Benign appendageal tumors with combined features, such as sebaceous units in association with either eccrine and/or apocrine elements, are occasionally a source of diagnostic confusion. Differentiation is probably influenced not only by the endogenous genetic potential of the stem cells giving rise to the tumor, but also by exogenous stromal effects such as regional vascularity and molecular attributes of the epidermis, basement membrane, dermis, or subcutis (7). In human skin, keratinocytes with markers of stem cells appear to reside in the “bulge” and basal layer of the epidermis (9). Cells with the capability of proliferating and differentiating along appendageal lines (progenitor cells) appear to traffic from this site to the basilar epidermis, the germinative layer of the sebaceous gland, and the hair follicle (6,8,10).
Carcinomas of the epidermal appendages occur and have the potential to metastasize. Three types of glandular carcinoma are recognized: carcinomas of sebaceous glands, eccrine glands, and apocrine glands. Other epithelial carcinomas with varying follicular differentiation have been reported, such as pilomatrix carcinoma, malignant proliferating trichilemmal tumor, trichilemmal carcinoma, and trichoblastic carcinoma (Table 30-1).
As noted, the histopathology of a tumor often can be interpreted by comparing it to the histology of normal skin appendages. For example, clear or pale cell change due to cytoplasmic glycogen may recapitulate the embryonic acrosyringium in a nodular hidradenoma or the glycogenated follicular outer root sheath in a trichilemmoma. Cytoplasmic fat vacuoles that indent the nucleus are typical of sebaceous differentiation, which is seen in sebaceous glands, sebaceous adenomas, and sebaceous carcinomas. Many adnexal tumors contain a distinctive, eosinophilic hyaline stroma that is associated with epithelial elements. This stroma assists in differentiating many basaloid adnexal neoplasms from basal cell carcinoma. Eccrine tumors commonly exhibit a nearly acellular hyalinized eosinophilic stroma with dilated thin-walled vessels (e.g., nodular hidradenoma, eccrine poroma). The duct and tubule formation and focal keratinization seen in eccrine tumors may differentiate them from renal cell or other metastatic clear cell carcinomas.
Historically, immunohistochemistry has been of little value in definitively distinguishing the phenotypic patterns of adnexal neoplasms (11). However, with the identification of specific genetic mutations associated with appendageal neoplasms, immunohistochemistry has become more useful (12). Further research on adnexal tumors using current technologies, especially genomic sequencing and proteomic studies, may yield additional molecular markers useful for classifying these lesions.
Lesional histology evolves with time and in response to local effects. A trichilemmal cyst, possibly stimulated by inflammatory mediators, may transform into a proliferating trichilemmal cyst and ultimately into a malignant proliferating trichilemmal tumor. It is almost certain that tumor progression, the “process whereby tumors go from bad to worse,” operates in appendageal tumors as it does in most tumor types. However, from a clinical point of view, appendageal neoplasms should not always be regarded as precursors of malignancy.
Classification of Appendageal Tumors
The four groups of appendageal tumors with differentiation toward hair, sebaceous glands, apocrine glands, and eccrine glands can be further subdivided according to a gradient of decreasing differentiation. The three major subgroups are (a) hyperplasias, hamartomas, and cysts; (b) benign tumors; and (c) malignant tumors (Table 30-1). This classification is similar to the approach of the World Health Organization International Histological Classification of Tumours monograph (13).
The hyperplasias, hamartomas, and cysts are composed of mature or nearly mature structures. The benign neoplasms generally show less complete differentiation than the hyperplasias, but well-developed, differentiated, or partially differentiated structures are present. The malignant neoplasms exhibit a lower degree of differentiation, and these lesions may not produce differentiated cells or well-formed appendageal structures. Although most of the appendageal tumors fit well into one of the entities listed in Table 30-1, tumors manifesting intermediate degrees of differentiation are occasionally encountered and may be difficult to classify (14).
Development of the Appendageal Tumors
Appendageal tumors develop either from primary epithelial germs, from multipotent stem cells, or from cells of preexisting structures. In 1948, it was proposed that cutaneous tumors differentiating toward hair, sebaceous glands, or apocrine glands developed from primary epithelial germ cells and were primary epithelial germ tumors. Hyperplasias, adenomas, and benign epitheliomas are thought to arise therefore from primary epithelial germ cells that had differentiated before the onset of neoplasia (15).
