita (CMTC – OMIM 219250), also known as congenital generalized phlebectasia, is a rare sporadic condition, although over 300 cases have now been reported. 103 It is characterized by a persistent patterning of the skin by a reticulate network of dark violet-blue vessels (cutis marmorata), spider nevus-like telangiectases, and venous abnormalities variously described as phlebectasia, venous lakes, or venous hemangiomas.104.105.106.107.108. and 109. Lesions are usually located on the trunk or extremities where they may be localized or generalized. They are often unilateral and have a mosaic pattern.110. and 111. The skin changes appear in the neonatal period, and in many cases have a tendency to improve with time, often in the first two years.112. and 113. Lesions sometimes persist but there are no features predictive of this outcome. 113 In addition to cutaneous atrophy and ulceration or hypertrophy of the involved tissues, a range of other associated vascular and skeletal abnormalities has been described.106.114.115.116.117.118.119. and 120. Hypospadias has been present in several cases. 121 There is no involvement of internal organs by the vascular abnormalities. 104
Widespread CMTC can also be seen in the Adams–Oliver syndrome (OMIM 100300). The coexistence of CMTC and extensive, large Mongolian spots has been classified as type V phacomatosis pigmentovascularis.13. and 103. The syndrome characterized by macrocephaly and CMTC (OMIM 602501) is now regarded as a type of capillary malformation and not related to CMTC. 122 The name macrocephaly–capillary malformation has been suggested. 123
The etiology is uncertain. The condition may be a genodermatosis with autosomal dominant inheritance and reduced penetrance of the gene. It has been suggested that it may represent a functional disturbance resulting from reduced α-adrenergic innervation of cutaneous terminal vessels. 124 A translocation was present in one case. 122
Various changes, including dilated capillaries and veins in the dermis and subcutis, have been described in this condition.104. and 125. A recent review of all cases of CMTC in the English-language literature found that the most common finding was dilatation of capillaries and veins in the dermis. 126 In two cases, and in the additional case reported, there was a proliferation of vascular channels. The implications of this proliferation on the course of the disease were not clear. 126 Focal acantholytic dyskeratosis has been reported as an incidental, but closely associated abnormality in one patient. 127
The glomulovenous malformation (GVM – OMIM 138000) was known in the past as a glomangioma, and regarded as a variant of glomus tumor.128.129.130.131.132.133.134. and 135. Clinically they may simulate a venous malformation but they are histologically distinct. 136 They accounted for 5% of venous anomalies in one vascular unit. 137 A majority of cases are familial with autosomal dominant inheritance but a significant number are sporadic. As relatives may have inconspicuous lesions, they may be missed. Familial GVM is due to a mutation in the glomulin gene on chromosome 1p21–p22.138. and 139. Numerous mutations have been described; they appear to be loss of function mutations.101. and 140.
Typically GVMs are blue or violaceous lesions which may occur at any site or rarely the mucosa. They mostly occur on the extremities, but some occur on the trunk.137. and 141. Facial lesions are rare.142. and 143. GVMs are usually solitary, but multiple lesions are not uncommon. Lesions limited to one side of the body have been reported. 144 They may have a segmental distribution reflecting mosaicism. 145 They usually exhibit so-called ‘type 2 mosaicism’, which is characterized by conspicuous lesions locally superimposed on ‘milder’, disseminated lesions.146. and 147. Clinically the lesions may be macular, nodular or thickened, sometimes disfiguring plaques. 101 In this latter group are the congenital, disseminated, plaque-type variants which are often mistaken for venous malformations.148. and 149. They may extend deeply and involve muscle. 136 Smaller lesions may have hypertrichosis. 150 A case that clinically mimicked granuloma annulare has been reported. 151 They are usually painless, unless palpated.
The GVM presents at an earlier age and is less common than its histological simulant, the glomus tumor. Unlike blue rubber bleb nevus which it may clinically resemble, there is no gastric involvement in GVM. 152
Surgical excision is usually used for isolated lesions but vascular lasers and carbon dioxide lasers can be used if lesions are superficial. Sclerotherapy is another treatment option. 153 Large, disfiguring lesions remain difficult to treat. 101
Glomulovenous malformations have more prominent vessels and less conspicuous glomus cells than the glomus tumor proper. GVMs are poorly circumscribed and unencapsulated and consist of irregular ectatic vascular channels irregularly surrounded by small numbers of glomus cells (Fig. 38.3). Glomus cells may be so sparse that the lesions can be difficult to distinguish from conventional hemangiomas. Glomangiomatosis refers to diffuse angiomatosis with a histological excess of glomus cells. 154 The exact nosological position of the glomangiomyoma is unknown. Familial generalized variants have been described. 155 Their histology suggests a variant of GVM with smooth muscle cells in the wall.155.156. and 157. A variant derived from glomus tumors may also exist.
(A) Glomulovenous malformation. (B) Large vascular channels are surrounded by a few layers of glomus cells. (H & E)
The term ‘cystic lymphatic malformation’ has been suggested as a more appropriate designation than lymphangioma for localized malformations of lymphatics. 158 Most are present at birth or arise in infancy or early childhood.159. and 160. It has been suggested that lymphangiomas represent sequestrated lymphatic vessels that have failed to link up with the rest of the lymphatic system or with the venous system during embryological development.161. and 162. Histologically identical lesions arising because of acquired obstruction of lymphatics, often in association with lymphedema, are classified as lymphangiectases (see p. 897). There have been several attempts to classify lymphangiomas: the classification of Flanagan and Helwig159 divided them into superficial and deep types. Lymphangiomatosis is an additional category. Mulliken and colleagues, 71 who prefer the term ‘cystic lymphatic malformation’, have subdivided them into microcystic (lymphangiomas, verrucous hemangiomas, and angiokeratoma circumscriptum) and macrocystic (cystic hygromas and cavernous lymphangiomas). Combined macrocystic and microcystic lesions may occur.
The superficial lymphangioma is also known as superficial microcystic lymphatic malformation and lymphangioma circumscriptum. Although these lesions may occur on almost any part of the body, they are most common on the proximal parts of the limbs and in the limb girdle regions.160. and 163. It rarely involves the vulva. 164 Penile lesions are also rare.165.166. and 167. Both vulval and penile lesions may mimic venereal lesions. Secondary infection is a complication of lymphangioma of the penis, and of other sites. 168 There are typically multiple scattered or grouped translucent vesicles and papulovesicles in an area of skin; single small lesions composed of a group of vessels also occur.160.169. and 170. The lesions have been likened to frogspawn. Secondary hemorrhage and thrombus formation in vesicles may produce red or purple coloration in the lesions. Some lesions have a warty appearance owing to epidermal hyperplasia and hyperkeratosis. 171 There may be an underlying deep lymphangioma or other abnormality of lymphatic drainage, resulting in lymphedema and enlargement of the limb.172. and 173. The underlying muscle is sometimes involved. 174 Most of the lesions are present at birth or develop in early infancy or childhood. Occasionally the lesions appear first in adult life: 175 this is most common in the small localized form. 160 In the typical extensive lesion the superficial vessels communicate through deep vessels with large closed lymphatic cisterns in the subcutaneous or deeper tissues; 176 the superficial ectatic lymphatic vessels appear to result from raised pressure in these cisterns.161. and 177. Magnetic resonance imaging has been used to demonstrate the full extent of these lesions. 178 This underlying abnormality may explain the tendency of the lesions to recur after superficial excision. Lesions may enlarge and spread with time, and they may persist indefinitely. The development of lymphangiosarcoma has been reported in an area of superficial lymphangioma treated with radiotherapy. 179 Squamous cell carcinoma may also develop in these lesions. 180
Treatment is usually by surgical excision but non-surgical techniques such as cryotherapy, carbon dioxide laser, electrodesiccation, and bleomycin have been used. Results with these non-surgical therapies have not been satisfactory.164. and 169. OK-432, a new sclerosant, produced satisfactory results when used on an extensive lesion of the vulva that was inoperable. 164 Hypertonic saline sclerotherapy can also be used. 175 The pulsed dye laser has also given good results. 181
The epidermis is elevated above the general level of the skin by solitary or grouped ectatic lymphatics located in the papillary dermis (Fig. 38.4). This accounts for the raised vesicles seen clinically. These channels abut closely on the overlying epidermis and are thin walled, consisting predominantly of an endothelial lining. The vessels may contain eosinophilic proteinaceous lymph or blood or thrombus, and occasionally foamy histiocytes or multinucleate giant cells. 161 Scattered lymphoid cells are sometimes seen in the dermis. There is atrophy of the epidermis directly over the vessels, with elongation of the rete ridges such that the vessels may appear to be intraepidermal, the picture resembling that of angiokeratoma. Deep irregular lymphatics are sometimes seen beneath the surface vessels in the dermis and subcutis, particularly in the extensive lesions. 161 The presence of these subcutaneous lymphatic cisterns with small amounts of smooth muscle in the wall are features that distinguish superficial lymphangioma (lymphangioma circumscriptum) from acquired lymphangiectasia, in which they are absent. Both share saccular dilatation of the superficial lymphatic channels. 182
Superficial lymphangioma. There are dilated lymphatic channels in the upper dermis. (H & E)
The channels are highlighted by the new lymphatic endothelial marker D2-40.
Deep lymphangioma (macrocystic lymphatic malformation) includes the lesions known as lymphangioma cavernosum (cavernous lymphangioma) and cystic hygroma. 159 The term ‘cystic hygroma’ has generally been used for large deep lymphangiomas in the neck or axilla which consist of single or multiloculate fluid-filled cavities. Cystic hygromas of the posterior triangle of the neck are associated with hydrops fetalis and fetal death, as well as obstructed labor. 183 There is an association with the 45,XO karyotype (Turner’s syndrome), other congenital malformation syndromes, and several varieties of chromosomal aneuploidy. 183 A coexisting capillary malformation (nevus flammeus) has been reported overlying a cystic hygroma. 184 It is thought that deep lymphangiomas represent failed connection between the jugular lymph sac and the internal jugular vein.162. and 183. There is no clear-cut distinction between other deep lymphangiomas and classic cystic hygromas, 159 and it has been suggested that the appearance of the tumor is determined by the site and nature of the tissues in which it arises. 185 Deep lymphangiomas present as soft swellings in the skin and subcutaneous tissues. Progressive extension into deeper structures, such as muscle, is said to be an unfavorable sign. 159 The overlying epidermis is normal except in those cases in which there is an associated superficial lymphangioma. When cut across, these tumors vary from a spongy mass of small vascular spaces to large and ‘multicystic’. Most are present at birth or arise in the first few years of life.159. and 185. Lymphangioma scroti is the term used for deep and cavernous lesions of the scrotum. The adjacent inner thighs are often involved as well. 186
The histological picture of these tumors is inconstant. There are irregular dilated lymphatic channels of variable size in the dermis, subcutis, and deeper tissues. These structures vary from an endothelium-lined channel with no obvious supporting stroma to vessels with thick fibromuscular walls. The intervening dermis or subcutis may be unaltered, or there may be loose or compact fibrous stroma. 159 Blood may be present in some channels. In larger lesions collections of lymphocytes are sometimes present in the stroma and cause the endothelium to bulge into the vascular lumen. Lymphatic channels can be confirmed using the D2-40 antibody.
Lymphangiomatosis, a rare disorder occurring mainly in children, may have skin lesions, although it primarily involves bones, parenchymal organs, and soft tissue.187.188. and 189. The prognosis in this form is related to the extent of the disease. A variant limited to bone, soft tissue, and skin, and with a good prognosis, has been described. 187
In lymphangiomatosis involving skin, the dermis and subcutis are infiltrated by dilated lymphatic channels which dissect dermal collagen and surround pre-existing structures, a pattern seen in well-differentiated angiosarcoma. Vessels are lined by a single layer of flat endothelium which stains positively for factor VIII-related antigen, Ulex europaeus-1 antigen and, variably, with CD31 and CD34. 187 Thrombomodulin is a more recent marker, which is said to be specific for lymphatic endothelium. 190 Podoplanin, detected by the antibody D2-40, is now the most specific marker of lymphatic endothelium.
Verrucous hemangioma is a vascular malformation which appears to arise at birth or in childhood, and enlarges and spreads in later life.191.192. and 193. Lesions occur predominantly on the legs, and consist of bluish-red soft papules, plaques, and nodules which become wart-like as the patient ages, or following trauma. 194 Satellite nodules may develop. Lesions may measure up to 8 cm in diameter. 195 Recurrence is frequent after removal of the lesions because of involvement of the deeper tissues. An eruptive form with multiple disseminated cutaneous lesions and a linear variant have also been described.196. and 197.
Fully evolved lesions consist of dermal and subcutaneous foci of small and large vessels, with overlying verrucous hyperplasia of the epidermis. There is irregular papillomatosis with acanthosis and hyperkeratosis. Angiokeratomatous areas may be present; however, unlike angiokeratomas, which are ectasias of superficial vessels (see p. 896), verrucous hemangiomas involve deeper levels.191. and 192. Verrucous hemangiomas have been included in some classifications with the microcystic lymphatic malformations. 158
Chronic ulceration of the vermilion border of the lips is sometimes associated with the presence of an artery of abnormally large caliber running in a very superficial location beneath the squamous epithelium. These vessels have been called ‘caliber-persistent arteries’ because of the failure of normal narrowing of the lumen as the vessel approaches the mucosal surface.198. and 199. Some cases have been misdiagnosed clinically as squamous cell carcinoma. 200 The vessel may show fibroelastotic intimal thickening. 201 Multiple sections may be necessary to demonstrate the vessels. 198 Brisk hemorrhage may follow the inadvertent biopsy of such a lesion. 202 The diagnosis can be made by Doppler ultrasonography. 203
In this group, the vascular channels of which the lesion is composed are predominantly pre-existing blood vessels which have undergone dilatation.
In hereditary hemorrhagic telangiectasia (HHT), also known as Osler–Rendu–Weber disease (OMIM 187300), multiple punctate telangiectases occur in the skin and mucous membranes. The respiratory tract, gastrointestinal tract, and urinary tract may be involved. 204 The nasal mucosa, lips, mouth, and face are frequently affected, and epistaxis is the most common presenting symptom. Although lesions may be present in childhood, they do not usually appear until puberty. The number and size of the lesions increase with advancing age, and with pregnancy. The lesions may clinically mimic the telangiectases seen in the CREST syndrome. 205 Fibrovascular abnormalities of the liver, cerebral arteriovenous fistulae, and pulmonary arteriovenous fistulae are associated abnormalities.206.207. and 208. Vitiligo and autoimmune thyroiditis have been reported but they may have been coincidental. 209 Cerebral abscesses are occasionally a complication. 210
The incidence of the disease is approximately 1–2/10 000. 211 Inheritance is by an autosomal dominant trait. HHT is caused by mutations in either the endoglin (ENG) gene on chromosome 9q34 (HHT1), or the activin A receptor type II-like kinase-1 (ACVRL1 or ALK1) gene on chromosome 12q13 (HHT2).14.211. and 212. Both are transmembrane receptors for transforming growth factor β (TGF-β), which plays a significant role in angiogenesis. Mutations in the SMAD4 gene can cause a syndrome consisting of both HHT and juvenile polyposis. 211 This mutation can also occur in uncomplicated HHT. Two further loci linked to chromosome 5 (HHT3) and chromosome 7 (HHT4) have been detected. 211 Hundreds of different mutations in ENG and ALK1 have been reported including the deletion of the entire ALK1 gene. 211
Telangiectatic macules and diffuse erythematous areas likewise composed of a fine meshwork of ectatic vessels are seen in this condition, which occurs most frequently in women, appearing in early childhood. 215 Lesions appear first on the lower extremities and spread gradually to involve the trunk and arms. 216 Its diagnosis depends on the exclusion of other primary and secondary telangiectasias. 217 Gastrointestinal bleeding from an associated ‘watermelon’ stomach has been reported. 218 Conjunctival involvement is rare. 219
Hereditary benign telangiectasia (OMIM 187260) is inherited as an autosomal dominant trait. The disorder has been mapped in one family to the CMC1 locus on 5q14, but this gene has been specifically excluded in one large family. 220 A non-hereditary variant has also been reported. 221 It is characterized by widespread cutaneous telangiectases, which commonly appear in childhood. 222 In some of the pedigrees, the telangiectases are limited to sun-exposed areas, but in others the lesions are widespread. 220 They may be separate entities. Congenital lesions also occur. 223 Many lesions on the face may resemble spider nevi. The lesions are more prominent during pregnancy. There are no systemic vascular lesions associated with this form of telangiectasia.224. and 225. The lesions are characterized by dilatation of the horizontal subpapillary venous plexus. 223
Unilateral nevoid telangiectasia is also known as unilateral dermatomal superficial telangiectasia. The telangiectases have a dermatomal distribution and particularly involve the trigeminal and the third and fourth cervical and adjacent dermatomes. 226 The lesions may be present at birth227 or they may be acquired at times of physiological or pathological estrogen excess, including puberty and pregnancy in females, and chronic liver disease.228. and 229. The condition has been reported in association with metastatic carcinoid tumor, 230 and also in two males with hepatitis C but without evidence of cirrhosis or abnormal hormone levels. 231 It has also developed in a young man after chemotherapy. 232 Increased numbers of receptors for estrogen and progesterone have been reported in the lesional area compared to normal skin. 233
Pulsed dye laser is a useful modality for treating this condition, although reversible pigmentary disturbances often occur in persons of color. 234
Ataxia-telangiectasia (Louis–Bar syndrome – OMIM 208900) is an inherited, progressive neurological disorder resulting from cerebellar degeneration. 235 Cutaneous telangiectases are a constant but clinically unimportant part of this syndrome.236. and 237. Telangiectases appear in childhood in the bulbar conjunctiva and in the skin of the face, pinnae, neck, and limbs. They arise from the superficial vascular plexus. These changes are followed by progressive cerebellar ataxia from cerebellar cortical atrophy. Other skin changes have been described including granulomas, segmental pigmentation, progeric changes, and seborrheic dermatitis.238.239.240.241. and 242. Profound dysfunction of both cell-mediated and humoral immunity results in decreased resistance to viruses and recurrent sinus and pulmonary infections. 235 Thymic aplasia or hypoplasia and a decrease in the lymphoid tissue in lymph nodes, spleen and elsewhere are associated with deficiency of IgA, IgE and IgG, and abnormal function of T lymphocytes. An increased sensitivity to ionizing radiation and a markedly increased risk of developing cancers, particularly lymphomas and leukemias, are other facets of this syndrome. Affected persons have a shortened life expectancy, with death usually occurring in the second or third decade of life.
Inheritance is autosomal recessive with variable penetrance. Mutations occur in the ataxia–telangiectasia mutated (ATM) gene on chromosome 11q22.3. ‘Homozygotes’ (strictly speaking, many are compound heterozygotes because each of their chromosomes 11 carries a different mutation) express the full syndrome. Heterozygotes have an elevated risk of cancer, especially female breast cancer, of ischemic heart disease, and of early mortality. 243 The ATM protein has several important roles. It may be associated with dysregulation of the immunoglobulin gene superfamily, and has a role in signal transduction and cellular responses to DNA damage. 38 Accelerated telomere loss may be responsible for other manifestations.