Given that multipotent stem cells give rise to cutaneous appendageal structures and are present in the skin, appendageal tumors likely arise from stem cells that possess the potential of differentiating into tumors that manifest hair, sebaceous gland, eccrine gland, or apocrine structures (6). Appendageal tumors associated with genetic syndromes, such as multiple cylindromas, multiple trichoepitheliomas, and the nevoid basal cell carcinoma syndrome are probably derived from abnormal multipotent stem cells that are genetically predisposed to proliferate and form tumors with altered appendageal structures. This scenario of tumor formation is likely to apply to sporadic occurrences of the same appendageal tumors. In some instances, multipotent stem cells may manifest more than one differentiation pattern, as is seen most commonly on the scalp, where a syringocystadenoma may exhibit three different lines of appendageal differentiation.
Terminology
The terms nevus, hamartoma, and carcinoma used in the classification of the appendageal tumors require definition.
The term nevus is used in the literature in two different ways, referring (a) to a tumor composed of nevus cells derived from melanocytes (nevocellular nevus, melanocytic nevus, pigmented nevus) or (b) to a lesion that is usually present at birth and is composed of mature or nearly mature structures, such as nevus sebaceous, eccrine nevus, nevus verrucosus, and nevus flammeus. To avoid confusion, it is advisable to use the term nevus with a qualifying adjective and to assume that nevus without a qualifying adjective designates a tumor composed of melanocytic nevus cells.
The term hamartoma applies to those nevi that have no melanocytic nevus cells and, like congenital hyperplasias, are composed of mature or nearly mature structures. Hamartoma, derived from the Greek word hamartanein (to fail, to err), was chosen as the designation for “tumorlike malformations showing a faulty mixture of the normal components of the organ in which they occur” (16).
The term epithelioma has been used by many authors as a synonym for carcinoma. However, because the literal meaning of the word is “tumor of the epithelium,” the term may be employed as a designation of benign as well as of malignant tumors of the epithelium, provided that a qualifying adjective is added (17). Because the term has been used for both benign and malignant neoplasms, it seems prudent to avoid its use except in a clearly defined context. The term carcinoma is used for malignant epithelial tumors, including those that locally invade and cause tissue destruction as well as those that have the capacity for distant metastasis. As in previous editions, we have selected terms that are widely understood and used, not necessarily those that are above all semantic dissection (18).
TUMORS WITH DIFFERENTIATION TOWARD HAIR FOLLICLE COMPONENTS
Hair Follicle Nevus
Clinical Summary. The hair follicle nevus, also called a congenital vellus hamartoma, presents most commonly at birth as a small nodule on the face. It is a rare, small tumor (19–22). A few reported cases have raised the possibility of a novel neurocutaneous syndrome (23,24). Features of hair follicle nevus may be noted in porokeratotic adnexal ostial nevus (25–27). A mucinous variant of hair follicle nevus has also been described (28).
Histopathology. There are focally increased vellus hair follicles, occasionally accompanied by a few small sebaceous glands. The lesion classically lacks the cartilage seen in accessory tragus and the central dilated pore seen in trichofolliculoma (Fig. 30-1) (19,29,30).
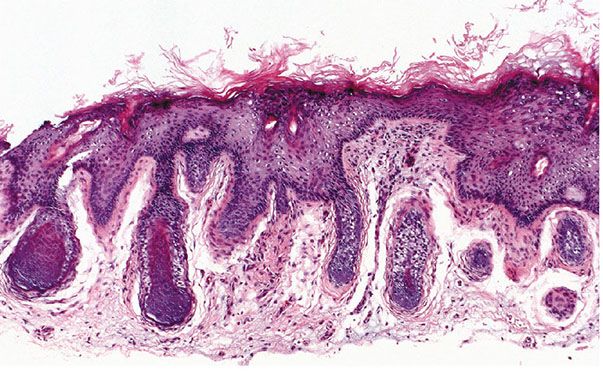
Figure 30-1 Hair follicle nevus. Increased numbers of small hair follicles are present in the dermis.