One or more ‘spider’ angiomas (nevi) are present in 10–15% of normal adults. 244 The face, neck, upper part of the trunk, and arms are the regions usually involved; it is very uncommon for lesions to occur below the level of the umbilicus. There is a higher incidence in pregnant women, and in patients with chronic liver disease. Lesions may regress following the pregnancy. In children, ‘spider’ nevi tend to arise on the hands and fingers. 245
‘Spider’ angiomas consist of a central punctum within a generally circular area of erythema. Fine branching vessels or ‘legs’ radiate from the punctum.
Sclerotherapy is often used to treat these lesions. 246
These lesions are rarely biopsied or excised. They consist of a central ascending spiral thick-walled arteriole which ends in a thin-walled ampulla just beneath the epidermis (Fig. 38.6). From the ampulla, thin-walled branching channels radiate peripherally in the papillary dermis. Glomus cells have been described in the wall of the central arteriole. 244
‘Spider’ angioma (nevus). A deep dermal vessel has given rise to a vertically oriented vessel leading into a superficial ampulla. There are thin-walled vessels in the upper dermis. (H & E)
Venous lakes are common vascular ectasias. They appear as soft, dark-blue, often multiple, papules a few millimeters in diameter, which occur on the ears, face, lips, or neck of the elderly.247. and 248. Minor trauma to the lesions may produce persistent bleeding.
The lesions reported as capillary aneurysms probably represent venous lakes. 249 The clinical similarity of these lesions to malignant melanomas has been highlighted, particularly if thrombosis of the vascular lumen occurs.
Many venous lakes are removed by punch or surgical excision. They may also be treated by cryosurgery and electrodesiccation. Lasers, particularly the pulsed dye laser, are also effective. 250
Usually only a single large dilated vascular channel is present, in the upper dermis (Fig. 38.7). It has a very thin fibrous wall and a flat endothelial lining. A thrombus is sometimes present in the lumen or part thereof. These lesions appear to represent a dilated segment of a vein or venule. 247
Venous lake. There is a solitary large vascular channel in the upper dermis. (H & E)
The angiokeratoma is characterized by ectasia of superficial dermal blood vessels, with associated epidermal changes. Dermoscopy is helpful in improving the diagnostic accuracy of solitary angiokeratomas. The presence of dark lacunae on dermoscopy has a specificity of 99%. 251 Five clinical variants have been recognized: all have similar histopathological features. 252 The variants are discussed below:
1. The Mibelli type develops in childhood and adolescence, with warty lesions over the bony prominences of the hands, feet, elbows, and knees.252. and 253. The term ‘hyperkeratotic vascular stain’ has been used for congenital lesions. 254 It is more common in females and it may be associated with pernio. A thrombosed solitary lesion may mimic malignant melanoma. 255 Laser treatment is a common method of treatment. 254
2. The Fordyce (scrotal) type arises as early as the second and third decades, but is seen most commonly in elderly men.256. and 257. The penis, upper part of the thighs, and lower part of the abdomen may also be involved. 258 The lesions are single or multiple, red to black papules, occurring along the course of the superficial scrotal vessels. Scrotal angiokeratomas may be associated with varicoceles, 259 inguinal hernias, an oral angiokeratoma, 260 and thrombophlebitis. 256 Spontaneous regression has been reported following the surgical treatment of an associated varicocele. 261 An equivalent lesion occurs on the vulva in young adult females. Increased venous pressure associated with pregnancy, vulval varicosities, or hemorrhoids has been implicated in the pathogenesis of the vulval lesions. 262 An association with the contraceptive pill has also been suggested. 263
3. Solitary and multiple types occur on any part of the body, but the lower extremities are most commonly affected. In one series the lesions were solitary in 83% of cases and multiple in 17%. 252 A zosteriform distribution has been described. 264 Angiokeratomas have been reported in a patient with juvenile dermatomyositis. 265 It was postulated that the lesions developed as a compensatory response to the obliterative angiopathy of the dermatomyositis. They have also been reported in association with cerebral cavernous malformations. 79
4. Angiokeratoma circumscriptum is the least common variant. 266 It consists either of a plaque composed of small discrete papules, or of variable hyperkeratotic papules and nodules with a tendency to confluence.267. and 268. A linear lesion has been described. 269 Lesions are almost always unilateral, and they occur predominantly on the leg, trunk, or arm. The neck and the penis are rare sites.269. and 270. Lesions develop in infancy or childhood, predominantly in females.
5. Angiokeratoma corporis diffusum consists of multiple papules, frequently in clusters, and usually in a bathing-trunk distribution. Originally thought to be synonymous with Anderson–Fabry (Fabry) disease (OMIM 301500), it is now evident that this vascular lesion may occur in association with other enzyme disorders and also in people with normal enzyme activity (see p. 485).271.272.273.274. and 275. Anderson–Fabry disease is an X-linked recessive disorder characterized by a deficiency of the lysosomal enzyme α-galactosidase A and the accumulation of the neutral glycolipid ceramide trihexidose in lysosomes in many types of cell. 276 Homozygous male patients generally, but not always, develop the lesions of the disease. 277 The skin lesions are usually present by adult life; the changes are often absent or slight in childhood. 278 An eruptive form is exceedingly rare. 279 Females with the genetic abnormality may also develop the lesion, but this occurs in less than 25% of cases. 278 Other enzyme deficiencies associated with angiokeratoma corporis diffusum include α-l-fucosidase deficiency (fucosidosis – OMIM 230000), β-galactosidase deficiency (OMIM 256540), α-N-acetylgalactosaminidase deficiency (Kanzaki disease – OMIM 609242),280. and 281. β-mannosidase deficiency (OMIM 248510),282. and 283. aspartylglucosaminidase deficiency (OMIM 208400), 284 and neuraminidase deficiency (OMIM 256550).285.286. and 287. A dominantly inherited form, associated with arteriovenous fistulae but no metabolic disorder, has been reported. 288
Treatment will depend on the number, size, and extent of the lesions. Surgical excision is an option for smaller lesions. Cryotherapy and electrocautery can be used but recurrence and/or scarring may occur with these two procedures. Laser therapy with CO2, argon, or pulsed dye laser may also be used.
In angiokeratomas there is marked dilatation of papillary dermal vessels to form large cavernous channels. There is associated irregular acanthosis of the epidermis with elongation of the rete ridges which partially or completely enclose the vascular channels (Fig. 38.8). A collarette may be formed at the margins of the lesions and there may be thrombosis of the vessels. The surface epidermis may show varying degrees of hyperkeratosis. The occurrence of a deep dermal hemangioma has been reported in association with angiokeratoma circumscriptum.267. and 289. This combination may represent a verrucous hemangioma. In patients with Anderson–Fabry disease there is vacuolation of smooth muscle in arterioles and arteries and in the arrectores pilorum. Frozen sections of lesions may show PAS-positive and Sudan-black-positive granules in endothelial cells, pericytes, arrectores pilorum, and eccrine sweat glands. The vessels do not usually express CD34. 289
Angiokeratoma. The elongated rete ridges partly surround the vascular channels in the papillary dermis. (H & E)
Examination of lesions or of normal skin from patients with Anderson–Fabry disease shows electron-dense lipid bodies in the cytoplasm; they are either membrane bound or free in the endothelial cells, pericytes, smooth muscle cells, and fibroblasts. These bodies may show a characteristic lamellar pattern. They are not seen in the other types of angiokeratoma or in the lesions in cases of angiokeratoma corporis diffusum with normal enzyme activities. 271
The ultrastructure of the vessels in scrotal angiokeratomas and in Anderson–Fabry disease is similar to that of the small valve-containing collecting veins at the junction of the dermis and subcutaneous fat. 60 The possible role of raised intravenous pressure in the formation of scrotal and vulval angiokeratomas has been mentioned. It has been suggested that the lesions in Anderson–Fabry disease may follow weakening of vessel walls and subsequent dilatation, the result of lysosomal storage of lipid and consequent cellular damage. 290
Numerous skin disorders are associated with telangiectases. These include such disparate conditions as collagen vascular disease, cutaneous mastocytosis (OMIM 248910), and chronic graft-versus-host disease. Telangiectases may appear following trauma, including repetitive injury from the use of a keyboard (‘computer palms’), 291 or be associated with skin damage due to solar and other forms of radiation (Fig. 38.9).
Secondary telangiectasia occurring in sun-damaged skin. (H & E)
Several syndromes are associated with cutaneous telangiectases, such as Cockayne’s syndrome (OMIM 216400), Bloom’s syndrome (congenital telangiectatic erythema – OMIM 210900), and Rothmund–Thomson syndrome (poikiloderma congenitale – OMIM 268400). 236 Bloom’s syndrome is a rare autosomal recessive genodermatosis consisting of photosensitivity, telangiectases, growth retardation, and an increased incidence of malignancies. 292 The defect has been mapped to chromosome 15q26.1.
A unique case, characterized by telangiectasia with marked collagen deposition around the basal lamina of the vessels resembling amyloid, has been reported as cutaneous collagenous vasculopathy.293. and 294.
The nature of the two cases with dome-shaped papules resembling hemangiomas that eventually resolved leaving pigmented areas is unknown. 295 The eruptions were recurrent, but took longer to heal than the lesions of eruptive pseudoangiomatosis (see p. 908). The lesions were regarded as being telangiectatic in type. 295
Iatrogenic telangiectases have been produced by lithium, isotretinoin, and interferon-α. Photodistributed telangiectases have been reported following the use of cefotaxime (a cephalosporin), the calcium channel blockers nifedipine, felodipine and amlodipine,296.297.298.299. and 300. and the serotonin-norepinephrine reuptake inhibitor venlafaxine. 301
Reticulate telangiectatic erythema is an asymptomatic telangiectatic and erythematous plaque that develops some time after the implantation of a device, usually a pacemaker or cardiac defibrillator.302. and 303. It has also been reported overlying a hip prosthesis. 304 The cases reported as sternal erythema, a distinctive thoracic surgical wound eruption that developed after coronary bypass grafting, 305 appear to be a related entity. 306 So too are the erythematous patches, sometimes quite large, that develop on the breast following a surgical procedure. Patch testing in all cases has been negative, but it is still postulated that the lesion represents a reaction to implanted hardware, which may simply be sternal wires in the cases overlying bypass surgery wounds. It has also been suggested that it may be an anatomic variant of costal fringe (see below).
All cases have shown variable, but usually mild epidermal atrophy and capillary telangiectasia in the upper dermis. The infiltrate, which is perivascular and lymphocytic, is usually sparse, but it was a little heavier in the case overlying the hip prosthesis. 304
Costal fringe is an acquired lesion in elderly men and, less frequently, elderly women. It consists of a band-like pattern of telangiectases across the anterior aspect of the thorax, usually near the costal margin. It is uncommon in young adults. 307 The telangiectases represent dilated postcapillary venules of the superficial vascular plexus. 308
Lesions that are clinically and histologically similar to superficial lymphangiomas (cystic lymphatic malformations) may develop in areas of skin affected by obstruction or destruction of the lymphatic drainage.171. and 309. The interference with the lymphatics may result from radiotherapy or surgery,310.311. and 312. and has been described in the chest and arm following radical mastectomy and radiotherapy,313.314.315. and 316. in the penis and scrotum following surgery for a sacrococcygeal tumor, 317 and on the vulva and on the thigh following surgery and radiotherapy for carcinoma of the cervix.318.319.320.321. and 322. Chylous reflux may rarely present with milia-like lymphangiectasia of the thighs323 or scrotum. 324 It has also occurred following genital mutilation performed as a cultural practice. 325
Lymphangiectasia has also developed on the abdomen in a patient with cirrhotic ascites and previous liver transplantation. Peritoneal mesothelial cells refluxed into the cutaneous lesions. 326
Cutaneous lymphangiectases have also been reported in association with severe photoaging and topical corticosteroid application. 327 Facial lymphangiectases are a rare complication of porphyria. 328
Lymphedema, a chronic condition characterized by swelling of one or more limbs or other parts of the body, is due to a defect in lymph transport. It contributes to local infections, such as cellulitis in the affected limb.329. and 330. Severe lymphedema of the extremities, genitalia, and face is associated with intestinal lymphangiectasia in Hennekam syndrome (OMIM 235510). 331 Lymphangiectasia is a feature leading to the lymphedema. Learning disability is also a feature of this syndrome.
This group of vascular tumors includes a variety of lesions in which there is a hyperplasia or a benign neoplastic proliferation of blood vessels of different types. 332
Infantile hemangioma (hemangioma of infancy), a benign proliferation of blood vessels, has also been called in the past, ‘strawberry’ nevus, infantile capillary hemangioma, and benign infantile hemangioendothelioma.333. and 334. They are the most common tumors of infancy, with an incidence in the newborn population of approximately 2%. They are especially common among infants born prematurely.335. and 336. They can affect up to 10% of white children within the first year of life. 20 A recent systematic review of the medical literature led the authors to conclude that 10% was probably an overestimation and that the incidence was closer to 4–5%. 337
In most series there is a female preponderance of cases.338. and 339. Familial cases are rare. 340 Some of these cases (OMIM 602089) have autosomal dominant inheritance with genes mapped to the 5q region. One or more lesions may be present on any part of the body, but the head, neck, and trunk are the most commonly affected sites. 341 In a recent series from New York University, 379 hemangiomas from 316 patients were studied. Of these hemangiomas 57% occurred on the head and neck, 19% on the trunk, 14% on the extremities and nearly 10% on the perineum. 342 Of the 216 hemangiomas on the head and neck, 190 were focal and 26 were segmental. The parotid gland and overlying skin may be involved, this being the most common tumor of the parotid gland in children. 343 Lesions are often not visible at birth but appear in the first few weeks of life. Rarely, a faint erythematous macule or an area of pallor with telangiectasia is present at birth. 344 The lesions evolve and enlarge over a period of months to become raised and bright red in color, with a smooth or irregular surface. Sometimes they mimic a bruise. 345 A small number are abortive and show minimal growth. 346
Other hemangiomas which are rapidly involuting or non-involuting occur.347. and 348. Interestingly, these lesions are usually both GLUT-1 negative.347. and 349. The rapidly involuting congenital hemangiomas (RICH) may present with thrombocytopenia, low fibrinogen, and elevated fibrin degradation products. It is transient and dissimilar to the Kasabach–Merritt phenomenon. 350 RICHs are fully developed at birth and involute rapidly during the first month of life. They are sometimes quite large and exophytic. They seldom require treatment. 350 The non-involuting capillary hemangiomas (NICH) grow proportionately with the child. 347 The vessel walls in these tumors are often thicker and the endothelium may have a hobnail appearance. 351
Most infantile hemangiomas have reached maximum size by the age of 3–6 months.341. and 344. A brief plateau period then ensues. Total or partial regression then occurs in the majority of lesions and is usually maximal by 5–7 years of age.339. and 341. This regression appears to be mediated by apoptosis accompanied by reduced proliferation of cells. 352 Most cases therefore require no surgical intervention,344. and 353. although newer laser techniques and even cryosurgery are being used with success.344.353. and 354. Pulsed dye laser treatment should be confined to superficial, ulcerating variants, and to residual telangiectasia of involuted hemangiomas only. 20 It has been suggested that embolization is an important mechanism in the laser destruction of cutaneous lesions. 355 Periorbital lesions may warrant active therapy because of the association of visual complications.344.356.357. and 358. Complete involution is less likely to occur where there is a deep cavernous component or more complex vascular malformation.359.360. and 361.
Ulceration, which is the most common complication of hemangiomas, occurs in 5–21% of cases. Treatment is mandatory, as significant pain, potential for bleeding and infection, and increased risk of scarring exist. 342 Treatment of such cases usually involves a combination of local wound care, barrier creams, topical antibiotics, and systemic or intralesional corticosteroids.342. and 362. Pulsed dye laser has also been used (see above).
A locus for an autosomal dominant predisposition to hemangiomas has been identified on chromosome 5q. A similar locus may be involved in the formation of sporadic hemangiomas.363. and 364. Molecular studies of vascular tumors have found abnormalities of different tumor suppressor genes located on various chromosomes. Multiple hemangiomas were reported recently in a patient with a t(3q;4p) translocation. 365
It has been suggested that the incidence of infantile hemangiomas is increased following chorionic villus sampling during pregnancy. 366 Other ischemic placental injuries are also associated with an increase in infantile hemangiomas. In a comparative study of 13 neonates who developed hemangiomas with 13 who did not, there were major differences in placental morphology in the two groups. Gross lesions with disturbances of the utero-placental circulation were found in all 13 cases with infantile hemangiomas while only three cases in the control group had any placental changes at all, and in two they were mild. 367 Interestingly, these hemangiomas share a phenotype with the placental microvasculature,368. and 369. but a recent study has shown that the endothelial cells originate from the child and not from the mother. 370
Multiple disseminated cutaneous hemangiomas are sometimes associated with multiple visceral hemangiomas (diffuse neonatal hemangiomatosis). This is usually a fatal disorder (see below). Solitary, segmental hemangiomas of the skin can also be associated with visceral hemangiomatosis.371. and 372. They are frequently associated with other developmental abnormalities as well.373.374.375. and 376. Ulceration is more likely in these segmental (facial) lesions when compared to the focal, tumor-like type. 377 Systemic corticosteroids prevent the further growth of these lesions and reduce the pain. 378 Care must be exercised with corticosteroids in infants as complications of therapy are common. 379
The simultaneous occurrence of infantile hemangiomas and congenital melanocytic nevi has been reported. The intimate relationship of some of these lesions led the authors to speculate that their concurrence was not coincidental. 380 Congenital pseudoclubbing of the fingernail may accompany subungual hemangioma. 381
Other work has shown that the endothelial cells of these hemangiomas have the cell morphology and protein expression of embryonic endothelial cells, indicating a dysfunction in maturation of the endothelial cells in these lesions. 382 Another study has shown that the endothelial cells of these hemangiomas have high levels of erythrocyte-type glucose transporter protein (GLUT-1), but this is not found in vascular malformations. 383 Cellular adhesion molecules are also involved in the formation and maturation of hemangiomas. ICAM-3 appears to play a role in the early stages of vessel formation. 384 The finding of hypoxia inducible factor 2α (HIF-2α) in the nuclei of hemangiomas is interesting in view of the relationship of this tumor to hypoxic events. 385 This results in down-stream overexpression of vascular endothelial growth factor (VEGF). 385
The finding that the endothelial cells in this tumor are clonal has major implications for our understanding of this lesion. 386 It supports their separation from venous malformations. 387 This finding is consistent with the possibility that these tumors are caused by somatic mutations in one or more genes regulating endothelial cell proliferation. 386
The treatment of infantile hemangiomas has been discussed, in part, above. Newer therapies being tried for superficial lesions include imiquimod 5% cream.388.389. and 390. Misdiagnosis of vascular lesions is still a problem; it can lead to inappropriate treatment.391. and 392.