Differential Diagnosis. The small vellus hair follicles present in the central face resemble the hair follicles in a hair follicle nevus. Other entities to consider in the differential diagnosis include accessory tragus and hair follicle hamartoma (19,23,29,31). Some authors consider these entities to be on a spectrum of similar hamartomas (29,32).
Principles of Management. These lesions may be removed primarily for cosmetic reasons (19,20). There is no report of these lesions behaving in a biologically aggressive manner.
Trichofolliculoma
Clinical Summary. Trichofolliculoma occurs in adults as a solitary lesion, usually on the face but occasionally on the scalp or neck (33). More rarely, the lesions can be multiple or present on the extremities or genitalia (34). Trichofolliculoma consists of a small, skin-colored, dome-shaped nodule. Frequently, there is a central pore. If such a central pore is present, a wool-like tuft of immature, often white hairs may be seen emerging from it, which is a highly diagnostic clinical feature (33).
Histopathology. On histologic examination, the dermis contains a cystic cavity lined by squamous epithelium; this cyst contains keratinized material and frequently fragments of hair shafts that are birefringent on polaroscopic examination (Fig. 30-2) (35). In cases with a visible central pore, the cystic space is continuous with the surface epidermis, an indication that the lesion is analogous to an enlarged, distorted hair follicle. In some cases, multiple cystic spaces are present in the dermis.

Figure 30-2 Trichofolliculoma. In the dermis, there is a keratin-filled cyst lined by squamous epithelium with associated follicular structures.
Radiating from the wall of these cysts are many small, yet well-differentiated hair follicles (Fig. 30-3). Well-developed secondary hair follicles often show a hair papilla (Fig. 30-3). These follicles usually have an outer and an inner root sheath, the latter of which may contain eosinophilic trichohyaline granules and a central small hair shaft (Fig. 30-3). These small, fine hairs are visualized best where the secondary hair follicles appear in cross-sections. Small groups of sebaceous gland cells may be embedded in the walls of the secondary hair follicles (36).

Figure 30-3 Trichofolliculoma. Emanating from the cyst wall are numerous small “secondary” hair follicles, some of which show, in addition to a hair and an outer root sheath, an inner root sheath with trichohyaline granules.
In some of the more rudimentary secondary follicles, one observes a central horn cyst in place of a hair, as seen also in trichoepithelioma (37). Glycogen can be demonstrated in the outer root sheath of the secondary hair follicles, just as it is seen in mature hair follicles (35). The size of the secondary hair follicles can range in size from vellus to terminal hairs. Abnormal tertiary follicles have been identified in association with involuting secondary follicles (38).
Trichofolliculoma stains positively with CK16 and CK17 in the primary cystic structure and the secondary hair follicles, confirming outer root sheath differentiation. The basal layer of the primary follicle and the secondary follicle are positive for CK15, a marker for hair follicle stem cells (38). In all trichofolliculomas, epithelial strands interconnect the secondary hair follicles. Because these epithelial strands differentiate in the direction of the outer root sheath, the peripheral cell row is palisaded, and because of their glycogen content, the cells within the strands may appear large and vacuolated (39).
The lesional stroma of trichofolliculoma is rich in CD34-positive fibroblastic cells. These cells are oriented in parallel bundles that encapsulate the epithelial proliferations similar to that of the normal fibrous root sheath (38). S100-positive cells may be seen in the stromal component (38). Rarely, the stroma of trichofolliculoma can have a striking myxoid appearance (40). Trichofolliculoma has been reported in association with angiomyxoma, basal cell carcinoma, and nevus lipomatosus superficialis (41–43).
Additional Studies. Experimental evidence suggests that trichofolliculomas undergo morphologic changes corresponding to the normal hair follicle cycle (44). Trichofolliculoma is thought to arise from a single dilated pore, which then differentiates toward the lower segment of the hair follicle via induction from the perifollicular stroma (45). Transgenic mice that overexpress a stabilized form of β-catenin develop trichofolliculomas (46). Therefore, increased signaling through the β-catenin pathway may play a role in the development of trichofolliculomas in humans. Transgenic mice expressing increased epidermal expression of the Noggin gene, an inhibitor of bone morphogenetic protein signaling, exhibit appendageal tumors resembling trichofolliculomas, likely via the Wnt and Shh signaling pathways (47–49).