Multiple cutaneous hemangiomas may occur with or without disseminated visceral hemangiomas. 393 Segmental hemangiomas often have similar visceral associations. The cutaneous lesions are capillary hemangiomas, which are present at birth or appear in infancy. Sometimes they have been called miliary hemangiomatosis. 394 In those cases with visceral involvement, any organ may be affected. Visceral involvement is a poor prognostic sign, death occurring in the majority of severe cases, usually within a few months of birth.395. and 396. Common causes of death include high-output congestive cardiac failure associated with arteriovenous shunts (particularly in the liver), central nervous system complications, and bleeding associated with the Kasabach–Merritt syndrome.395.397.398.399.400. and 401. This syndrome has been cured by the surgical excision of a complex vascular lesion in an infant. 402 It was segmental rather than diffuse. Other developmental abnormalities have been reported in association with this condition. 403
Hemangiomas of the head and neck region are sometimes associated with anomalies of the major blood vessels, including aortic coarctation. 404 They are not strictly diffuse, but rather segmental in distribution. One such syndrome is PHACES (posterior fossa malformations, hemangiomas – especially large, plaque-like, facial lesions – arterial anomalies, cardiac anomalies, eye abnormalities, and sternal cleft and/or supraumbilical raphe).335.405.406.407.408. and 409. In PHACES (OMIM 606519), nearly 90% of affected individuals are females. In 70% of cases there is only one extracutaneous manifestation.410. and 411. Forme frustes may occur. 412 Linear hypopigmentation has also been reported. 413 Other syndromic presentations include lumbosacral hemangiomas of infancy associated with spinal dysraphism414 or genitourinary abnormalities, and hepatic hemangiomas with cutaneous and sometimes thyroid abnormalities. 411 Large perineal hemangiomas are another segmental (rather than diffuse) form with associated defects. PELVIS syndrome refers to the present of perineal hemangioma, external genitalia malformations, lipomyelomenigocele, vesicorenal abnormalities, imperforate anus, and skin tag. 415 Becaplermin gel has been used to treat ulcerated perineal hemangiomas in a small series. 416
A purely cutaneous form of diffuse neonatal hemangiomatosis has also been reported, and has been called ‘benign neonatal hemangiomatosis’.417.418. and 419. The prognosis in this group is good. In these patients, and in some survivors of those with visceral hemangiomas, spontaneous regression of lesions occurs.417.418. and 420. In one such case with only cutaneous lesions, the lesions had all resolved by 2 years of age. 421
Early lesions are highly cellular and involve the dermis, but extension into the subcutis may occur. Vascular lumina are small, often slit-like and unapparent, and lined by plump endothelial cells (Fig. 38.10). Moderate numbers of normal mitotic figures are present, and mast cells and dermal dendrocytes are frequent in the intervening stroma. 422 It has been suggested that the mast cells may play a role in angiogenesis and therefore in the formation of these lesions. 423 The vascular proliferation often has a marked lobular configuration; this is often more obvious in the subcutis. Here fat lobules are partly or completely replaced and the appearance may resemble that of angiolipomas. 341 Perineural infiltration can be present. 424 This was in a cellular infantile variant. This finding is unusual in the light of a more recent finding that nerve bundles are absent in hemangiomas but present in vascular malformations. 425 Lesions with vessel proliferation, in and around sweat glands, have also been described. 426 As the lesions evolve, vascular lumina become larger and more obvious. A central draining lumen may become evident in each lobule. The endothelium lining the vessels becomes flatter. With regression of lesions there is disappearance of vessels, interstitial fibrosis, and fat replacement of vascular tissue in the lobules of the subcutis. 341 Immunohistochemical studies have demonstrated endothelial (CD31+) and pericytic (SMA+) differentiation in cells. 427 The early involution phase is characterized by ICAM-1 (CD54) expression and a sparse infiltrate of CD8+ cells. 334 The endothelial cells express GLUT-1, in contrast to malformations. They also express Lewis Y antigen (LeY), merosin, and CD15.334. and 428. The transcription factor encoded by the Wilms’ tumor 1 (WT1) gene is expressed in the endothelium of hemangiomas but not in vascular malformations. 429 It is also expressed in angiosarcomas. 430
(A) Hemangioma of infancy. (B) The lumina are slit-like and unapparent. (H & E)
Ultrastructural studies of capillary hemangiomas have shown plump endothelial cells surrounded by a basement membrane and pericytes. 102 Intracytoplasmic vacuoles are present in endothelial cells, and are thought to represent an early stage in lumen formation. 431 Crystalloid inclusions have been identified in endothelial cells in early cellular lesions. 432 Vessels within a hemangioma may have features of capillaries, venules, or arterioles. 102
Sinusoidal hemangioma is an uncommon benign vascular tumor with some similarities to a venous malformation (‘cavernous’ hemangioma). However, it occurs as an acquired lesion in adults, rather than in children. 433 This variant is most common in females, the trunk (including the breast) and limbs being the most common sites. The tumor involves the subcutis and deep dermis.
It is an acquired lesion and has therefore not been included with the ‘Hamartomas and malformations’.
A lobular architecture is characteristic of sinusoidal hemangioma (Fig. 38.11). Lobules are composed of thin-walled interconnecting vascular channels forming a sinusoidal pattern. Vessels are closely approximated, with little intervening stroma. Tangential sectioning of vessel walls produces a pseudopapillary pattern. Unlike typical ‘cavernous’ hemangiomas, lining cells may appear focally atypical with nuclear hyperchromatism. Mitotic figures are not seen. Calcification is a rare complication. 434
Sinusoidal hemangioma. There is a lobular pattern, with interconnecting vascular channels. (H & E)
Hemangioblastomas are intracranial or intraspinal tumors arising sporadically or in association with von Hippel–Lindau syndrome (OMIM 193300). Two cases have been reported in the skin, one on the nose, 435 and the other in the soft tissue of the inner ankle. 436 The lesion from the nose did not recur, despite its treatment by shave excision.
The lesions were composed of sheets of vacuolated436 or clear cells435 with intervening vessels ranging from small capillaries to large ectatic, thin-walled vessels. In the case with vacuolated cells there was some resemblance to a lipoma. 436 This case also showed focal necrosis and some stromal hemosiderin. The tumor cells stained for neuron-specific enolase and calponin but not for HMB-45, epithelial membrane antigen, or glial fibrillary acid protein (GFAP). Some cells were S100 positive in one case, but not the other. The vascular endothelial cells expressed CD31 and CD34.
‘Cherry’ angiomas (senile angiomas, Campbell de Morgan spots) are very common single or multiple bright red papules, up to a few millimeters in diameter, which occur predominantly on the trunk and proximal parts of the limbs. There is typically a pallid halo surrounding the lesions. Rare before puberty, the incidence rises sharply in the fourth decade, such that they are almost universal in old age. 437 They were common in Iranian veterans, many years after exposure to sulfur mustard poisoning. 438 A case with early-onset lesions has been reported in association with nevus flammeus (capillary malformation). 439
Treatment options are many. It can be removed by surgical, punch, or shave excision. Cryosurgery and electrodesiccation are other options. Laser-treated lesions undergo inflammation, necrosis, and eventual healing by 4 weeks. 440
In small early lesions, one or more dilated interconnecting thin-walled vascular channels are present in the dermal papillae. In older lesions there is loss of rete ridges and atrophy of the superficial epidermis, with formation of a polypoid lesion composed of a network of dilated communicating channels with scant intervening connective tissue (Fig. 38.12). A collarette may be present at the periphery of the lesions.
‘Cherry angioma’. This polypoid lesion is composed of dilated vascular channels and scant intervening stroma. A collarette is present at the periphery. (H & E)
These lesions appear to be the dilated and interconnected segments of venous capillaries and postcapillary venules in the dermal papillae. The vessels of the upper horizontal plexus are not involved. The non-replicating nature of the endothelial cells comprising these lesions indicates that they are probably not true neoplasms. 441 The high incidence of these lesions in old age suggests that their occurrence is an age-related degenerative phenomenon.
A case with the histological appearances of this entity, but clinical features of discrete agminated red papules coalescing into a plaque has been reported. It was called ‘acquired agminated acral angioma’. 442
Chan and colleagues coined the term ‘glomeruloid hemangioma’ for a characteristic benign vascular tumor which they considered a cutaneous marker for POEMS syndrome (Crow–Fukase syndrome).443.444.445.446. and 447. POEMS syndrome is a multisystem disorder characterized by polyneuropathy (peripheral sensorimotor neuropathy, papilledema), organomegaly (hepatosplenomegaly, lymphadenopathy), endocrinopathy (impotence, gynecomastia, amenorrhea, glucose intolerance, hypothyroidism, adrenal insufficiency), M-protein (paraproteinemia, osteosclerotic myeloma, marrow plasmacytosis), and skin changes (hyperpigmentation, hypertrichosis, sclerodermoid features, and hemangiomas).448. and 449. Diffuse skin hyperpigmentation is the most common skin finding, seen in more than 90% of patients. 450 Multicentric Castleman’s disease and POEMS syndrome are overlapping conditions.451. and 452. HHV-8 has not been present in several cases of this combined disease.453. and 454.
Vascular tumors in POEMS syndrome are eruptive, multiple, red, or purple in color, and distributed on the trunk and limbs. Lesions are usually small papules from pinhead in size to a few millimeters, although larger, bluish subcutaneous compressible tumors have also been described. 455 One larger lesion with cerebriform features has been reported. 445 The tumors have variously been described as showing features of cherry angioma, capillary hemangioma, cavernous hemangioma, lobular hemangioma (pyogenic granuloma and tufted angioma), and targetoid hemangioma. 456 Lesions may show overlap features.303.455.457. and 458. Glomeruloid hemangiomas appear to be more common in the Japanese than in other races or ethnic groups.
There have now been several reports of glomeruloid hemangiomas occurring in the absence of POEMS syndrome. In each case they presented as solitary lesions.459.460. and 461.
In the characteristic glomeruloid hemangiomas described by Chan et al there are dilated (sinusoidal) dermal vascular spaces filled by grape-like aggregates of small capillary vessels, resulting in structures resembling renal glomeruli. Between these small vessels are plump cells which appear to be endothelial cells, being positive for factor VIII-related antigen. These cells and endothelial cells lining vessels contain PAS-positive eosinophilic globules, which stain for immunoglobulins at their periphery. In the cases reported without accompanying POEMS syndrome these eosinophilic globules expressed both kappa and lambda in a polytypical fashion. 460 Electron microscopy has suggested that the globules are enlarged secondary lysosomes (thanatosomes). 462
A recent study has shown endothelial cells with two different immunophenotypes. 444 The capillary-type endothelium had a CD31+/CD34+/UEA 1+/CD68− phenotype, while the sinusoidal endothelium had a CD31+/CD34−/ UEA 1−/CD68+ phenotype. 444 There is increased expression of vascular endothelial growth factor (VEGF). 463 In cases reported recently without concurrent POEMS syndrome, the endothelial cells and plump, vacuolated cells were positive for CD31 and CD34. 462 Isolated cells containing PAS-positive globules were positive for CD68. 459
A morphologically distinct vascular tumor has been reported in association with a solitary bone plasmacytoma. It is mentioned here because of the association of some cases of plasmacytoma with POEMS syndrome or Castleman’s disease. 464 The lesion was characterized by an extensive plaque with vascular hyperplasia and dermal mucin deposition. 464 The acronym AESOP syndrome, reflecting the component parts, has been used.
Papillary hemangioma was reported by Suurmeijer and Fletcher in 2007. Their 11 cases presented as a solitary bluish papule in the head and neck region. 465 Most lesions had been present for several years. None of the patients had systemic disease, including POEMS syndrome (see above). Only one lesion recurred locally, although surgical margins were involved in several cases.
It has been suggested recently that papillary hemangioma and glomeruloid hemangioma represent the same pathophysiological process. 462
The lesions were primarily situated in the dermis but extension into the subcutis occurred in six cases. Discontinuous areas of papillary branching were often present. There were multiple ectatic vessels with predominantly intravascular papillary growth. The papillae had cellular cores containing pericytes and stromal cells, arranged around normal small capillaries. 465 The surfaces of the papillae were covered by focally swollen endothelial cells containing numerous hyaline globules consistent with dysfunction of the autophagocytic–lysosomal pathway. 465
Arteriovenous hemangioma (acral arteriovenous tumor) presents as a solitary, red or purple papule with a predilection for the lips, the perioral skin, the nose, and the eyelids of middle-aged to elderly men.466.467. and 468. Less than 10% are multiple.469. and 470. It is usually asymptomatic. Lesions measure 0.5–1.0 cm in diameter. It has an association with chronic liver disease,471. and 472. epidermal nevus syndrome, and other vascular malformations. Some lesions are probably examples of vascular malformations. 80
Surgical excision is the treatment of choice for superficial lesions. Deep lesions which are also large may require embolization. 469 An acquired digital lesion (see below) has been treated successfully with Nd:YAG laser. 470
There is a well-circumscribed non-encapsulated collection of large, thick-walled vessels in the upper and mid dermis (Fig. 38.13). These vessels are lined by endothelium and have a fibromuscular wall which contains elastic fibers but no definite elastic laminae. Most vessels have the characteristics of veins. 473 In approximately one-third of cases there are thin-walled dilated angiomatous capillaries superficial to the large tumor vessels. The stroma is often myxoid. Estrogen receptors have not been detected in the vessels. 472
(A) Arteriovenous hemangioma. (B) It is composed of thick-walled vascular channels. (H & E)
Acquired digital arteriovenous malformations are a distinct entity thought to result from shunts between an artery and a vein in a fingertip.474.475. and 476. Their extent can be determined by ultrasound imaging. 470 They are composed of thick- and thin-walled vessels, some of the former having small amounts of smooth muscle in their walls. 477 The thin-walled channels are venules. The lesions lack the tumor-like qualities of the arteriovenous hemangioma. Shunts are visible between the two classes of vessels. 474
The symplastic hemangioma is akin to other symplastic tumors which are characterized by bizarre, hyperchromatic cells, which represent a degenerative phenomenon.351. and 478. It is composed of thick-walled and variably dilated vessels. The atypical cells, with markedly hyperchromatic nuclei, are located within the vascular smooth muscle wall, or in the interstitium (Fig. 38.14). They are spindle or epithelioid in shape; their nuclei may be multinucleate. 478 Perivascular hemorrhage, vascular thrombosis, and focal papillary endothelial hyperplasia are often present. Rare atypical mitoses may be present. 478 The pleomorphic cells show focal smooth muscle positivity. 478
(A) Symplastic hemangioma. (B) Bizarre cells are in the wall of vessels and in the interstitium. (H & E)
Microvenular hemangioma, a benign vascular tumor, occurs predominantly in young and middle-aged adults as a single, slow-growing lesion on the trunk or limbs. Lesions are purple to red papules or nodules, usually less than 1 cm in diameter.479.480. and 481. Larger plaque forms have been reported. 482 An association with systemic immunosuppression has been suggested, but there is little evidence to support this view. 483
The main characteristic of this tumor is a proliferation of thin uniform branching collapsed-looking vessels with inconspicuous lumina. The vessels involve the dermis and occasionally the superficial subcutis. The intervening stroma is collagenous, sometimes with a desmoplastic appearance. Endothelial cells are sometimes plumper than normal, but not atypical; they stain for factor VIII-related antigen, CD34, CD31, and Ulex europaeus-1 lectin. A peripheral layer of pericytes stains for smooth muscle actin.482. and 484. Eosinophilic globules, commonly seen in Kaposi’s sarcoma, are not seen in this tumor.
Targetoid hemosiderotic (hobnail) ‘hemangiomas’ are single lesions occurring in young or middle-aged persons, with a male predominance. They involve the trunk or limbs.332.485.486.487.488.489. and 490. The face is rarely involved. 491 Characteristically the lesion has a ‘targetoid’ appearance with a violaceous central papule surrounded by an area of pallor and an ecchymotic or brown ring, 492 which expands and subsequently disappears. The central papule persists. It has been reported in pregnancy,486. and 487. and in a father and son. 493 Rare cases have been reported that altered during the menstrual cycle, becoming larger and painful prior to menstruation. 494 Lesions may be monitored by dermoscopy. 495
A lymphatic origin has been confirmed by the new lymphatic endothelial cell marker D2-40. 496 Previously it was thought to have been a traumatized angiokeratoma, 490 although a lymphatic origin had been suggested by others. 489 HHV-8 has not been demonstrated in this lesion. 497
The term ‘hobnail hemangioma’ has been used for a group of vascular lesions related to targetoid hemosiderotic hemangioma, but encompassing tumors with morphological features of retiform hemangioendothelioma, progressive lymphangioma, and Dabska’s tumor.498. and 499. The term is being used currently as a synonymous nomenclature. 500 It is the preferred term by some authors, despite its dubious origin. Both terms are now misnomers.
Initially there are ectatic vascular channels lined by plump (hobnail) endothelial cells in the upper dermis. The vessels have intraluminal papillary projections. In the deep dermis and subcutis the channels dissect collagen bundles and surround sweat glands. 499 There is a variable mild inflammatory infiltrate about vessels and considerable extravasation of erythrocytes and hemosiderin (Fig. 38.15). Fibrin thrombi may be seen in the superficial vessels, a feature not present in the early lesions of Kaposi’s sarcoma. Plasma cells are also uncommon; eosinophilic globules have not been reported. Old lesions are composed of collapsed thin-walled anastomosing vascular channels with hemosiderin. Staining with D2-40, the new lymphatic endothelial marker, shows microshunts between neoplastic lymphatic channels and small blood vessels, explaining the aneurysmal microstructures and the erythrocytes and hemosiderin. 500
Targetoid hemosiderotic hemangioma. (A) There is no superficial vascular dilatation in this case. (B) There are small vessels and abundant hemosiderin. The lesion was clinically typical. (H & E)
In one series, the tumor cells stained for CD31 in all cases tested, whereas only 3 of the 28 cases stained completely for CD34. In addition, 4 out of 8 cases stained positively for vascular endothelial growth factor receptor-3 (VEGFR-3), suggesting lymphatic differentiation. 489 This has been confirmed by the presence of lymphatic marker D2-40, and the absence of CD34 staining of endothelial cells, and the lack of actin-labeled pericytes. 500
Although spindle-cell hemangioendothelioma was originally believed to be a vascular tumor of low-grade malignancy, 501 there is convincing evidence that this entity may in fact represent a non-neoplastic process.502.503.504.505.506.507.508.509. and 510. Accordingly, Requena and Ackerman believe it should be renamed ‘spindle-cell hemangioma’. 509 Fletcher et al have suggested that these lesions represent a reactive vascular process arising in association with a local abnormality of blood flow, because the lesions are often associated with local malformation of blood vessels, have an integral smooth muscle component, occasionally regress spontaneously, and do not metastasize. 502 A similar view has been expressed by Perkins and Weiss. 506 This tumor has been reported in Maffucci’s syndrome and in association with venous malformations and the Klippel–Trenaunay syndrome.501.503.506.511. and 512.