Principles of Management. No treatment is necessary. For cosmetic reasons, these lesions are often treated by excision, electrodesiccation, or curettage. Complete removal is curative.
Folliculosebaceous Cystic Hamartoma
Folliculosebaceous cystic hamartoma exhibits many features of a trichofolliculoma. While the two lesions may have a similar pathogenesis through disordered epithelial–mesenchymal interactions, folliculosebaceous cystic hamartoma does not arise from trichofolliculoma (50,51). The folliculosebaceous cystic hamartoma consists of a folliculosebaceous proliferation with a cystlike infundibular dilation and a mesenchymal stroma with variable fibroplasia. It typically manifests a more prominent sebaceous differentiation and a more prominent surrounding stroma than trichofolliculoma (52). The folliculosebaceous cystic hamartoma may have rare focal follicular differentiation, but it typically lacks the secondary and tertiary hair follicles seen in trichofolliculoma and sebaceous trichofolliculoma (45,51).
Sebaceous Trichofolliculoma
Clinical Summary. Sebaceous trichofolliculoma shares features of folliculosebacous cystic hamartoma and trichofolliculoma (51). It occurs in anatomic regions rich in sebaceous follicles, such as the nose. This tumor exhibits a centrally depressed, fistula-like opening from which terminal hairs and vellus hairs protrude (53).
Histopathology. There is a rather large, irregularly shaped, centrally located cavity lined by squamous epithelium. Many radially arranged pilosebaceous follicles connect to the cavity. These follicles contain sebaceous ducts and numerous well-differentiated, large sebaceous lobules, as well as hair follicles containing partially terminal and partially vellus hairs (53).
Differential Diagnosis. Unlike folliculosebaceous cystic hamartoma, which presents as a solitary papule or nodule, sebaceous trichofolliculoma clinically presents with a central porelike opening that exhibits protruding hairs. Histopathologically, folliculosebaceous cystic hamartoma is additionally characterized by stromal mesenchymal prominence, a finding not seen in the previously reported cases of sebaceous trichofolliculoma (54).
Principles of Management. These benign lesions do not require removal unless there are symptomatic or cosmetic reasons to do so. Simple excisions have been used to treat these lesions.
Dilated Pore and Pilar Sheath Acanthomas
Clinical Summary. Dilated pore and pilar sheath acanthomas clinically share with trichofolliculoma and sebaceous trichofolliculoma the presence of a central pore. Histologically, this central pore is seen as a large cystic space that is continuous with the surface epidermis, lined by squamous epithelium, and filled with keratinous material.
The dilated pore, originally described in 1954 by Winer, occurs primarily on the face as a solitary lesion, predominantly in adult males (55). It can also occur on the eyelid (56). It has the appearance of a giant comedone and typically does not possess palpable induration (55).
The pilar sheath acanthoma, first described in 1978, is usually found on the skin of the upper lip of adults. It is seen elsewhere on the face only rarely (57). It occurs as a solitary skin-colored nodule with a central porelike opening (58).
Histopathology. The dilated pore differentiates toward the infundibulum both architecturally and cytologically (59). It shows a markedly dilated pilar infundibulum lined by an epidermis that is atrophic near the ostium but hypertrophic deeper in the cystic cavity. In the deeper cystic cavity, it shows many rete ridges and irregular thin proliferations into the surrounding stroma (Figs. 30-4 and 30-5). The keratin-filled cystic cavity may extend into the subcutaneous fat. Small sebaceous gland lobules and vellus hair follicles may be attached to the lining epithelium, particularly in deeper portion of the cavity (55).