Although tumors may occur at any age from birth to adulthood, approximately 50% of cases arise before the age of 25. Lesions may be single or multiple; multiple lesions tend to occur in a single area. Occasionally lesions are widespread. The hands and feet are the most common sites, but lesions also occur on the trunk.501. and 513. Oral lesions are rare. 514 Tumors are circumscribed, hemorrhagic nodules. They may increase in size and number with time, often over a long period. Local recurrence of lesions is common after removal. In one study ‘recurrences’ were noted to arise in adjacent, previously unaffected tissue, suggesting a new lesion. Such recurrences may arise from contiguous spread along or multifocal involvement of a vessel. 506 There is only one documented case of metastasis by this tumor and that followed multiple recurrences, radiotherapy, and transformation to a conventional angiosarcoma. 501
Tumors are situated in the dermis and subcutis; rarely, they occur in the lumen of a vein. There are three main components. The first is a vascular component of thin-walled cavernous channels which may contain thrombi or phleboliths. The second is a solid area of spindle cells with slit-like vascular spaces; these areas may resemble Kaposi’s sarcoma (Fig. 38.16). The third component is plump endothelial cells, either in groups or lining vascular channels. Some of these cells have intracytoplasmic vacuoles. Fletcher et al have also noted the frequent presence of large, malformed, thick- or thin-walled vessels adjacent to the tumor, as well as bundles of smooth muscle cells near cavernous vessels or in solid areas. 502 Nuclear atypia is minimal; mitoses are rarely seen. Lymphocytes and siderophages may be associated with the lesions, but eosinophilic globules, as seen in Kaposi’s sarcoma, have not been reported. The spindle cells in this tumor are generally more bland-looking and regular in appearance than those in Kaposi’s sarcoma. Furthermore, the spindle-cell fascicles are not as well-formed as those in Kaposi’s sarcoma. 515
(A) Spindle-cell hemangioendothelioma. (B) There are thin bundles of spindle cells between the vascular channels. (H & E)
Immunohistochemical studies have confirmed the endothelial nature of the cells lining cavernous channels which stain for CD31, CD34, factor VIII-related antigen, and Ulex europaeus-1 lectin. Solid epithelioid and spindle-cell areas are negative for these antigens. 502 Interleukin-8 (IL-8) is also expressed. 516 Lesions do not contain HHV-8. DNA flow cytometric and immunohistochemical studies have shown that this tumor has low proliferative activity and is diploid, consistent with a reactive lesion. 503
Ultrastructural studies have shown a heterogeneous cell population in solid areas; however, occasional cells contain Weibel–Palade bodies, confirming that some cells are showing endothelial differentiation.
Acquired elastotic hemangioma arises on the sun-damaged skin of middle-aged to elderly persons. Although all six cases in the original report by Requena and colleagues were women, two of the five cases seen by the author have been in males. 517 Clinically, lesions present as solitary plaques on the dorsal aspect of the forearms, and less commonly the lateral neck. 517 They are rarely diagnosed as vascular lesions clinically.
There is a broad, band-like proliferation of capillary blood vessels in the superficial dermis, arranged parallel to the surface (Fig. 38.17).351. and 517.
Elastotic hemangioma. There is a proliferation of small blood vessels that are arranged parallel to the surface. (H & E)
Angioma serpiginosum is a rare condition in which multiple pin-sized vascular puncta occur either singly or in clusters on any part of the body except mucous membranes and the palms and soles.518. and 519. The legs are the most common site. Extensive cutaneous involvement and linear lesions along the lines of Blaschko have been reported. 520 Lesions appear before puberty and progress by the development of further puncta at the periphery of the involved area. Late onset has been recorded. 521 The lesions do not regress. Females are more commonly affected, and a familial grouping of cases has been reported,522. and 523. but the mode of transmission is still unclear, with the likelihood of X-linked (OMIM 300652) and autosomal dominant forms (OMIM 106050).
Microscopic examination shows single or grouped ectatic, congested, thin-walled capillaries in the papillary dermis. Thick-walled capillaries and downgrowth of the rete ridges between groups of vessels have also been described.524. and 525. The latter lesions resemble angiokeratomas. Inflammation is not usually present. Although previously regarded as a telangiectatic process, an element of vascular proliferation appears to be present. 524
Only a few cases of giant cell angioblastoma, an exceedingly rare tumor of skin and soft tissues, have been reported. 526 The tumor is present at birth or noted soon after. It is locally infiltrative, but grows slowly. The morphology suggests an unusual form of neoplastic angiogenesis. 526 Metastases have not been recorded.
There are concentric arrays of oval-to-spindle cells around small endothelium-lined channels. These primitive cells arranged around the vessels tend to differentiate toward pericytes and express smooth muscle actin while the endothelial cells express CD31 and factor VIII-related antigen. Large mononuclear and multinucleate giant cells are also present. Perineural and intraneural involvement by small vessels is common. A stromal infiltrate of lymphocytes and plasma cells is another feature.
Epithelioid angiomatous nodule is a very rare vascular tumor described by Brenn and Fletcher in 2004. 527 The lesions are mostly solitary and involve the trunk, the head and neck, 528 and less often, the extremities. 529 Rare cases present as eruptive, recurring nodules. Despite vague resemblance to epithelioid hemangioendothelioma the lesion is quite benign. 530 There is an absence of microsatellite instability.
Surgical excision is curative. The eruptive cases (see above) cleared with corticosteroids, only to recur later. 529
The lesion is often situated in the upper dermis. Sometimes it involves the deep dermis. It is a circumscribed, mainly solid proliferation of large polygonal epithelioid cells with eosinophilic cytoplasm, with enlarged nuclei and prominent nucleoli.527. and 529. Vascular channels with epithelioid endothelium are either focal or diffuse. There are extravasated erythrocytes and hemosiderin pigment in over half. There is a scant inflammatory infiltrate, predominantly of lymphocytes. Eosinophils have been prominent in a few cases. Mild fibrosis may be present.
Angiolymphoid hyperplasia with eosinophilia (ALHE) is a tumor of skin and subcutaneous tissues composed of vessels, a proliferation of a distinctive type of endothelial cell, and a variable component of inflammatory cells. 532 Whether it is a true neoplasm or a reactive process is at present undecided. It appears to be identical to the lesions known as pseudopyogenic granuloma, atypical pyogenic granuloma, epithelioid hemangioma, histiocytoid hemangioma, intravenous atypical vascular proliferation, and nodular angioblastic hyperplasia with eosinophilia and lymphofolliculosis.533.534.535.536.537. and 538.
Kimura’s disease (eosinophilic lymphogranuloma),539.540.541.542. and 543. although previously included by some in this group of conditions, is a separate entity.544.545. and 546. It is clinically different, typically presenting as large subcutaneous masses in young to middle-aged Asian men. It has been reported in non-Asians. 547 Coexistence of the two conditions in the same patient suggests they may be part of a spectrum. 548 The majority of lesions are located around the ears or in the parotid gland. 549 A case presenting with widespread prurigo nodularis-like lesions has been reported. 550 Elevated serum immunoglobulin E levels and peripheral blood eosinophilia are also common. 551 The etiology of Kimura’s disease is unknown but it may be an aberrant immune reaction to an as yet unknown stimulus. Epstein–Barr virus DNA has been detected in lesional tissue in one case. 552 Clonal populations of T cells have been detected in some patients. 553 Treatment includes surgical resection and regional or systemic corticosteroid therapy. Cytotoxic therapy, cyclosporine (ciclosporin),554. and 555. and radiation have also been used. 551 The disease has a good prognosis but it may recur locally. Death from Kimura’s disease is rare. 547
In angiolymphoid hyperplasia, lesions involve the subcutaneous tissue or the dermis or both. Single or multiple pink to red-brown papules or nodules occur, predominantly on the face, scalp, and ears; they are uncommon on the limbs and trunk. There is a predilection for the retroauricular area. 556 Widespread lesions and multiple lesions limited to one limb have been reported.557.558. and 559. Cases have been reported in a port wine stain, 560 over a deep vascular malformation, 561 and on the vulva,536. and 562. penis,563. and 564. lip, 565 inner canthus, 566 and oral mucosa.567. and 568. It has been reported in association with pregnancy,569. and 570. HIV infection, 571 lichen amyloidosus, 572 and the nephrotic syndrome. 573 It may mimic bowenoid papulosis. 556 Symptomatic lesions may be painful, pruritic, or pulsatile. 574 In some series there is a female predominance. 575 Young to middle-aged people are most commonly affected. Some cases are associated with a blood eosinophilia. Lesions may remain for years without evidence of involution, and they may recur after excision. 537 Peripheral T-cell lymphoma and severe atherosclerosis developed in an adolescent with multiple lesions of ALHE. 576 The authors of the paper believed that the pathology of these conditions was related. 576
The true nature of this lesion is uncertain. A few examples appear to be associated with trauma, 577 oral contraceptives,574. and 578. HIV infection, 571 or pregnancy, but not HHV-8 infection.558.579. and 580. It has been suggested that the lesions are a reactive hyperplastic process secondary to an underlying arteriovenous malformation or traumatic pseudoaneurysm.332.581. and 582. The interleukin-5 levels are increased, which may explain the eosinophilia.582. and 583. Clonal populations of T cells have been detected in some, but not all of the cases tested.579.584. and 585.
Many different treatments have been used for this condition, suggesting that no treatment method is universally successful. Many are surgically excised. They can also be treated by laser therapy, 586 most often the pulsed dye laser.559.587.588. and 589. Cryosurgery590 and electrosurgery are other options. Intralesional corticosteroids, interferon-α,591. and 592. and bleomycin593 have also been used. Tacrolimus ointment594 and the anti-interleukin-5 antibody mepolizumab595 have both produced cures.
The lesions consist of circumscribed collections of vessels and inflammatory cells. The vascular component comprises thick- and thin-walled vessels lined by plump endothelial cells (Fig. 38.18). These cells also occur in clumps that appear solid or sometimes contain small lumina. They are ‘epithelioid’ in appearance, with a large nucleus and abundant eosinophilic cytoplasm, and are characteristic of this condition. Prominent cytoplasmic vacuoles are seen in some cells. Normal mitotic figures are sometimes present. Intravascular proliferations of these cells may be seen in the lumina of larger vessels. 536 Exuberant examples may be confused with epithelioid hemangioendothelioma and epithelioid angiosarcoma. 563 There is one report of multinucleated cells, some of which were endothelial sprouts and others fibrohistiocytic cells. 596 Associated with the vascular and endothelial proliferations is a stromal cellular infiltrate which varies in intensity and consists of lymphocytes (sometimes with lymphoid follicle formation), eosinophils, and mast cells. The stroma may be fibrous or myxoid in character. Sometimes the fibrotic reaction is excessive. In one case it was associated with a florid granulomatous reaction with many multinucleated giant cells, often of Touton type. 597
(A) Angiolymphoid hyperplasia with eosinophils. (B) The vascular channels are lined by plump, partly vacuolated endothelial cells. There are scattered eosinophils in the stroma. (H & E)
A rare intra-arterial variant has been described. There was a prominent lymphocytic rim-like component. 598
Although these epithelioid cells share some enzymes with histiocytes599 they do not contain lysozyme; they have ultrastructural features of endothelial cells, including Weibel–Palade bodies. 574 The epithelioid cells stain for CD31, CD34, factor VIII-related antigen, and Ulex europaeus-1 lectin, but not for cytokeratins or epithelial membrane antigen.600. and 601. The vacuoles in their cytoplasm possibly represent primitive vascular lumina.
Kimura’s disease is composed of reactive lymphoid follicles with a dense infiltration of eosinophils, sometimes forming eosinophilic abscesses.547.602.603. and 604. An eosinophilic epithelioid granulomatous reaction with central eosinophilic abscesses has been reported. 605 Mast cells are increased.604. and 606. Vessels (postcapillary venules) are increased in number, but their endothelial cells are usually flat. 547 Vascular cords are unusual. 557
‘Angiolymphoid hyperplasia with high endothelial venules’ is the term suggested for a cutaneous lesion with a characteristic admixture of lymphoid hyperplasia (without eosinophils) and a vascular proliferation previously called APACHE – acral pseudolymphomatous angiokeratoma of children (see p. 1003). 607 APACHE continues to be the designation attached to this entity. It appears to be a ‘pseudolymphoma’.608.609. and 610.
The term ‘lobular capillary hemangiomas’ refers to a group of vascular tumors characterized by the presence of capillary-sized vessels arranged in lobules.611. and 612. Included in this category are pyogenic granuloma and its variants, acquired tufted angioma (angioblastoma, progressive capillary hemangioma), 611 and glomeruloid hemangioma. The infection-related angiomatoses (bacillary epithelioid angiomatosis and verruga peruana) may also have a lobular pattern.
Pyogenic granuloma is a common benign vascular tumor of mucous membranes and skin. Studies suggest that it represents a hemangioma and not simply a florid proliferation of granulation tissue.613.614. and 615. Common sites include the gingiva, lips, fingers, and face.616. and 617. Periungual lesions may develop, sometimes along Beau’s lines.618. and 619. Less common sites include the trunk, vulva, 620 arms, legs, and conjunctiva. 621 There is a 3 : 2 male preponderance. 617 These tumors are commonly polypoid and pedunculated, but they may be sessile. Most are red or red-brown in color. Some darker lesions may clinically mimic nodular malignant melanoma. The lesions typically evolve rapidly to maximum size over a period of weeks. In one series this varied from 0.5 to 4 cm, with a mean diameter of 1.1 cm. 622 The surface is often ulcerated and bleeds easily. Dermoscopy is a useful tool for improving the recognition of these lesions. There is a reddish homogeneous area surrounded by a white collarette. 623
Spontaneous involution of lesions is uncommon but has been reported in cases of disseminated pyogenic granuloma, 624 and post partum in women who develop lesions during pregnancy (epulis gravidarum). 625 The lesions may develop at any age, and both sexes are affected. There is a female predominance in some reports, 626 but not in others. 627 In one series of oral and nasal mucosal lesions there was a marked male predominance in the first two decades and a female predominance during the childbearing years. 613 Children most commonly develop this tumor after the age of 1 year; rarely, congenital lesions occur. 621 Occasionally, the tumors may be multiple or disseminated.624.628.629.630.631. and 632. Multiple satellite recurrences sometimes occur after treatment of the primary lesion, particularly when the latter was on the trunk, an uncommon site for pyogenic granuloma.633.634. and 635. A giant recurrent pyogenic granuloma has been reported on the face. It had satellitosis. 636 Satellite vascular lesions resembling a vascular malformation have been reported following removal of a nevocellular nevus. 637 Pyogenic granulomas have been reported in a pre-existing nevus flammeus, 638 a port wine stain,621.639. and 640. unilateral dermatomal superficial telangiectasia, 641 and a ‘spider’ angioma. 642 Recurrent intrapapillary endothelial hyperplasia has developed at the site of a previously excised pyogenic granuloma. 643 Pyogenic granulomas developed in the mouth in a patient with oral pemphigus vulgaris. 644
In the majority of cases there is no apparent cause for these lesions; a minority follow trauma,613.621.622. and 645. retinoid therapy (both topical and systemic),646.647.648.649.650. and 651. granulocyte colony-stimulating factor (G-CSF) therapy, 652 reverse transcriptase inhibitor therapy, 619 insect bite, 621 burn,600. and 632. scald, 653 or cryotherapy. 654 Pyogenic granulomas of the nail bed region have been reported following the use of capecitabine,655. and 656. gefitinib, 657 and systemic 5-fluorouracil. 658 The higher incidence in women during the childbearing years, the occurrence in pregnancy, an association with use of the oral contraceptive pill, and the spontaneous regression of lesions following parturition suggest that a hormonal factor is involved in their genesis. 625 This theory has been disputed for cutaneous pyogenic granulomas, but not for mucosal ones.627.659. and 660. Unlike bacillary angiomatosis, there is no association with Bartonella infection. 661 It has also been suggested that pyogenic granulomas represent small acquired arteriovenous fistulae. 616 Immunohistochemical and ultrastructural studies have confirmed that pyogenic granulomas are tumors of vessels and endothelial cells.662. and 663. Recent work indicates that, like angiosarcomas, they express various activators of specific transcription factors. 664
Pyogenic granuloma can be treated by a single shave and electrocautery, 665 curettage with electrodesiccation, or surgical excision. A randomized trial found that curettage followed by electrodesiccation gave better cosmetic results and required fewer treatments than did cryotherapy. 666 Laser therapy has also been used. Topical imiquimod 5% cream has been used for recurrent lesions, 665 and for focal facial lesions. 667 It may be used under occlusion. 668 A novel treatment for polypoid lesions is the use of a suture around the base. 669
Pyogenic granulomas are lobular capillary hemangiomas (Fig. 38.19). 613 The lobular arrangement of the lesions is distinct from the pattern of capillaries in granulation tissue and, unlike granulation tissue, the capillaries do not usually involute with time. The underlying morphology is often obscured by secondary ulceration, edema, hemorrhage, and inflammatory changes. In uncomplicated lesions there is a lobulated proliferation of capillary-sized vessels. The deep lobules are compact and cellular, with small indistinct lumina. Occasional mitotic figures may be seen within the cellular lobules. The apoptotic rate is low, possibly a reflection of its rapid growth. 670 Toward the surface, the lobules are larger and less tightly packed, and have distinct capillaries with larger branching lumina. The lobules are separated by myxoid or fibrous connective tissue septa. 671 The rare presence of spindle cells may mimic Kaposi’s sarcoma. 672 A lobulated variant of Kaposi’s sarcoma mimicking pyogenic granuloma has been reported. 673 Extramedullary hematopoiesis has been reported in the stroma in two cases.674. and 675.
Pyogenic granuloma. There is a well-developed collarette at the margins of this lobular vascular proliferation. (H & E)
The surface epithelium is attenuated, and at the margins of the lesion there is often an epidermal collarette formed by elongated rete ridges or sweat ducts. Surface ulceration and inflammation are secondary events, and sometimes lead to the formation of true granulation tissue near the surface of the lesions. Mast cells are not increased in pyogenic granuloma, unlike proliferative-phase hemangiomas.423. and 621.
Lesions with the same lobular hemangiomatous pattern have also been described within veins676 and in subcutaneous tissues.677. and 678. Intravenous pyogenic granuloma occurs predominantly on the neck, arms, and hands.676.679.680. and 681. The lesions are composed of a lobular proliferation of capillaries set in a fibromyxoid stroma. A fibrovascular stalk usually connects the lesion to the intima of the involved vein. 682Subcutaneous pyogenic granuloma occurs predominantly on the upper extremities. 677
Immunohistochemistry is rarely carried out because the diagnosis can easily be made on light microscopy. It may be performed in atypical and multiple lesions. The vascular markers CD31, CD34, and factor VIII are positive. Lesions are negative for the glucose transporter GLUT-1. 652
Wilson Jones first gave the name acquired tufted angioma to an unusual acquired vascular proliferation683. and 684. that had previously been reported as progressive capillary hemangioma, 685 and ‘angioblastoma (Nakagawa)’ in the Japanese literature.686. and 687. The Japanese cases have some clinical differences and may not be identical to acquired tufted angioma, although the current consensus is that they are the same entity. 688 Over 200 cases have now been reported. The condition is characterized by slowly spreading erythematous macules, plaques, and nodules; rarely, there are multiple lesions. 335 Raised papules resembling pyogenic granulomas are sometimes seen within the lesion.611. and 689. This vascular tumor occurs predominantly in children and young adults, but has also been reported in older individuals.690.691. and 692. Congenital cases are rare.688. and 693. Lesions arise most commonly on the neck and upper trunk, but also at other sites;694.695. and 696. they are sometimes painful.694.697. and 698. Lesions can become quite large.699.700. and 701. Hypertrichosis has been present in several cases.698. and 701. Platelet trapping in the lesions, producing the Kasabach–Merritt syndrome, is an uncommon complication.702.703.704.705. and 706. There are rare associations with pregnancy, 707 liver transplantation, 697 healed herpes zoster, 708 and port wine stain (nevus flammeus).709. and 710. Familial cases have also been described. 711 Most cases do not regress, as do ‘strawberry nevi’ of infancy and childhood, but there are reports of partial or complete regression and the recurrence of lesions.691.707.712.713.714. and 715. Regression appears more common in congenital and early-onset lesions. 688
Treatment of large lesions can be difficult. Smaller lesions are often treated by surgical excision but recurrences are common. Intralesional steroids, cryotherapy, and radiotherapy have not been successful. 699 Subcutaneous interferon-α and the pulsed dye laser have given mixed results. 716 One case was aggravated by the use of a long-pulsed Nd:YAG laser. 717
There are multiple separated cellular lobules within the dermis and subcutis. Some lobules bulge the walls of dilated thin-walled vascular channels which are within the lobules or at their periphery. This sometimes gives the vessels a semilunar profile. 611 These larger vessels have a distinct endothelial lining.