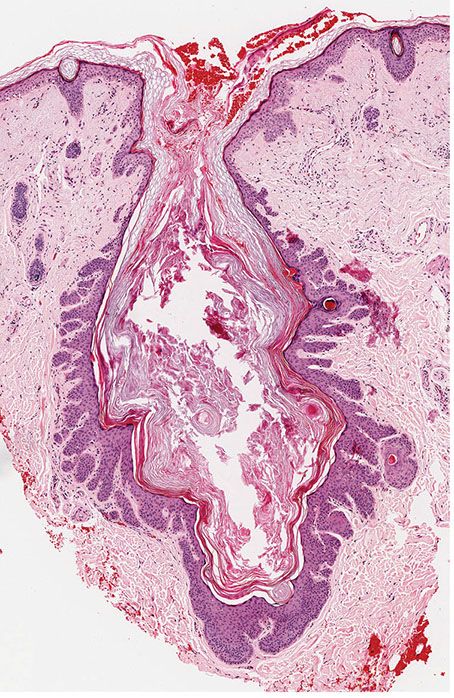
Figure 30-4 Dilated pore of Winer. An irregular cystic cavity containing keratin is present in the dermis.
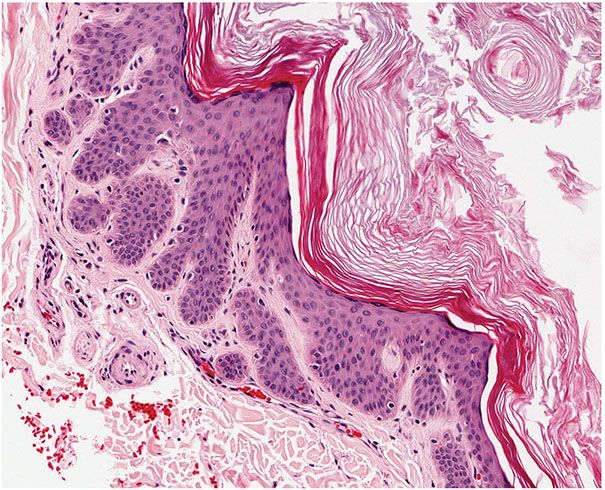
Figure 30-5 Dilated pore of Winer. Small projections of epithelial cells extend from the wall of the cyst into the surrounding dermis.
The pilar sheath acanthoma differs from the dilated pore by showing a larger, irregularly branching cystic cavity. In place of thin proliferations, as seen in the dilated pore, numerous lobulated masses of cells radiate from the wall of the cystic cavity into the dermis and the subcutaneous tissue (Fig. 30-6) (58). The pilar sheath acanthoma can exhibit some features of the outer root sheath, in that portions of the lesion may show peripheral palisading and contain varying amounts of glycogen (Fig. 30-7) (60). Abortive hair follicles may be seen, though no hair shafts should be seen in the central cystic cavity (57). It has been suggested that pilar sheath acanthomas can manifest differentiation of all components of the pilosebaceous unit.
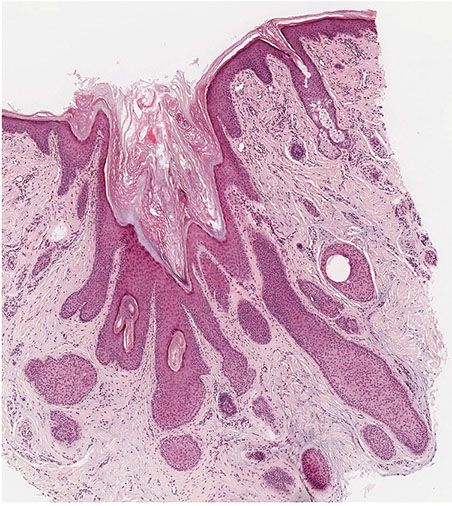
Figure 30-6 Pilar sheath acanthoma. Lobulated masses of cells extend from the cystic cavity into the surrounding dermis.

Figure 30-7 Pilar sheath acanthoma. The lobulated masses are composed of bland cells resembling infundibular keratinocytes.
Principles of Management. These lesions can be treated with simple excision.
Fibrofolliculoma and Trichodiscoma
Clinical Summary. Fibrofolliculomas and trichodiscomas consist of 2- to 4-mm, yellow-white, smooth, dome-shaped lesions often on the face. Formerly these lesions were considered separate entities, but are now recognized as the same lesion and appear different on the basis of sectioning plane, anatomical site, and “age” of the lesion (61).