Each lobule is composed of cells with spindle-shaped and oval nuclei (Fig. 38.20). Mitotic figures may be seen but there is no cellular atypia. Small capillary-sized vascular lumina are present in these areas. The morphology of the lobules resembles those seen in pyogenic granulomas. Hemosiderin may be present in the lesions; inflammation and edema are not usually seen. Some authors have noted proliferation of eccrine sweat glands near the vascular lobules.707. and 718.
Acquired tufted angioma. The multiple vascular lobules are composed of spindle-shaped and polygonal cells. (H & E)
The pattern of cellular nodules with a peripheral dilated channel superficially resembles Kaposi’s sarcoma, but the lobules lack the characteristic interlacing bundles of spindle cells and slit-like vessels, and usually lack an inflammatory infiltrate with plasma cells. In endovascular papillary angioendothelioma of childhood, papillary processes lined by atypical endothelial cells protrude into vascular lumina. In Masson’s ‘vegetant intravascular hemangioendothelioma’, papular processes, composed of hyperplastic endothelium supported by fibrous stalks, are confined within vascular lumina.
Tumor cells are positive for CD31 and CD34, but stain only weakly or not at all for factor VIII-related antigen. 719 They are negative for GLUT-1. 720 The dilated vessels partly stain with D2-40, but the proliferative capillaries are negative. 720
Glomus tumors are neoplasms which resemble elements of the glomus apparatus in the skin. 721 The glomus apparatus contains a central coiled canal, the Sucquet–Hoyer canal, which is lined by endothelium and several layers of glomus cells. Glomus tumors combine cells resembling glomus cells and vascular structures.
The glomus tumor is almost always a solitary, purple dermal nodule on the extremities, particularly the fingers and toes. It may have a subungual location and lie within a slight depression in the underlying phalanx. This variant usually presents in adults and occurs with an equal sex incidence. The tumors are almost always painful. The pain varies in intensity but may be severe and paroxysmal, and occur spontaneously or be induced by pressure or cold. Rarely, a small cluster of tumors may occur. 722 Of the 56 cases of extradigital glomus tumor reviewed recently at the Mayo Clinic only one case involved an extracutaneous site (the trachea). 723 The forearm and knee were the most common extradigital sites of involvement. 723
Although they almost always occur in the skin, rare lesions have been reported at other sites, including the deep soft tissues, 724 bone,725. and 726. vagina, 727 trachea, 728 lung, 729 gastrointestinal tract,730. and 731. oral cavity, 732 nasal cavity,733. and 734. within veins,710. and 735. and in cutaneous nerves.736. and 737. There is no evidence of an aggressive clinical behavior with the intravascular lesions. 738 It is uncertain how many of these extracutaneous cases would now be reclassified as glomerulovenous malformations (GVMS). Most of the cases reported in the past as disseminated or regional glomus tumors would now be regarded as glomulovenous malformations. They are considered with the vascular malformations on page 891.
Benign lesions are occasionally locally infiltrative; 739 they may recur after removal. 740 Atypical glomus tumors are discussed further below.
The glomus tumor proper is a well-circumscribed or encapsulated dermal tumor which may extend into the subcutis. It is composed of solid aggregates of glomus cells surrounding inconspicuous vessels. Glomus cells are rounded, regular cells with eosinophilic cytoplasm and darkly staining round to oval nuclei (Fig. 38.21). Tumor cells and vessels are embedded in a fibrous stroma. Some tumors contain large amounts of myxoid stroma (Fig. 38.22). 741 The tumor matrix contains small unmyelinated nerve fibers and mast cells. The uniformity of the cells and their lack of pleomorphism are features of these tumors. Oncocytic and epithelioid variants have been described.742. and 743. The latter are composed of large polygonal to spindle-shaped cells with abundant eosinophilic cytoplasm and large irregular nuclei. The cytological atypia is thought to represent cellular senescence. 743 The term ‘symplastic glomus tumor’ has been used for these lesions, which have a high nuclear grade in the absence of any other malignant features. 154 Another variant is the infiltrative glomus tumor, characterized by an infiltrative growth pattern at the periphery of the lesion. 744 Some of these lesions have been regarded in the past as malignant glomus tumors. This category of infiltrative glomus tumor is not included in the recent classification proposed by Folpe et al. 154 Extensive glomus cell hyperplasia has been reported in the vicinity of a cluster of multiple glomus tumors. 722
Glomus tumor. There are sheets of glomus cells and a few blood vessels. (H & E)
Glomus tumor. The stroma between some of the nests of glomus cells contains mucin. (H & E)
Immunohistochemical techniques have demonstrated vimentin, muscle-specific actin and α-smooth muscle actin in the cytoplasm; laminin and type IV collagen are present in the basal lamina-like material.130.745. and 746. The cells lack the cell markers of endothelial cells (CD31, CD34). The absence of desmin from the tumor cells is a feature shared with some cells of vascular smooth muscle.130. and 746.
Multiple nerve fibers containing substance P have been identified within solitary glomus tumors. 747 Substance P is known to be a sensory afferent neurotransmitter for mediating painful stimuli.
The glomus coccygeum is a vestigial structure sometimes found in skin and deeper specimens removed from the lumbosacral region. It is a heavily innervated modified arteriovenous anastomosis akin to the Sucquet–Hoyer canals of the distal fingers and toes. 748 Their function at this site is unknown.
They are usually an incidental finding in resected specimens of a pilonidal sinus.748. and 749. They average only 1 mm in diameter, although they are reported to reach 5 mm in size.748. and 750.
It is composed of clusters and nests of small to medium-sized epithelioid cells intimately associated with small vascular channels. There is a superficial resemblance to a glomus tumor or paraganglioma. 750 Small nerve bundles are present in the stroma. A complex hamartoma composed of elements resembling a neurovascular hamartoma, a lipomatous hamartoma, and glomus bodies has been reported in the intergluteal region. 751
The cells express muscle-specific actin, vimentin, and neuron-specific enolase. The small nerves are highlighted by an S100 preparation. The epithelioid cells are weakly positive for smooth muscle actin, and negative for desmin, synaptophysin, and chromogranin. The endothelial cells of the vessels stain for CD31. 748
Prose and colleagues reported three children who, during an apparent viral illness, developed angiomatous papules that resolved spontaneously. 752 Other similar cases have been reported. 753 A subsequent case has been characterized by spontaneous healing in 2 weeks, followed by a recurrent eruption 3 weeks later, that again healed. 754 Another report described an outbreak of the condition in nine adult patients in a mental institution. 755 Subsequent cases have not always been associated with an obvious viral illness, suggesting multiple etiologies.754.756. and 757. They have been associated with graft-versus-host disease,758. and 759. cyclosporine, and insect bites. 760
The pathogenesis of these lesions is unknown but it may involve an increase in vascular growth factors. 758 As the lesions usually resolve spontaneously, no treatment is necessary. Cases associated with graft-versus-host disease are larger and probably a different (persistent) entity (eruptive angiomatosis).758. and 759.
The lesions are composed of dilated blood vessels in the upper dermis, lined by plump ‘hobnail’ endothelial cells. 752 The endothelial cells have not always been enlarged. 756 A mild perivascular infiltrate of lymphocytes has been present in most cases.755. and 761.
The two recorded cases of papular angioplasia were in elderly patients who presented with vascular papules of the face and scalp in which the dermal vascular proliferation contained atypical bizarre endothelial cells. 762 The lesions, which were termed ‘papular angioplasia’, were thought of as a ‘pseudomalignancy’. Their relationship to pyogenic granuloma and angiolymphoid hyperplasia is uncertain.
Multinucleate cell angiohistiocytoma was first described by Smith and Wilson Jones. 763 It is characterized by grouped, red to violet papules that may resemble Kaposi’s sarcoma: 764 almost all reported cases have been in females over 40.764.765.766. and 767. Cases in males have been reported. 768 The legs, particularly the calves and thighs, are most commonly involved; 769 the hands are the second most common site.764. and 766. Occasional cases have occurred on the chest764 and face.764.770. and 771. Papules develop over several months and then growth ceases. Some lesions are pruritic. 769 Spontaneous resolution does not often occur. The etiology is not known; HHV-8 has not been detected. 772 Requena et al have regarded it as a variant of dermatofibroma, 769 a view also expressed by Puig et al. 773 In one case, the lesions were successfully treated by cryosurgery. 769 Surgery and argon laser have also been used.
It is not clear whether multinucleate cell angiohistiocytoma is primarily a vascular or stromal cell tumor. The two main components are increased numbers of ectatic or narrow vessels in the upper and mid dermis, and large angulated multinucleate cells (Fig. 38.23). In addition, there is a less conspicuous component of ‘fibrohistiocytic’ (dendritic) cells scattered between the collagen bundles in a horizontal pattern. 768 These cells express factor XIIIa. 773 The vessels are small venules and capillaries, lined by endothelial cells with prominent nuclei. Angulated multinucleate cells have 3–10 nuclei arranged in a ring or clumped together; they are basophilic. These large cells stain with vimentin but not with macrophage markers (Mac387, CD68, lysozyme), CD31, CD34, S100, or factor XIIIa. 772 A recent paper did show staining of these multinucleate cells with factor XIIIa. 769 Other interstitial cells appear to be indeterminate or Langerhans cells (CD1a positive) or macrophages (lysozyme, CD68, and Mac387 positive).764. and 770. Discrete foci of inflammatory cells are also present. The ultrastructural features have also been described. 765
Multinucleate cell angiohistiocytoma. There are atypical cells and an increase in small vessels in the papillary dermis. (H & E)
Reactive angioendotheliomatosis (reactive angiomatosis) is a cutaneous vascular proliferation that presents as infiltrated, red-to-blue patches and plaques, often with purpura. Necrosis and ulceration may sometimes develop. 774 The lesions may occur at any body site. They measure from 1 to 3 cm or more in diameter. This vascular proliferation may be associated with a variety of conditions, many of which have in common luminal obstruction by thrombi or abnormal proteins. It may occur in association with chronic disseminated intravascular coagulation (DIC), 775 cryoglobulinemia, 776 infections, paraproteinemia with myelomatosis, leukemia, 777 dermal amyloid angiopathy, 778 intravascular immunoglobulin deposits associated with a monoclonal gammopathy, the antiphospholipid syndrome, 779 the lupus anticoagulant, sarcoidosis, 780 systemic diseases, 781 hepatopathy, and arteriovenous fistulae used for hemodialysis.774.782.783. and 784. Some patients are iatrogenically immunosuppressed. 781 A similar lesion has been reported overlying an implanted nickel nail for delayed union of a fracture, and as an idiopathic phenomenon in a child.785. and 786. The lesions usually resolve after the withdrawal or the cure of the initiating process. It must be distinguished from malignant angioendotheliomatosis, an angiotropic lymphoma, and from the intravascular simulant seen in patients with rheumatoid arthritis and variously called reactive angioendotheliomatosis, cutaneous histiocytic lymphangitis, and intravascular histiocytosis (see p. 961), 787 although a recent paper suggested that reactive angioendotheliomatosis may be intravascular histiocytosis. 788
In 2003, Rongioletti and Rebora suggested the concept of the ‘cutaneous reactive angiomatoses’ characterized histologically by different patterns of intravascular or extravascular lobular or diffuse hyperplasia of endothelial cells, pericytes, and sometimes histiocytes. 789 They included the following conditions under this umbrella term:
• reactive angioendotheliomatosis
• diffuse dermal angiomatosis
• acroangiodermatitis (pseudo-Kaposi’s sarcoma) – see page 910
• reactive intravascular histiocytosis – see page 961
• glomeruloid reactive angioendotheliomatosis (with cold agglutinins)
• angiopericytomatosis (angiomatosis with cryoproteins).
This latter condition, described by LeBoit et al, 776 and included here as an example of reactive angioendotheliomatosis actually had a periluminal proliferation of pericytes and intraluminal thrombi. The case referred to as glomeruloid reactive angioendotheliomatosis had lobular proliferations of dilated vessels filled with clusters of closely spaced capillaries with a glomeruloid pattern. 789
Reactive angioendotheliomatosis is characterized by a benign intraluminal proliferation of endothelial cells which may occlude the vascular lumina. The vessels are dilated. A proliferation of vessels also occurs which may have a diffuse, lobular, or mixed pattern. 781 Sometimes the proliferated endothelial cells produce a glomeruloid appearance. The cells may be large and mildly atypical but they are not malignant. Thrombi and protein deposits are sometimes found. The dermis may show reactive (fasciitis-like) alterations. 781 There is usually only minimal inflammation, if any. The cells express CD31, CD34, and factor VIII-related antigen. In one case, the intraluminal cells stained with CD68, a feature seen in intravascular histiocytosis. 788
Granular deposits of IgA, IgM, and complement were found around the vessels and at the dermal–epidermal junction in two cases. 790
Diffuse dermal angiomatosis is a variant of reactive angioendotheliomatosis associated with severe atherosclerosis causing vascular narrowing.791.792. and 793. Clinically there are poorly circumscribed, violaceous and livedoid plaques with frequent ulceration. 794 They may be painful. Lesions are usually on the lower legs. Several cases have been reported on the breast.790.795.796. and 797. The breasts are usually pendulous, and there may be a further factor such as heavy smoking, anticardiolipin antibodies, or subclavian artery stenosis. 797 The term diffuse dermal angiomatosis is a misnomer for these cases and segmental or localized (reactive) angioendotheliomatosis would be a better term. 798 Lesions resolve completely following revascularization of the limb. 794 Interestingly, two cases of vascular proliferation have been reported in the skin distal to an arteriovenous fistula made for hemodialysis: one case resembled reactive angioendotheliomatosis and the other diffuse dermal angiomatosis, indicating the close relationship between these two entities. 783
Cases reported on the breast have responded to isotretinoin,796. and 797. although in one of these cases correction of a subclavian artery occlusion was also carried out. 797 Reduction mammoplasty was successful in a patient with macromastia. 795
In diffuse dermal angiomatosis the hyperplastic endothelial cells diffusely infiltrate the papillary and reticular dermis, sometimes forming small vascular lumina.791. and 792. The cells stain with CD31 and CD34. 799 This contrasts with the intravascular proliferation seen in reactive angioendotheliomatosis.
Acroangiodermatitis (pseudo-Kaposi’s sarcoma) is a vasoproliferative disorder that resembles Kaposi’s sarcoma both clinically and histologically.332.800.801. and 802. The lesions arise in a background of increased venous pressure due to chronic venous insufficiency,803. and 804. paralysis of a limb, 805 congenital arteriovenous malformation,806.807.808. and 809. Klippel–Trenaunay syndrome,810. and 811. activated protein C resistance, 812 or acquired arteriovenous fistula.813. and 814. Acroangiodermatitis has also been reported in an above-knee amputation stump.815.816.817. and 818. The majority occur on the lower part of the legs and on the feet; in cases associated with arteriovenous malformations they are found over the site of the malformation. In most cases affecting the legs there is also stasis dermatitis. The lesions consist of purple papules and nodules with variable surface scale. 819 Those cases associated with venous insufficiency or paralysis of the legs have a characteristic distribution, occupying a triangular region on the extensor surface of the foot and toes, with the most prominent lesions on the first and second toes. 803 Lesions may be unilateral or bilateral, depending on the cause. There is a male preponderance. 820
Rongioletti and Rebora have included acroangiodermatitis with the cutaneous reactive angiomatoses (see above). 789 Ischemia with an increase in vascular endothelial growth factor (VEGF) underlies many of these conditions. In acroangiodermatitis, some of the predisposing factors result in the arteriovenous steal syndrome with distal ischemia and a localized increase in VGF. 811 The treatment is very dependent on the underlying cause. 802
The papules and nodules consist of a proliferation of small dilated vessels in an edematous dermis (Fig. 38.24). The vessels have fairly regular profiles and lack the jagged outline and ‘promontory’ sign seen in early lesions of Kaposi’s sarcoma. 821 Plump endothelial cells, without atypia, line the vessels. The cells are positive for CD31 and CD34, 823 but negative for HHV-8, and D2-40. 824 The perivascular cells do not stain for CD34. 811 A slight perivascular fibroblastic proliferation is also seen but is not marked. Some lesions show nodular collections of vessels with narrow lumina. 822 Extravasated red blood cells, hemosiderin, and a variable round-cell infiltrate are seen around the vascular proliferation. Plasma cells are usually not present. Mast cells may be numerous. 811 The overlying epidermis may show hyperkeratosis.
(A) Acro-angiodermatitis. (B) There is a proliferation of small vessels in an edematous dermis. Fibroblasts are also increased. (H & E)
Rarely, patients with chronic disseminated intravascular coagulation (DIC) develop plaques of purplish discoloration which on biopsy show an apparent increase in small vessels arranged in leashes in the dermis and subcutaneous fat (Fig. 38.25). The vessel proliferation represents organization of thrombosed vessels. 775 A similar ‘angiomatosis’ has been reported in cryoproteinemia. 776
Vascular proliferation in a patient with chronic disseminated intravascular coagulation (DIC). Thrombi are present in some of the vessels. (H & E)
The penile myointimoma is a rare myointimal proliferation described by Fetsch et al in 2000. 825 It affects the corpus spongiosum of the glans penis or corona. It affects all ages including children. The lesions average 1 cm in diameter.
Pierre Masson first described this vascular proliferation in hemorrhoidal veins; he regarded it as a neoplastic process. He named it ‘hémangioendothéliome végétant intravasculaire’. 828 Its importance lies in its histological resemblance to angiosarcoma: the name ‘Masson’s pseudoangiosarcoma’ has also been proposed.829.830.831.832. and 833. The condition is now generally regarded as an unusual pattern of organization of a thrombus within a vein,332.830.832. and 834. or within one or more of the component vessels of various vascular abnormalities. These include cavernous hemangiomas,829. and 831. pyogenic granulomas,643.829. and 835. and lymphangiomas. 836 Organizing hematomas of soft tissues may also show this pattern. 837 In most cases there is a single lesion, but multiple lesions have also been described. 838 They usually present clinically as firm, sometimes painful nodules which appear blue or purple through the overlying skin. 832 On section, typical lesions appear encapsulated and cystic; they contain variable amounts of thrombus. Although lesions can occur at any site, including the tongue, 833 they are most commonly found on the fingers, head and neck, and trunk.829.832. and 839. There is a female predominance in most series.829. and 832. Local excision is curative.
A history of trauma is sometimes present. The lesion is thought to result from the liberation of vascular growth factors in the area. 840
In most examples the proliferation is limited to the lumen of an identifiable vein or vessel in a vascular abnormality (Fig. 38.26). Occasionally there is only a fibrous capsule lacking definite features of a vessel wall. Rarely, the proliferation extends outside the lumen, possibly due to rupture of the wall of the vessel. Masses of papillary processes are present within the lumen and they are almost always associated with some thrombus. Each papillary frond is covered by a single layer of plump endothelial cells. Mitotic figures may be present but they are never frequent. There is no multilayering of the cells, and solid cellular areas, cellular tufts, atypia, and necrosis are not usually evident; however, Renshaw and Rosai have described severe cytological atypia in lesions from the lip. 841 The core of the papillae consists of fibrin or collagenous connective tissue, depending on the stage of organization.