The lesions can be solitary or multiple. Multiple fibrofolliculomas have been described in association with kidney neoplasms, lung cysts, and spontaneous pneumothorax in Birt–Hogg–Dubé syndrome (62). In patients with the Birt–Hogg–Dubé syndrome, the lesions are numerous and present mainly on the face and neck. Other less common mucocutaneous features of Birt–Hogg–Dubé include multiple epidermal cysts, severe facial sebaceous hyperplasia, and oral papules (63). Multiple fibrofolliculomas may present outside of Birt–Hogg–Dubé as well. In a patient reported with a large connective tissue nevus, fibrofolliculomas were present in large numbers within and surrounding the connective tissue nevus (64).
Multiple trichodiscomas also occur without fibrofolliculomas as small papules either widely disseminated or localized to one area (65,66). Familial multiple discoid fibromas, formerly termed familial multiple trichodiscomas, is a distinct syndrome unrelated to mutations in folliculin, whereby patients develop lesions around their pinnae at a young age, and lack the systemic features seen in Birt–Hogg–Dubé. Discoid fibromas lack the follicular epithelial features of fibrofolliculomas commonly seen in Birt–Hogg–Dubé (67,68).
Histopathology. Fibrofolliculomas exhibit a central distorted hair follicle that is surrounded by a mantle of basophilic, fibrous stroma (Fig. 30-8) (64,69). Numerous thin, anastomosing bands of follicular epithelium extend into this stroma (Fig. 30-9).
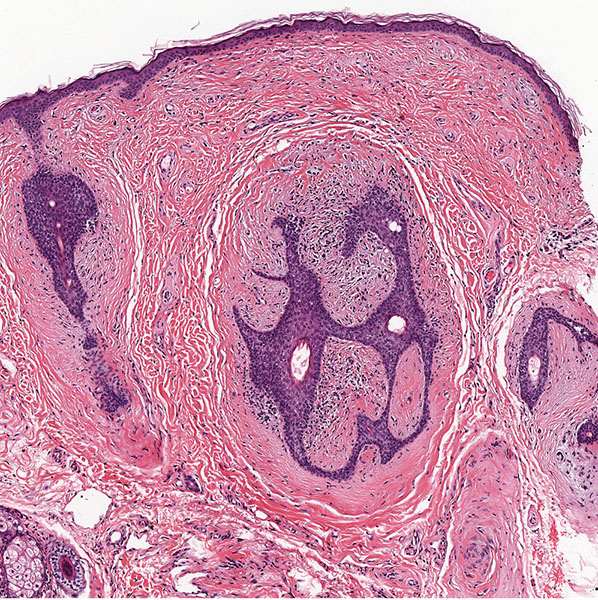
Figure 30-8 Fibrofolliculoma. In the dermis, there are distorted follicular structures associated with epithelial cords and a fibrous stroma.

Figure 30-9 Fibrofolliculoma. Numerous thin, anastomosing bands of follicular epithelium extend from a central cystic structure into the bland fibrocellular stroma.
Trichodiscomas are seen in the dermis as an area of fine fibrillary connective tissue containing ectatic blood vessels (Figs. 30-10 and 30-11). Epithelial hyperplasia resembling follicular structures, including those seen in fibrofolliculomas, is often seen. A hair follicle is usually found at the margin of the lesion (64,69). Trichodiscomas have been regarded as hamartomas of the mesodermal component of hair disks (65). A symplastic variant of trichodiscoma with an increased number of spindled cells demonstrating multinucleation and “ancient change” has been described (70).
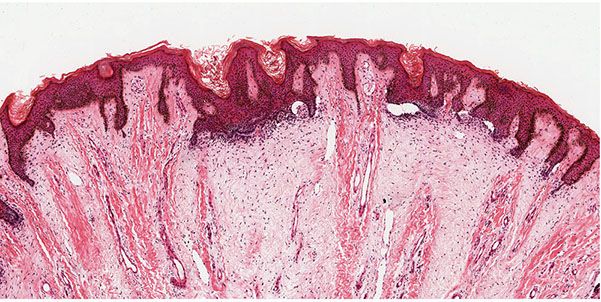
Figure 30-10 Trichodiscoma. Present in the dermis are areas of fine, fibrillar connective tissue.