Intravascular papillary endothelial hyperplasia. Vascular channels are present within the lumen of a dilated vein. (H & E)
Electron microscopy confirms the endothelial nature of the cells and demonstrates that they lie on a basement membrane, outside of which are pericytes. 842
Bacillary angiomatosis was first described by Stoler et al in 1983. 843 It is a systemic disorder which was first identified because of cutaneous lesions resembling Kaposi’s sarcoma. 844 Bacillary angiomatosis is caused by two closely related Gram-negative coccobacilli, Bartonella (Rochalimaea) henselae and Bartonella (Rochalimaea) quintana.845.846.847.848. and 849. Bartonella henselae is also a common cause of cat-scratch disease; it has also been associated with a bacteremic syndrome and peliosis hepatis. 845Bartonella quintana is the causative agent of trench fever.
Bacillary angiomatosis occurs primarily in those with HIV infection,844.850.851. and 852. but has also been reported in organ transplant recipients, 845 in patients with leukemia,853. and 854. in patients on systemic steroid therapy, 855 and even in immunocompetent individuals, including children.856.857.858.859.860. and 861. Up to two-thirds of cases are associated with exposure to cats; lesions sometimes follow a bite or scratch. 862 Cutaneous tumors are usually multiple and take the form of pyogenic granuloma-like lesions, subcutaneous nodules or, uncommonly, hyperpigmented indurated plaques, the last form occurring particularly in black people. 863 Leg ulcers may follow scratches from cats. 864 Pyogenic granuloma-like lesions are dusky-red in color, often pedunculated, frequently bleed and may be mildly tender. They resemble the lesions of verruga peruana. Spontaneous regression sometimes occurs. The lesions of bacillary angiomatosis also occur on respiratory and gastrointestinal tract mucosa, and in the heart, liver, spleen, bone marrow, muscle, soft tissue, and brain. Patients often have constitutional symptoms, particularly when extracutaneous lesions are present. 845 Angiomatous and papillomatous lesions of the oral cavity have been reported from Bartonella infections associated with allogeneic bone marrow transplantation and oral graft-versus-host disease. 865 They are a related entity.
Although bacillary angiomatosis is treatable and curable, it may be life-threatening if not addressed. 861 It usually responds to treatment with erythromycin, ciprofloxacin, third-generation cephalosporins, tetracyclines, or aminoglycosides. Erythromycin is the treatment of choice. 864
Superficial lesions are characterized by small round blood vessels in an edematous stroma. Deeper lesions are more cellular and compact. In both forms blood vessels are lined by plump, epithelioid endothelial cells. A background inflammatory cell infiltrate of lymphocytes, histiocytes, and neutrophils is also present. Neutrophils are plentiful in deeper lesions. A peripheral collarette and ulceration are seen in superficial lesions (Fig. 38.27). Pseudoepitheliomatous hyperplasia is a rare event. 866 Organisms are seen as clumps of amphophilic granular material, particularly near neutrophils; they are readily demonstrated by a Giemsa, Warthin–Starry, or Grocott–methenamine silver stain.850. and 867. Immunohistochemical techniques and PCR-based methods have also been used to identify organisms.848. and 868. Ultrastructurally, organisms are seen in the interstitium of lesions and are pleomorphic structures with a trilaminar wall and coarsely granular cytoplasm. 869
(A) Bacillary angiomatosis in a patient with AIDS. (B) Neutrophils, nuclear dust and a clump of granular material (representing bacteria) are present in the stroma between the small vascular channels. (H & E)
(Microscopic slide kindly provided by Dr Philip LeBoit, Department of Pathology and Dermatology, University of California, San Francisco, USA)
Bacillary angiomatosis is usually easy to differentiate from Kaposi’s sarcoma because of the presence of epithelioid endothelial cells, neutrophils, and organisms in the former, and spindle cells and slit-like vessels in the latter. The two conditions may occur concurrently, 870 but bacillary angiomatosis is much less common than Kaposi’s sarcoma in patients with AIDS. Neutrophils are confined to the surface of ulcerated lesions of pyogenic granuloma; this lesion is usually more obviously lobulated. Organisms are not seen in pyogenic granuloma.
The skin lesion of the eruptive phase of Carrión’s disease (bartonellosis), caused by Bartonella bacilliformis, is known as verruga peruana (Peruvian wart).871. and 872. Carrión’s disease is endemic at altitudes between 800 and 2500 meters in parts of Peru, Ecuador, and Colombia. Multiple, miliary superficial hemangioma-like lesions or larger, deeper, sometimes ulcerated lesions occur in this condition. They resolve spontaneously over weeks to months.871. and 872.
Superficial lesions consist of a proliferation of capillary-like vessels in the papillary dermis, with the formation of a collarette. There is an associated inflammatory infiltrate composed of lymphocytes and plasma cells. In nodular lesions there is a multilobular proliferation or more solid aggregation of cells in the dermis, and sometimes also in the subcutis. Vascular lumina are fewer and smaller. The ‘tumor cells’ are large and epithelioid; groups of them are surrounded by a reticulin network. Mitotic figures are frequent. A spindle-cell element is occasionally seen and may make distinction from Kaposi’s sarcoma extremely difficult. Immunohistochemical studies have identified these cells as endothelial in nature, as they are positive for both factor VIII-associated antigen and Ulex europaeus-1 lectin. 871 In regressing lesions there is involution and necrosis of the vascular elements, associated with a heavy infiltrate of lymphocytes, histiocytes, and neutrophils. There is subsequent fibrosis.
The causative organism is not seen by light microscopy but may be found on ultrastructural examination, predominantly in an extracellular location but occasionally in phagosomes. 871 Rocha Lima inclusions, consisting of conglomerates of apparently intracellular cytoplasmic granules that are colored red by Romanowsky–Giemsa stains, may be seen within the endothelial cells. Ultrastructurally these appear to consist of phagosomes containing organisms and interstitial matrix-like material as well as a labyrinth of cisternal channels with similar contents. 871
Lymphangioendothelioma (acquired progressive lymphangioma) is a rare, benign vascular proliferative lesion of lymphatic origin.873.874. and 875. It has not always been accepted as being a lymphatic lesion. 876 The cases reported recently as multifocal lymphangioendotheliomatosis were characterized by hundreds of congenital red-brown skin plaques as large as a few centimeters, with similar lesions throughout the gastrointestinal tract. 877 The thrombocytopenia that accompanied these cases was due to platelet trapping in the channels. The vessels expressed the lymphatic marker LYVE-1. 877 This term has now been proposed as an alternative name for widespread lesions. 875 There are histological similarities to cutaneous angiosarcoma, but the course of the reported cases has been benign. The lesion presents as an erythematous patch or plaque which gradually enlarges, often over many years. 878 Multifocal lesions with thrombocytopenia occur, as already mentioned. There is no site predilection, cases having been reported on the abdomen, leg, arm, and head. 878 Most tumors reported have been in adults841 but this lesion may also arise in children;873.876.878. and 879. rarely, it is congenital.880. and 881. It has been associated with previous femoral arteriography, 882 trauma to a ‘vascular birthmark’, a tick bite, and radiotherapy.883.884. and 885. One case resolved spontaneously. 886
The lesions reported as benign lymphangiomatous papules of the skin (BLAP) are related lesions occurring at sites of radiotherapy.887. and 888. Other vascular tumors may occur in irradiated skin.889.890.891. and 892. In a recent review of 32 cases, Patton, Deyrup, and Weiss used the term ‘atypical vascular lesions’ (AVLs) in preference to BLAP for these cases. 891 A similar view was taken by Mattoch et al, who acknowledged the close clinical and histopathological relationship of these lesions to angiosarcoma. 890 Five of their cases were subsequently shown to have angiosarcoma, indicating that all such cases should have wide local excision with clinical follow-up. 890 Unfortunately none of these cases was stained with D2-40. Brenn and Fletcher also abandoned the term BLAP for these cases and referred to them as AVLs (see p. 920). 893 In summary, it would seem that the concept of BLAP needs reconsideration in the light of long-term follow-up and recent reports. It will be interesting to see whether the angiosarcomas that develop exhibit lymphatic markers. Further follow-up of the benign vascular proliferations in irradiated skin reported by Requena et al in 2002 would also be instructive. 892 They have also been reported in association with an untreated ovarian fibroma. 894 The lesions almost disappeared after removal of the tumor. The lesions present as solitary or multiple papules or vesicles. Disruption of the lymphatic drainage is the probable pathogenesis of these acquired papules. 894
The main feature is the presence of thin-walled interconnecting vascular channels at various levels of the dermis or subcutis. These vessels tend to be arranged horizontally, particularly in the upper dermis. Vessels are smaller, more irregular, angular and cleft-like at deeper levels, and ‘dissect’ the dermal collagen bundles. The vessels are lined by flat to plump endothelial cells with some focal nuclear hyperchromasia and crowding. No frank atypia or mitotic figures have been reported. No intraluminal cell clumps are seen. Vessels contain proteinaceous material, a few red blood cells, or are empty. The vessels are usually surrounded by chronic inflammatory cells, often including a few plasma cells. 895
Immunohistochemical studies are conflicting: endothelial cells have been reported as staining positively or negatively for factor VIII-related antigen and Ulex europaeus-1 lectin, and positively for CD34 and CD31.884. and 896. The cells stain strongly with the lymphatic endothelial marker D2-40, but further cases should be studied before accepting that positivity with this marker is applicable to other cases. 897 They are negative for HHV-8. 898 Basement membrane has been shown to be present by positive staining for type IV collagen and laminin. It has been absent by electron microscopy. A smooth muscle component has also been demonstrated focally, around the vascular spaces.841.876.880.882.883. and 886.
Histologically, this lesion may mimic angiosarcoma and patch-stage Kaposi’s sarcoma. The distinction from well-differentiated angiosarcoma, particularly from the D2-40 positive cases can be very difficult; usually there is more cellular atypia or cellular tufts in angiosarcoma. The long clinical history and the site of the lesion may be helpful. The face and scalp are the usual sites for cutaneous angiosarcoma. Early Kaposi’s sarcoma may be impossible to distinguish from lymphangioendothelioma, but in the former there may be red blood cells, hemosiderin, and an inflammatory cell infiltrate which includes plasma cells. Inflammation is minimal in lymphangioendothelioma. 841 The patch stage of Kaposi’s sarcoma is usually seen in AIDS-related cases, and lesions are often multiple. Although it is also D2-40 positive, Kaposi’s sarcoma stains for HHV-8, which does not occur in lymphangioendothelioma.
Benign lymphangiomatous papules of the skin (BLAP) consist of markedly dilated vascular spaces in the upper dermis exhibiting atypical features which include endothelial cells with plump or flattened nuclei, and numerous small papillary projections. As the lesions descend into the deeper dermis, the spaces become smaller and their lumina irregular, focally dissecting the collagen. 887 Some of the postradiation cases reported as angiosarcomas in the past may be this benign entity, 889 but as Sharon Weiss and colleagues have demonstrated recently, these atypical vascular lesions (as they prefer to call them) may be a precursor, rather than a risk factor for the development of angiosarcoma. 891 The endothelial cells are positive for Ulex europaeus-1 lectin and weakly positive for CD31, CD34, and factor VIII-related antigen. They stain with the lymphatic endothelial marker D2-40. 899
This category of vascular tumors has evoked considerable controversy, particularly the use of the term ‘borderline’ malignancy to describe the behavior of some tumors in this group. 900 The term ‘hemangioendothelioma’ has also attracted criticism because it has been applied to both benign and malignant vascular tumors. 509 While acknowledging the validity of this comment, the designations originally reported will be used here to avoid confusion.
The following tumors have a variable behavior and outcome:
• Kaposi’s sarcoma
• hemangiopericytoma
• Kaposiform hemangioendothelioma
• endovascular papillary angioendothelioma of childhood.
Kaposi’s sarcoma (KS) – OMIM 148000, composed of vessels and spindle-shaped cells, was first described by Kaposi in 1872 as ‘idiopathic multiple pigment-sarcoma of the skin’. 901 The epidemic type of KS, first reported in the United States in 1981, has provoked considerable interest in this previously uncommon condition.902.903. and 904. More than 10 000 articles are currently referenced on the Medline database. 905 There are four clinicopathological types: classic, African (endemic) type, a variant associated with immunosuppressive therapy, and an HIV-associated (epidemic) type. 906 Human herpesvirus type 8 is the etiological agent in all types. 907
The classic type affects predominantly men in the fifth to seventh decades. It is exceedingly rare in children, 908 and uncommon in adolescents. 909 There is an increased incidence in Jews and eastern Europeans, and in people of Mediterranean origin.910.911. and 912. It has also been reported in other races.913.914.915.916.917. and 918. In most cases the lesions are limited to the skin of the extremities, particularly the lower part of the legs and feet; occasionally lymph nodes and other organs are involved. The penis is sometimes involved.919.920. and 921. The incidence of visceral involvement varies in different parts of the world, possibly reflecting the incidence of the different serotypes of the HHV-8 virus. 922 Mild immune suppression and genetic factors may also contribute to the development of KS in some patients with the classic type of the disease.923.924.925.926. and 927. Idiopathic CD4+ lymphocytopenia has been present in rare case of KS.928. and 929. Lymphedema sometimes precedes or follows the appearance of the lesions.930.931. and 932. It has also been associated with lipedema, a form of lipodystrophy. 933 The classic type of KS has a chronic course, with the development of more lesions; death is usually due to other causes. Immunosuppression and an older age are associated with a poorer outcome. 934 There is an increased risk of developing other tumors, particularly malignant lymphomas.935.936.937.938.939. and 940. Spontaneous regression of the lesions may occur.
Seropositivity to HHV-8 has been found in 40% of first-degree relatives of patients with classic KS. 941 The specific relationship with the index person had no significant effect on the prevalence of seropositivity in the family members, suggesting a predominantly non-sexual horizontal transmission route of the virus. 941
The African type of KS is endemic in parts of tropical Africa, with the highest prevalence in eastern Zaire and western Uganda. 942 There are three main subtypes, 943 the most common being nodular disease, similar to classic Kaposi’s sarcoma with a benign clinical course. A more aggressive subtype is characterized by extensive florid and infiltrative skin lesions, which may involve soft tissues and underlying bone. 944 This subtype may present with large, fungating tumors. 945 A lymphadenopathic subtype occurs predominantly in children: in this variety there is involvement of lymph nodes, often without cutaneous lesions, and the prognosis is poor. 946 There is a marked male predominance in endemic African KS. 947 Epstein–Barr virus (EBV), as well as HHV-8, is frequently present in African cases.948. and 949. HIV-related cases are now being seen in parts of Africa, including in women.950. and 951. In one study from Togo, HIV serology was positive in 78.5% of patients with KS. 952 The mortality rate at 2 years was 45% for AIDS-associated KS and 5% for African KS (HIV negative). 952
Kaposi’s sarcoma is a rare complication of organ transplantation, chemotherapy for tumors, and long-term corticosteroid and other immunosuppressive treatment for a variety of dermatological and other conditions, including hepatitis C infection.936.953.954.955.956.957. and 958. It has been reported following corticosteroid treatment of patients with bullous pemphigoid959. and 960. and pemphigus,961.962. and 963. and in patients with lymphoma treated with interferon. 964 It has been seen in renal transplant recipients,965. and 966. but only rarely in cardiac transplant recipients. In two studies the prevalence in renal transplant patients was 0.3% and 1.6%.967. and 968. In cardiac transplant recipients the prevalence has been reported as 0.41%. 969 It may also follow bone marrow transplantation. 970 The male preponderance of cases is less marked than in the other forms of the disease; younger age groups are also affected. Tumors may appear within a short time of commencement of immunosuppressive therapy and may regress spontaneously following cessation of therapy. 954 Progression of classic KS has been associated with the use of rituximab, a monoclonal antibody directed against the CD20 antigen of B cells. 971 There may be a more aggressive clinical course than in classic KS. Death may follow widespread disease, particularly from gastrointestinal hemorrhage.