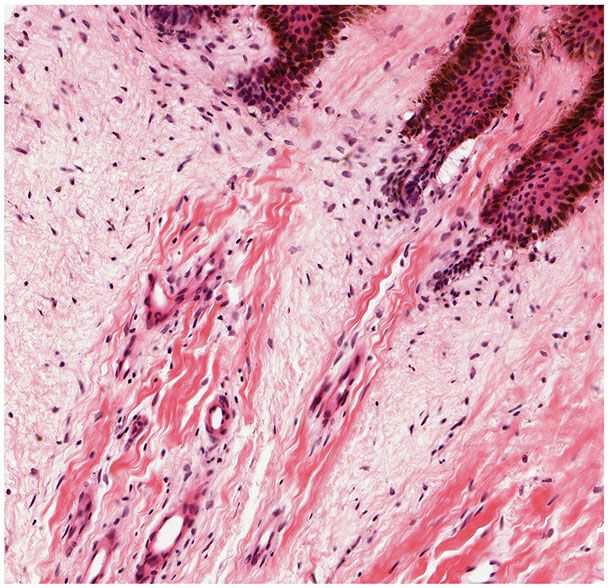
Figure 30-11 Trichodiscoma. The altered connective tissue exhibits ectatic vessels, a myxomatous appearance, increased numbers of mature fibrocytic cells, and basaloid follicular epidermal hyperplasia.
Additional Studies. The immunophenotypic characteristics of syndrome-associated and sporadic types of fibrofolliculoma/trichodiscoma are identical and consist of perifollicular spindle cells that are positive for vimentin and CD34 and negative for factor XIIIa. These findings suggest that these lesions originate from the hair follicle mantle (71).
Birt–Hogg–Dubé syndrome is associated with mutations in the folliculin gene on chromosome 17p. The folliculin gene product is believed to function as a tumor suppressor, which delays cell cycle progression through late S and G2/M phases (72). Aberrant folliculin expression may also directly impact adherens junctions, dysregulating the integrity of the epithelium (73).
Certain cases of Birt–Hogg–Dubé have been described with features overlapping with tuberous sclerosus, whereby patients develop renal angiomyolipomas as well as angiofibromas. Similarly, tuberous sclerosis patients have been reported with fibrofolliculomas (74). This phenotypic overlap is thought to occur because of folliculin and tuberous sclerosis complex proteins both acting in a common pathway: the mammalian target of rapamycin (75,76).
Principles of Management. Although recurrence is common, multiple ablative therapies have been used including excision, dermabrasion, laser therapy, and electrodesiccation.
Trichoepithelioma
Clinical Summary. Trichoepithelioma occurs either as a solitary lesion or as multiple lesions. The name trichoepithelioma is preferred over epithelioma adenoides cysticum and multiple benign cystic epithelioma, because it indicates that this tumor differentiates toward hair structures.
Multiple trichoepitheliomas are transmitted as an autosomal dominant trait (77). Generally, the first lesions appear in childhood and gradually increase in number (78). Numerous rounded, skin-colored, firm papules and nodules ranging in size from 2 to 8 mm are distributed mainly in the nasolabial folds, but also the nose, forehead, and upper lip. Occasionally, lesions are seen also on the scalp, neck, and upper trunk.
Ulceration of the lesions occurs rarely, and may indicate an occult malignancy. There have been a few cases of basal cell carcinoma arising in the setting of multiple trichoepitheliomas (79–82). Rarely, patients with multiple familial trichoepitheliomas can have malignant transformation of their lesions to trichoblastic carcinoma (83,84). Pigmented lesions, including malignant melanoma and blue nevus have been described in association with trichoepitheliomas (85).
The simultaneous presence of trichoepithelioma and cylindroma, the latter of which is also dominantly inherited, has been observed repeatedly (see Cylindroma section).
Solitary trichoepithelioma occurs more commonly than multiple trichoepitheliomas (78). It is not inherited and consists of a firm, elevated, flesh-colored nodule usually less than 2 cm in diameter. Its onset usually is in childhood or early adult life (86). Most commonly, the lesion is seen on the face, but it may occur elsewhere including the vulva (87,88). The presence within the same tumor of a solitary trichoepithelioma and an apocrine adenoma has been described (89). A solitary trichoepithelioma can also arise in the setting of a facial scar (90).