The epidemic type of KS is associated with AIDS, caused predominantly by the retrovirus human immunodeficiency virus type-1 (HIV-1).972.973. and 974. In West Africa, AIDS is also associated with a second retrovirus, HIV-2; KS is also reported in this group. 975 In Europe, the USA, and Australia, this form of KS is most common in homosexual and bisexual men. HIV-associated KS also occurs in women, children, hemophiliacs, and intravenous drug users. The prevalence of the tumor in risk groups other than homosexual and bisexual men varies. It is uncommon in hemophiliacs and in recipients of blood transfusions who develop HIV infection.976. and 977. There is an intermediate risk in intravenous drug users. 978 In women who have contracted HIV infection by sexual contact, the prevalence of KS is highest in those who have had a bisexual partner. 977 KS in HIV infection is rare before the age of 15. 977 Children of parents in high-risk groups for KS and those children who acquire HIV infection by blood transfusion have the highest risk of developing KS. 979 The proportion of patients with AIDS who develop KS has fallen from 33% in 1981 to about 10% in 2000. 980 It has fallen further in Western countries with the more widespread use of highly active anti-retroviral therapy (HAART), 981 but because 99% of the 40 million patients with AIDS in the world cannot afford HAART, KS is still a very common problem. 982 KS exhibits a less aggressive presentation in patients already receiving HAART. 983
The distribution of lesions in this form differs from that in classic KS. The trunk, arms, head and neck are frequently involved. Lesions are usually multiple. Skin lesions in one case were localized to areas of tacrolimus application. 984 There is frequent involvement of mucosal surfaces and internal organs. The musculoskeletal system may also be involved. 944 In one autopsy study the lungs were involved in 37% of cases, the gastrointestinal tract in 50%, and lymph nodes in 50%; 29% had evidence of visceral lesions without skin lesions. 985 The extent of cutaneous involvement does not correlate well with the extent of visceral disease. 986 The clinical course ranges from chronic to rapidly progressive. Most patients die from opportunistic infections or other complications of AIDS, rather than from KS. In one large series of AIDS patients, survival was best in those whose only manifestation was KS. 987 The lesions may regress spontaneously. 988
Skin lesions are similar in all groups, but in the epidemic form they tend to occur earlier and to be smaller and more subtle. Early lesions are brown to red macules or patches which may resemble a bruise. Papules, nodules, and plaques may be bluish or purple in color and may ulcerate. Unusual presentations include occurrence in a lymphedematous penis, 989 a subtle penile lesion in the classic variant, 990 zosteriform distribution, 991 the Koebner phenomenon,992. and 993. and localization to an area of previous radiation. 994 Conjugal Kaposi’s sarcoma has been reported, but as the cases occurred in an HHV-8 endemic area (both were HIV negative) it may have been a fortuitous occurrence. 995
In 1994, Chang et al identified DNA belonging to a novel virus in tissue affected by KS. This virus, human herpesvirus type 8 (HHV-8), was originally known as Kaposi’s sarcoma-associated herpesvirus (KSHV). 996 It has now been detected in all epidemiological forms of KS.997.998.999.1000.1001.1002. and 1003. The occasional negative case may be related to technical difficulties with the identification. 1004 HHV-8 has also been implicated in the pathogenesis of multicentric Castleman’s disease and primary effusion lymphomas.1005.1006.1007. and 1008. The virus is not ubiquitous in the community and seropositivity is limited to a small percentage of the population. 1009 However, there are small geographical areas in Italy with a high incidence of the classic type of KS; they appear to be ‘hot-spots’ for HHV-8 infection. 1010
The mode of transmission of the virus has attracted considerable attention. HHV-8 is not shed in appreciable amounts in seminal fluid or from the rectum.1010. and 1011. The virus is found in oral secretions. 1012 Saliva containing significant amounts of HHV-8 could be the source of most infections in the general population. 1012 It appears that penile–oral contact is a high-risk activity, but this is obviously not the entire ‘story’. 1010 The virus was found in 44% of the heterosexual partners of patients with classic KS, but this study did not categorize the sexual contact/practices involved.1013. and 1014. A more recent study concluded that a non-sexual horizontal transmission route of the virus seems likely in endemic cases. 941 Transmission through needle sharing in drug use is another documented method of spread. 1015
As with other cell-transforming DNA viruses, infection with HHV-8 alone is probably not sufficient for the development of KS; additional cofactors are probably required.911. and 1016. A good illustration of this is the absence of KS among Ethiopian immigrants to Israel despite high seroprevalence of HHV-8 (39.1%). 1017 Isolated cases have been reported in which HHV-8 has been associated with other viruses, such as EBV1018 and HPV (in the case of penile lesions of KS).1019. and 1020. Furthermore, the explosive incidence of HIV-associated KS in the early 1980s (later in some countries) resulted from colliding epidemics of HIV and HHV-8 infections in the homosexual and bisexual communities.1010. and 1021. KS is said to be 20 000 times more common in patients with HIV infection/AIDS than in the general population. 980 In most cases, no cofactors have been identified. 1022 Attention has focused on the role of iron as a cofactor. Some of the countries with a high prevalence of classic and endemic KS have areas with iron oxide-rich volcanic clays. Anecdotally it has been reported on the palm in a metallurgist with regular contact with iron filings. 1023 The p53 tumor suppressor gene is an inconstant finding and appears not to have a significant role as a cofactor.1024.1025.1026. and 1027. Studies of HLA subtype in patients with HIV-associated and the classic form of the condition have suggested a significant association with HLA-A2, B5, B8, B18, and DR5.967.1028. and 1029. Another study has found a significant association of the classic subtype with DRB1*1104 and DQB1*0604. 1030 The CCR5-promoter mutation A59029G, which protects patients from HIV infection, does not protect against KS. 1031
HHV-8 infects CD19+ B cells as well as T cells, monocytes, endothelial-derived spindle cells, 1010 and CD34+ cells in the peripheral blood of patients with Kaposi’s sarcoma. 1032 It induces the formation of interleukin-6 which stimulates the expression of vascular endothelial growth factor, creating an angiogenic state. 1033 Overexpression of bcl-2 occurs in the majority of cases. 1034 Five subtypes of HHV-8 (A, B, C, D, and E) have been recognized. The subtypes have specific geographic distributions. 1012 Type C infection tends not to be associated with extracutaneous disease compared with subtypes A and B. 907 Three classes of viral gene transcripts are present within the virus. They have different functions, one of which is viral replication. 915
It has been considered that disseminated KS represents a multifocal reactive process, rather than a true sarcoma with the potential to metastasize.1035.1036. and 1037. In one study the cell population was found to be polyclonal, but another has shown a monoclonal population in multifocal lesions. 1038 The controversy can be partly resolved by regarding KS as binomial (hyperplastic/neoplastic), beginning as a reactive proliferation but behaving as a multifocal neoplasm in advanced stages.930.1039.1040.1041. and 1042. Abnormalities have been detected in various angiogenic cytokines and cellular control systems.1039.1043.1044.1045.1046.1047. and 1048. One such example involves the matrix metalloproteinases (MMP), a family of 23 endopeptidases capable of degrading extracellular matrix of connective tissue thereby facilitating tumor invasion. 1049 Some are involved in angiogenesis. Lesional cells in KS are immunoreactive for MMP-1, 2, 3, 7, 9, and 13, but not MMP-14. 1049 This is the rationale for the clinical trials involving inhibitors of MMP (COL-3, a modified tetracycline) for the treatment of this disease. 1049 Interestingly, several anti-retroviral agents now in use have been shown to induce KS regression by directly blocking angiogenesis because of their ability to inhibit the activation of MMP-2. 1049 The spindle cells are thought to be the proliferating component, while the endothelial cell population is thought to undergo a reactive hyperplasia. Other evidence suggests that the spindle-cell elements show endothelial differentiation.1050.1051.1052.1053.1054.1055.1056. and 1057. While KS tissue shows an expression of endothelial markers, KS-derived cell cultures express mesenchymal, non-endothelial markers. This has led to much of the confusion relating to these cells. 1058 Chronic stimulation of endothelial cells (possibly by viral infection) can produce transdifferentiation to a spindle-shaped cell. 1059 The peripheral blood of patients with KS contains circulating cells capable of in-vitro differentiation towards KS-like spindle cells. Chemokines in tissues might lead to their targeted deposition and growth. 1060 Derivation from lymphatic endothelium has also been suggested. 1061 This is supported by the finding of podoplanin, a marker of lymphatic endothelium, in this tumor. It is detected by the antibody D2-40.8.1062.1063. and 1064.
The response of a disease to various treatments may provide a valuable insight into its etiology and pathogenesis. Treatment with interferon alfa-2a has been associated with regression of the lesions although HHV-8 DNA persists in lesional skin.1065. and 1066. However, with HAART there has not only been regression of the lesions but also undetectable levels of HHV-8 DNA.1067.1068. and 1069. The advent of HAART has led to a declining incidence of KS. 1070 Remission of lesions in renal transplant recipients may follow a reduction in the immunosuppressive therapy. 907
The treatment goals for KS were eloquently stated in a recent (2008) editorial in Cancer. 905 They are (1) symptom palliation; (2) shrinkage of tumor to alleviate edema, organ compromise, and psychological stress; (3) the prevention of disease progression; and (4) perhaps cure. Anti-retroviral therapy is the first-line treatment of AIDS-related KS. 1071 For patients with widely disseminated, 1072 symptomatic KS, anaplastic KS, 1073 or in elderly patients with significant disease and comorbidities, pegylated liposomal doxorubicin is the treatment of choice. 905 The taxane paclitaxel gave a significant improvement in quality of life in one patient with advanced HIV-related KS. 1074 In a recent (2008) trial of weekly paclitaxel for advanced aggressive classic Kaposi’s sarcoma in 17 patients, the results were promising, with the drug controlling the aggressiveness of the disease. 1075 The drug was well tolerated. Trials with the matrix metalloproteinase inhibitor COL-3 produced many side effects. 905 Side effects were also a problem in the clinical trial using 9-cis-retinoic acid. 1076 Sirolimus has been used in transplant patients to replace other immunosuppressive drugs.906. and 966. It has antiangiogenic properties.963. and 1077. So too does shark cartilage (oral) from which there is some (anecdotal) improvement. 1033
Radiotherapy has also been used for both disseminated and local disease. 944 In one report a cure rate of 98.7% for classic-type cases was reported at 13 years. 1078 It has also been used for localized penile disease. 920 The use of sildenafil citrate may decrease the fibrosis of the corpus cavernosum that usually follows radiotherapy to this organ. 919 Topical imiquimod 5% cream produced a response in only half the patients treated (HIV negative), and trials of interferon-α have shown some success.909. and 1079. Finally, it should not be forgotten that most solitary cases of classic, localized KS are treated by surgery.
Imaging with 99mTc-MIBI can be used to give more precise staging of the disease. It can be used to assess response to treatment. 361
The microscopic appearance of the lesions is identical in the different types of KS.1080. and 1081. They evolve through patch, plaque, and nodular stages. 1082 The earliest lesions, corresponding to the flat macule or patch stage, are predominantly vascular in nature. Within the dermis there is a proliferation of irregular, often jagged, vascular channels which partly surround pre-existing blood vessels in some areas. This characteristic appearance has been termed the ‘promontory sign’ (Fig. 38.28). 821 The vascular proliferation is also present about appendages and between collagen bundles. The vessels are thin walled and lined by plump or inconspicuous endothelial cells. Scattered groups of perivascular lymphocytes and plasma cells may also be present (Fig. 38.29). Extravasated erythrocytes and deposits of hemosiderin are also found in the dermis.
Kaposi’s sarcoma. Dilated irregular vascular channels surround a pre-existing vessel (‘promontory sign’). (H & E)
Kaposi’s sarcoma. Lymphocytes and plasma cells are present in the stroma adjacent to an irregularly shaped vascular channel. (H & E)
The papules, nodules, and plaques consist of a dermal proliferation of interlacing bundles of spindle cells and intimately related, poorly defined slit-like vessels (Fig. 38.30). The proportion of vessels and spindle cells varies. There is an associated inflammatory cell infiltrate consisting predominantly of lymphocytes and plasma cells. Dilated thin-walled vessels are found at the periphery of the tumor. The spindle-cell component shows variable nuclear pleomorphism. Mitotic figures are present, but not usually frequent. Erythrocytes can be seen within vascular lumina and extravasated in and around the lesion.
Kaposi’s sarcoma. (A) This variant is composed of vascular spaces and spindle-shaped cells. (B) Parts of another lesion show atypical spindle cells and less obvious vessels. (H & E)
Clusters of eosinophilic hyaline globules, varying in size from just visible with the light microscope to larger than an erythrocyte, may be seen within spindle cells and macrophages or in an extracellular location. These were first described in African cases, but are also seen in classic KS and in the epidemic form.1035. and 1083. They resemble Russell bodies and are PAS positive, stain bright red with Mallory’s trichrome stain (Fig. 38.31) and are autofluorescent. 1084 They are seen in early patch lesions in some cases. 1035 They appear to represent effete red blood cell fragments that have been phagocytosed. 1085 Erythrophagocytosis by tumor cells has been described in all stages of Kaposi’s sarcoma. 1086 Apoptosis of endothelial cells is seen quite often in plaques and nodules; it is seen less often in patch-stage lesions. 1087 Lesions regressing as a consequence of HAART become surrounded by a dense fibrous stroma which eventually obliterates the lesion.1070. and 1088. Pharmacologically induced regression is characterized by a complete loss or decrease in spindle cells, increased lymphocytes, and dermal siderophage deposition. 1089
Kaposi’s sarcoma. Small hyaline globules are present in the cytoplasm of some macrophages and spindle-shaped cells. (Mallory’s trichrome ×1500)
Lymphangioma-like KS is an uncommon variant accounting for less than 5% of all cases.1090.1091. and 1092. It consists of grossly dilated channels lined by flattened endothelial cells with a bland appearance (Fig. 38.32). 1093 There are irregular, anastomosing channels, closely applied to the dermal collagen, with slender papillary projections into the lumen. Most of the spaces are devoid of erythrocytes, further enhancing the lymphangioma-like features. Clinically, the lesions may have a bulla-like appearance. 1094 There is usually an admixture of more typical lesions. There is strong reactivity of the tumor cells with anti-HHV-8 latent nuclear antigen-1. 1095
Lymphangioma-like area in a Kaposi’s sarcoma. Deeper areas resembled the more usual tumor. (H & E)
Pyogenic granuloma-like KS is a recently described variant of KS which closely simulates pyogenic granuloma. 673 No distinguishing histological features are present. Only the presence of HHV-8 latent nuclear antigen-1 (LNA-1) allows a distinction to be made. 673
Intravascular KS is another histological variant. 1096 It does not seem to be associated with a more aggressive behavior. The vascular channels have the morphological features of veins. There is an intravascular growth of interlacing fascicles of spindle cells with formation of cleft-like spaces, hyaline globules, and a lymphoplasmacytic infiltrate. The cells stain for HHV-8, CD31, and CD34. 1096 The D2-40 marker was not used.
An anaplastic form has been reported in African and sporadic cases.943.1097. and 1098. The anaplastic lesions exhibit greater cellularity, nuclear pleomorphism, and more frequent mitotic figures, and may have areas resembling angiosarcoma or fibrosarcoma.930. and 1097. In several cases, anaplastic KS developed in a lymphedematous (postmastectomy) arm, thus mimicking Stewart–Treves syndrome.1073. and 1099. Epidermal changes vary with the lesion and include atrophy and ulceration over raised lesions. A peripheral epidermal collarette is sometimes present about papules and nodules. Lesions with prominent hyperkeratosis sometimes occur. 1100
Immunohistochemical studies have demonstrated the presence of CD31 and D2-40, the new marker for lymphatic endothelium in nearly every case tested.8.1062.1063. and 1101. CD34 and factor VIII-related antigen were noted in some of the earlier studies.1101. and 1102. HHV-8 can be detected by PCR in paraffin-embedded tissue, although the viral load appears to be low.1103. and 1104. It is much easier to perform immunohistochemistry using the commercially available antibody against HHV-8, latent nuclear antigen-1 (LNA-1). It detects HHV-8 in the majority of cases. 1105 It is largely confined to the spindle cells in the nodular phase, but it is found in the endothelial cells of the slit-like vessels in the early patch stage (Fig. 38.33). 917 Calcitonin receptor-like receptor, which plays an important role in angiogenesis, is strongly expressed in KS and other vascular tumors. 1106
Kaposi’s sarcoma. Stromal spindle cells stain with antibody to HHV-8. (Immunoperoxidase stain for HHV-8)
The immunohistochemical demonstration of HHV-8 DNA may be a useful adjunct in the diagnosis of KS by fine needle aspiration, 1107 and when difficult lesions are encountered. 1108
The early vascular lesions are subtle and must be differentiated from telangiectases, pigmented purpuric dermatosis, acroangiodermatitis, and low-grade angiosarcoma. The vessels in KS are usually more irregular than in most developmental or acquired telangiectases, pigmented purpuric dermatosis, or acroangiodermatitis. 822 An inflammatory infiltrate which includes plasma cells is found in some early lesions of KS. Pyogenic granuloma has now entered the list of differential diagnoses with the report of a pyogenic granuloma-like variant of KS (see above). In low-grade angiosarcoma there is usually some evidence of cellular atypia. Small intravascular endothelial buds are sometimes seen. Irregular jagged vessels, dissection of collagen bundles by vascular structures, and an inflammatory cell infiltrate may all be seen in low-grade angiosarcoma. The commercial availability of anti-HHV-8 test kits has removed much of the difficulty associated with the diagnosis of patch-stage lesions. A case combining features of KS and diffuse dermal angioendotheliomatosis was described prior to the availability of this test. Its exact nature is therefore speculative. 1109
Lesions with a spindle-cell component must be differentiated from cutaneous smooth muscle tumors, some forms of dermatofibroma (fibrous histiocytoma), 1110 and spindle-cell hemangioendothelioma. 501 Smooth muscle tumors lack the intimate mingling of spindle cells, slit-like vessels, and eosinophilic globules, and are usually positive for desmin intermediate filaments on immunoperoxidase staining. Aneurysmal fibrous histiocytoma can usually be differentiated from Kaposi’s sarcoma because of the presence of foamy macrophages, multinucleate giant cells, and the overlying epidermal changes, which range from epidermal hyperplasia to basal budding resembling hair differentiation.
The lesions of spindle-cell hemangioendothelioma closely mimic Kaposi’s sarcoma, but the component vessels are usually more cavernous and, focally, the endothelial cells are epithelioid and vacuolated. Eosinophilic globules have not been reported in these tumors.
Hemangiopericytoma is a tumor first delineated by Stout and Murray in 1942. 1111 Enzinger and Smith have divided this uncommon tumor into two groups. 1112 The usual form of hemangiopericytoma (adult type) occurs in adults and rarely in children. It arises in deep soft tissues, particularly the lower extremities, pelvis and retroperitoneum, and occasionally other organs.1112.1113. and 1114. It rarely arises in the subcutis.1112.1113. and 1115. A second form (congenital or infantile hemangiopericytoma) is present at birth or arises in the first year of life; it is more common in boys.1116.1117. and 1118. The tumors in this group are multilobulate and arise almost exclusively in the subcutis of the head and neck, extremities or trunk; occasionally they arise in the dermis.1112. and 1119. Most are solitary, but multiple lesions also occur.1116. and 1120. Conventional hemangiopericytomas have an unpredictable course and may recur after treatment; they may metastasize, predominantly to the lungs and bone. 1112 Fewer than 25% of cases behave as malignant tumors. 1121 Congenital and infantile tumors have a benign clinical course, although they may grow rapidly during infancy or be complicated by hemorrhage.1112.1122.1123. and 1124. Hemangiopericytoma in children older than 1 year does not differ in behavior from the adult type. 1125
The concept of hemangiopericytoma has been questioned. 1121 Many soft tissue sarcomas may have a ‘hemangiopericytoma-like pattern’. 1126 Fletcher questions the existence of this entity, because tumor cells have only a limited morphological similarity to pericytes. Furthermore, a diagnosis is made without positive criteria. 1121 Nappi et al have concluded that ‘hemangiopericytoma represents both a pattern and a pathological entity … and should be considered to represent an exclusionary interpretation’. 1127 Infantile hemangiopericytoma is now considered by most authorities to be the same entity as infantile myofibromatosis, or one end of a spectrum of infantile myofibroblastic lesions.1121.1128. and 1129.
The adult type exhibits the characteristic pattern of tightly packed cellular areas surrounding endothelium-lined ramifying vessels. The cell boundaries are poorly defined; the nuclei are round or oval. Tumor cells are separated from the endothelial cells by a basement membrane and are themselves surrounded by a meshwork of reticulin fibers. Histological variations include myxoid areas, fibrotic areas, and, rarely, osseous and cartilaginous metaplasia. 1112 The deep variant with mature fat reported as a lipomatous hemangiopericytoma1130 has been regarded by others as a fat-containing variant of solitary fibrous tumor. 1131 Epithelioid histiocytomas may exhibit hemangiopericytoma-like areas.1132. and 1133. Tumor cells express vimentin only; they are negative for α-smooth muscle actin and α-sarcomeric actin. 746 Normal vascular pericytes are uniformly reactive for vimentin and α-isoforms of actin, and sometimes stain for desmin and HLA-DR. Tumor cells also stain for factor XIIIa, CD34, CD57, and HLA-DR1134 but these are not specific markers. Pericytes express 3G5 ganglioside. 1135 Further studies are needed to see if this marker leads to the retention of hemangiopericytoma as a diagnostic entity. Several studies have stressed the difficulty in predicting the behavior of these tumors from their histological appearance, but frequent mitotic figures, necrosis, hemorrhage, and increased cellularity are features that indicate a poor prognosis. 1113 DNA flow cytometry and immunohistochemical stains for PCNA are not helpful in predicting outcome in these tumors. 1136
Congenital and infantile hemangiopericytomas have the typical pattern of cells and vessels as described above, but they are multilobulate, with perivascular and intravascular tumor outside the main tumor mass.1112. and 1120. Endothelial proliferation within vascular lumina has been described. 1112 Mitotic figures and necrosis may be seen but do not indicate a bad prognosis as these tumors have a benign course. 1116 Hemangiopericytoma-like areas are sometimes seen in infantile myofibromatosis (see p. 821), which affects infants and children and may involve skin and subcutis. 1137 Conversely, foci of cells having a spindled, myoid appearance typical of myofibroblasts can be seen in typical cases of infantile hemangiopericytoma. These cases show focal staining for actin. In another case smooth muscle actin stained numerous cells around the vascular spaces, and some endothelial cells. 1129 This is similar to infantile myofibromatosis. 1129
Ultrastructural studies of adult cases have shown that the tumor cells are partially or completely surrounded by well-formed basement membranes. Pinocytotic vesicles and cytoplasmic filaments, sometimes with dense-body formation, are seen within the cells. 1138 More than half the cases do not show pericytic features. 1139
Kaposiform hemangioendothelioma is a locally aggressive vascular proliferation in infants and young children that usually presents in the skin as a single lesion, although a multifocal congenital case has been reported.1140.1141. and 1142. Adult cases also occur. 1143 Requena and Ackerman believe it should be renamed Kaposiform hemangioma because there are no reports of unequivocal metastasis. 509 Weiss and colleagues, on the basis of their study of 33 cases in which two cases had regional lymph node involvement at the time of the primary excision, concluded that it should continue to be classified as a vascular tumor of intermediate malignancy. No distant metastases occurred. 1144
A variety of sites can be involved, including deep soft tissues of the upper extremities, thigh, chest wall, scalp, neck, and retroperitoneum.1144. and 1145. Earlier reports had emphasized peritoneal and retroperitoneal sites.1146. and 1147. It is often associated with Kasabach–Merritt syndrome (OMIM 141000) or lymphangiomatosis.335. and 1140. Cases without the Kasabach–Merritt phenomenon are usually less than 8 cm in diameter, suggesting that tumors that grow no larger than this size are less likely to trap platelets in sufficient quantity to cause thrombocytopenia. 1148 If present, the Kasabach–Merritt syndrome can be treated with a stepwise regimen of prednisolone, dipyridamole, and interferon. 1149 HHV-8 has not been detected in several cases. 1140 The outcome depends on the site and the extent of the disease. Deaths have been reported from the associated Kasabach–Merritt syndrome and lymphangiomatosis. 1144
As with Kaposi’s sarcoma, there are interconnecting sheets or nodules of spindled endothelial cells lining slit-like or crescent-shaped vessels. The spindle-cell fascicles are generally shorter and narrower than those found in Kaposi’s sarcoma. 515 Small rounded vessels may also be seen. Unlike Kaposi’s sarcoma, there may be nests of epithelioid-like endothelial cells with eosinophilic cytoplasm containing hemosiderin and cytoplasmic vacuoles. Cellular atypia is minimal and mitoses infrequent. Hemosiderin may also be present in the spindle cells. Hyaline eosinophilic globules similar to those in Kaposi’s sarcoma are also seen. An occasional finding is microthrombi in vessel lumina. In the skin and subcutis there is surrounding dense hyaline fibrosis. Spindle cells stain for CD34 and focally for CD31.1145. and 1150. In the series of 33 cases reported by Sharon Weiss and colleagues, the endothelial cells in the nodules expressed CD31, CD34, and FLI1, but not GLUT-1, LeY, or HHV-8. 1144 In a series from Japan, all four cases expressed D2-40. 720 In three cases the response was markedly reactive, and in the fourth it was reactive in the peripheral area of KS-like proliferative capillaries and negative in the surrounding dilated vessels. 720 In contrast, the staining was reversed in tufted angioma with partial positivity in the dilated vessels but no staining in the cannonball-like proliferative capillaries. 720 The tumor has some similarities to spindle-cell hemangioendothelioma, but the latter occurs predominantly in adults, is superficial, and may have cavernous vessels and phleboliths. Kaposi’s sarcoma is usually multifocal; it is very rare in children, except as the endemic form.
Endovascular papillary angioendothelioma of childhood has had a confusing taxonomic evolution. Dabska, in the first report of this very rare vascular tumor, described it as malignant endovascular papillary angioendothelioma because it was locally invasive and appeared to have the potential to metastasize. 1151 Subsequently, others suggested that this lesion should be classified as of borderline malignancy because of its good long-term prognosis, minimal cellular atypia, and controversial metastatic potential.1152. and 1153. It was next regarded as a low-grade angiosarcoma1154. and 1155. and, now, on the basis of its staining pattern, it has been renamed papillary intralymphatic angioendothelioma (PILA), 9 reflecting its presumed lymphatic origin.
The majority of reported cases have been in children; rarely, adults are affected. 1156 There is a report of this lesion developing in a pre-existing vascular malformation in an adult. 1156 A recent case arose in a lymphangioma circumscriptum, further evidence of a lymphatic origin for the Dabska tumor. 1157 A tumor showing some similarities to this entity has also been reported in a background of lymphedema. 1158 In the majority of cases the lesions are present at birth. They occur in a variety of sites, either as a diffuse swelling of the skin or as an intradermal tumor. The tumors enlarge and may eventually invade deeper soft tissues and bone. Metastasis to regional lymph nodes and lungs has been reported. 1151 One of the original cases reported by Dabska subsequently died of widespread pulmonary metastases, but it may have been a retiform hemangioendothelioma. 1155
As recent cases have all been benign, this lesion may need re-evaluation once larger series have been published. 351 For this reason this tumor is now discussed with tumors that have ‘a variable behavior and outcome’.
There is some variability.1151. and 1159. Irregular vascular channels are present in the dermis and subcutis. They are lined by endothelial cells ranging from flattened to columnar in shape. Some cells have a ‘hobnail’ appearance. Within the lumina of these vessels are papillary structures covered by similar cells. The cores of the papillae are avascular and consist of fibrous tissue or peculiar eosinophilic hyaline globules, some with central clearing. These features are not seen in retiform hemangioendothelioma.
Some of these vascular structures have a glomeruloid appearance. Lymphocytes are seen both within the lumina of the vascular channels, in intimate association with the endothelial cells, and also in the extravascular stroma. There is nuclear hyperchromatism and mitotic figures are present. 1151
Immunohistochemical and ultrastructural studies have confirmed the endothelial nature of the tumor cells and identified the hyaline globules as basal lamina-like material.1152. and 1153. Tumor cells are positive for vimentin, factor VIII-related antigen, CD31, and focally for CD34. They are negative for keratins, EMA, S100 protein, and desmin. 9 Vascular endothelial growth factor receptor type 3 (VEGFR3), a marker for lymphatic endothelia, was positive in all cases studied. The lymphatic endothelial marker D2-40 was positive in all three cases of Dabska tumor studied in one report, 1063 confirming the lymphatic nature of this tumor.
Several of the rare vascular tumors previously categorized as being of variable or uncertain behavior have been reclassified as malignant tumors, although it is acknowledged that they are usually associated with an excellent prognosis. The following tumors will be considered in this category:
• angiosarcoma and lymphangiosarcoma
• epithelioid hemangioendothelioma
• retiform hemangioendothelioma
• composite hemangioendothelioma
• malignant and atypical glomus tumors.
A review of cutaneous vascular tumors, including malignant lesions, was published in 2008 by Goh and Calonje. 351
The distinction between malignant tumors showing blood vessel differentiation (angiosarcoma) and those showing lymphatic differentiation (lymphangiosarcoma) has often been unclear in the past. The advent of the new marker D2-40 has allowed a more reliable distinction to be made. As it has been found that a subgroup of cases of apparent angiosarcoma, including the postradiation cases of the breast, express D2-40, it is proposed to discuss these tumors together, but a brief discussion of lymphangiosarcoma will occur at the end of this section.
About 60% of all angiosarcomas occur in the skin or soft tissue, with 50% of cutaneous cases occurring on the head and neck, 1160 although this was the site in 96% of cases in one series. 1161 It has a male to female ratio of 2.5 : 1.
There are three main clinicopathological subtypes: idiopathic cutaneous angiosarcoma of the head and neck, angiosarcoma complicating lymphedema, and postirradiation angiosarcoma. 1036 A miscellaneous category is sometimes added.
Idiopathic cutaneous angiosarcoma of the head and neck most commonly involves the upper part of the face or the scalp of elderly people. 1166 Periorbital or eyelid involvement is sometimes seen.1167. and 1168. Men are affected more frequently than women. 1169 The lesions are single or multifocal, bluish or violaceous nodules, plaques or flat infiltrating areas; they may occasionally bleed or ulcerate. In one series they measured 1–9 cm in diameter, but much larger cases are recorded. 1161 Rare clinical presentations include recurrent angioedema of the face, 1170 alopecia, 1171 an inflammatory process, 1172 a rosacea-like eruption, 1173 a rhinophyma-like lesion, 1174 and involvement of an area of radiodermatitis. 1175 A clue to the presence of a head and neck angiosarcoma may be the presence of a positive head-tilt maneuver. 1176 This simple test involves placing the head at a level below the heart. If a vascular tumor is present, the area of involvement becomes markedly more violaceous and engorged. 1176
Thrombocytopenia may develop as a consequence of enlargement of the primary lesion or the development of metastatic deposits. 1177 Extensive local growth is common and margins are difficult to define surgically. Metastasis to regional lymph nodes and to the lungs occurs, often after repeated surgical excision of the primary growth. A reduction in adhesion molecules, such as cadherin, has been implicated in the local invasiveness and metastasis of angiosarcoma. 1178 The prognosis is poor. In one series, only 15% of patients survived for 5 years or more after diagnosis. 1169 It was 34% in a more recent series. 1161 This may reflect the fact that clinical diagnosis is often delayed until the lesions are advanced. Prolonged survival was recorded in three cases on the nose, but their diagnosis was made early, which may have contributed to the better survival. 1179 Complete spontaneous regression of the tumor has been reported.1180. and 1181.
Since 1948, when Stewart and Treves recognized the syndrome of postmastectomy lymphangiosarcoma, the association of such tumors with chronic lymphedema has become well known. 1182 In their original series they described the appearance of purplish-red raised macular or polypoid tumors in the chronically edematous arm of women who had undergone radical mastectomy on that side. The tumors appeared on average 12.5 years after surgery. Similar tumors have been reported in men after mastectomy. They have also occurred in cases without lymph node dissection and without preceding edema. Much less commonly, similar tumors arise in the limbs of patients with chronic lymphedema due to other causes. 1183 These include congenital lymphedema (Milroy’s disease), lymphedema following other types of surgery, and chronic venous stasis.1184.1185.1186.1187.1188.1189. and 1190. Lymphedematous tissue is immunologically vulnerable for the development of infections and neoplasms. 1191 Lymphangiosarcoma has also been reported in association with chronic filarial lymphedema, but appears to be a rare complication of this condition. 1192 Rarely, cutaneous angiosarcoma has been reported in lymphedematous extremities which have developed as a complication of malignancies other than breast carcinoma, such as Hodgkin’s disease.1193. and 1194. An angiosarcoma has also developed in a lymphedematous abdominal pannus. 1195
Postirradiation angiosarcomas are rare and have been documented after radiotherapy for a variety of benign and malignant conditions.1196. and 1197. At least 10 cases of angiosarcoma have been reported following irradiation of benign hemangiomas; the median latent period from irradiation to diagnosis of angiosarcoma was 21.8 years.1198. and 1199. Angiosarcomas have also been reported following treatment of eczema, 1200 tinea capitis, and sinusitis. 1201 This form of angiosarcoma is more common following therapy of a variety of malignant tumors, 1198 including carcinoma of the breast1202. and 1203. and cervix. 1204 With the advent of breast-conserving surgery and radiation as a common method of treating breast cancer, a type of angiosarcoma is seen that differs from the angiosarcoma arising in Stewart–Treves syndrome by its localization to the breast, its lack of lymphedema, and its shorter latency period.1205. and 1206. This new variant is multifocal at presentation in nearly half of the cases. 1205 Multiple capillary lobules are a deceptively benign presentation of these postradiation angiosarcomas of the breast.1207. and 1208. Angiosarcomas seem to form a morphological spectrum with the radiation-associated lesions described by Brenn and Fletcher in 2005 as atypical vascular lesions (AVLs). 893 AVLs comprise cases that were included in the past as acquired progressive lymphangioma and benign lymphangiomatous papules of the skin (BLAP). 893 Although the benign-appearing lesions have behaved in a benign fashion, one case progressed to angiosarcoma. Both the AVLs and the angiosarcomas arising in this setting stained with D2-40, but the number of cases tested was small. 893 So too, do the lesions reported as BLAP. The latent period between treatment and diagnosis is reported to be shorter than for benign conditions – an average of 12 years in one report1196– but it may be much shorter.1202. and 1203.
Angiosarcomas have been reported to arise in pre-existing benign vascular tumors, 1209 including lymphangioma, 179 and ‘port wine’ stains, 1210 and in benign and malignant peripheral nerve sheath tumors. 1211 An epithelial variant has arisen in a deep-seated plexiform schwannoma. 1212 They have also occurred as a complication of varicose ulceration, 1213 trauma, 1214 arteriovenous fistulae, 1215 renal transplantation,1216. and 1217. hereditary epidermolysis bullosa, 1218 xeroderma pigmentosum, 1219 a gouty tophus, 1220 retained foreign materials such as shrapnel, surgical sponges, and adjacent to a Dacron vascular prosthesis. 1221 Angiosarcoma of the scrotum has arisen after radiation therapy for cancer of the rectum. 1222 HHV-8 has been detected in several cases, but specifically excluded in others.1215.1223. and 1224. It has been detected in the lesions of two patients with AIDS who developed angiosarcoma. 1225 Intravascular dissemination of an angiosarcoma, mimicking angioendotheliomatosis, has been described. 1226 These two conditions have been noted to arise in association with each other. 1227 A case that produced granulocyte colony-stimulating factor has been reported. 1228
Sharon Weiss and colleagues in 2008, reviewed 69 cases of sporadic cutaneous angiosarcoma. 1229 Because the group included not only head and neck tumors (49 of the group), but also tumors at other sites, this discussion has been placed at the end of the recognized subtypes. Tumors associated with radiation or lympedema were excluded. Recurrences developed in 18 patients (26%) and metastasis in 15 (22%). Thirty patients died of the disease. Older age, anatomic site (trunk and extremities cases did worse than head and neck cases), necrosis, and epithelioid features correlated with increased mortality. 1229 These features seemed to diminish in importance with increased tumor size.
Various treatments have been used for angiosarcoma with limited success. They include surgery, paclitaxel (a taxane with antiangiogenic and apoptotic effects),1230. and 1231. docetaxel, 1232 liposomal doxorubicin, 1233 liposomal doxorubicin and radiotherapy, 1234 liposomal doxorubicin and intralesional pegylated interferon alfa, 1235 combination interferon alfa-2a and 13-cis-retinoic acid, 1236 interferon alfa-2b with interleukin-2 and surface radiotherapy, 1237 arterial interleukin-2,1238. and 1239. and combined chemotherapy and radiotherapy in various schedules. 1240 Surgery is the preferred treatment for lesions on the scalp but obtaining negative margins can be difficult. 1241 Postoperative radiation is recommended for tumors at this site. 1241
The appearances are similar in the three groups.1162.1163.1164. and 1182. The lesions are poorly circumscribed dermal tumors which infiltrate subcutaneous fat and other tissues and often have a multifocal distribution. Angiomatous and solid patterns may be seen (Fig. 38.34). In the angiomatous areas a meshwork of anastomosing dilated vessels extends between pre-existing dermal collagen bundles and around skin appendages. The vessels are irregular and lined by crowded endothelial cells, which range in appearance from virtually normal-looking to plump atypical protuberant cells with enlarged hyperchromatic nuclei (Fig. 38.35). Papillary processes may extend into the lumen of the vessel. In the solid areas of the poorly differentiated tumors the cells vary from spindle-shaped to polygonal. A spindle-cell pattern may predominate. 1239 Some areas may resemble Kaposi’s sarcoma. 1242 Cytoplasmic vacuoles resembling primitive vascular lumina may be seen in some cells. Reticulin stains show that the cellular proliferation is on the luminal side of the reticulin fibers in the angiomatous areas. Generally, as architectural differentiation decreases, cytological atypia and cell size tend to increase. 1163 Lymphocytic infiltrates are commonly seen and may obscure the underlying lesion, particularly in well-differentiated tumors with minimal cellular atypia. 1163 Mast cells appear to be increased.1219. and 1243. Distinguishing the well-differentiated angiomatous areas from benign vascular proliferations depends on the recognition of cellular atypia, the presence of crowding of lining cells and of solid papillary clusters of cells, and an irregular interconnecting pattern of vessels. The use of antibodies specific for signal transduction pathways is a feasible method for distinguishing benign from malignant endothelial processes in paraffin-embedded tissue. There is strong expression of mitogen-activated protein kinase (MAPK) in benign endothelial tumors and a greatly decreased expression in angiosarcoma. 1244 Infectious endothelial lesions (Kaposi’s sarcoma and verruga peruana) stain strongly. 1244 Caveolin, a scaffolding cell membrane protein, has a higher level of expression in benign vascular tumor than in angiosarcomas, including the well-differentiated variants. 1245 It has been suggested that caveolin expression might be useful in separating benign and malignant vascular tumors. 1245 The differentiation of early lesions from early lesions of Kaposi’s sarcoma may be dependent on clinicopathological correlation.
(A) Angiosarcoma. (B) The vascular channels are lined by atypical endothelial cells. (H & E)
(A) Angiosarcoma. Telangiectatic spaces are present. (H & E) (B) The endothelial cells stain for CD31. (Immunoperoxidase stain)
Poorly differentiated tumors with polygonal and epithelioid cells can resemble carcinomas and even amelanotic melanoma. 1246 Immunohistochemical markers for keratins and S100 protein will help to distinguish between these tumors, although S100-positive angiosarcomas have been reported.1246. and 1247. Epithelioid, spindle and granular cell variants of angiosarcoma have also been described. 515 The epithelioid variant sometimes expresses keratin (see below).1183.1248.1249. and 1250. When it occurs on the breast, the epithelioid angiosarcoma may be mistaken for mammary carcinoma, although the latter tumor does not express CD34 and CD31. In epithelioid angiosarcoma up to a third of the neoplastic cells can express MIB-1. 1251 Another histological variant is the verrucous angiosarcoma, in which there are striking verrucous changes in the overlying epidermis. 1252 Another variant, which may be mistaken for cutaneous lymphoma, is the pseudolymphomatous variant. 1253 It consists of irregular anastomosing vascular channels with pleomorphic endothelial cells which express CD31, CD34, and D2-40. There is a dense lymphocytic infiltrate between the vessels obliterating and destroying some of the channels. Germinal centers may also be present. 1253
Many immunohistochemical markers for endothelial cell differentiation have been used, including factor VIII-related antigen, Ulex europaeus-1 lectin,601. and 1254. PAL-E, 1255 CD34, 1162 and CD31. 1102 Epithelioid angiosarcomas often express CK8 and CK18 (approximately 50%), whereas the non-epithelioid types express CK7, CK8, and CK18 in about 20% of cases. One case of CD30+-epithelioid angiosarcoma has been reported. 1256 Epithelial membrane antigen (EMA) is expressed in about a third of non-epithelioid angiosarcomas, 1250 although the figure was lower than this in a recent study. 1257 Thus caution is needed in interpreting EMA positivity as evidence for an epithelial tumor. The monoclonal antibody AE1, which does not react with cytokeratins 7, 8, and 18, is advantageous in the differential diagnosis of angiosarcoma and carcinoma.1250. and 1258. Angiosarcomas also express WT1. 430 A variety of vascular tumors, including angiosarcoma express annexin II, also known as p36. 1259