Giant solitary trichoepithelioma, which measures several centimeters in diameter, is a distinct variant of trichoepithelioma. It arises in later life and occurs most commonly on the thigh and the perianal region, but has also been reported on the face (91).
Histopathology. As a rule, multiple trichoepitheliomas are superficial dermal lesions. They appear well-circumscribed, small, and symmetric on histologic examination. Horn cysts are the most characteristic histologic feature, although they may be absent in some lesions. They consist of a fully keratinized center surrounded by basophilic cells that have the same appearance as the cells in the basal cell carcinoma. The basophilic cells in trichoepithelioma tend to lack the high-grade atypia and mitoses, which may be prominent in carcinomas (Figs. 30-12 and 30-13). The keratinization is abrupt and complete, in the manner of so-called trichilemmal keratinization, as opposed to the horn pearls of squamous cell carcinoma. Quite frequently, one observes one or a few layers of cells with eosinophilic cytoplasm and large, oval, pale, vesicular nuclei situated between the basophilic cells and the horn cysts (78).
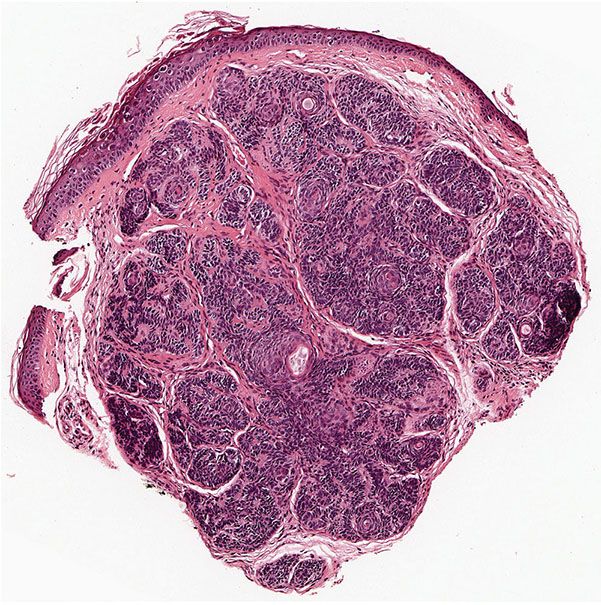
Figure 30-12 Trichoepithelioma. A dermal tumor with a prominent stroma, horn cysts of varying sizes, and basaloid epithelial formations.
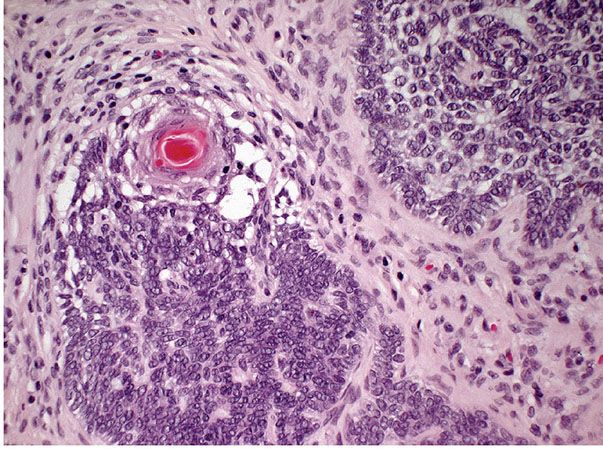
Figure 30-13 Trichoepithelioma. Fibroblasts encircle and are tightly associated with the basaloid epithelial islands, lacking the retraction artifact typical of basal cell carcinoma.
A second major component of multiple trichoepitheliomas are tumor islands composed of basophilic cells of the same appearance as epidermal or skin appendage basal cells. These islands are arranged usually in a lacelike or adenoid network, but may occasionally form solid aggregates (Fig. 30-13). These tumor islands show peripheral palisading of their cells and are surrounded by a stroma with increased numbers of fibroblasts. The fibroblasts encircle and are tightly associated with the basaloid islands, lacking the retraction artifact typical of basal cell carcinoma (Fig. 30-13). Both the adenoid and the solid aggregates show invaginations, which contain numerous fibroblasts and resemble follicular papillae, also known as papillary mesenchymal bodies (Fig. 30-14) (92).
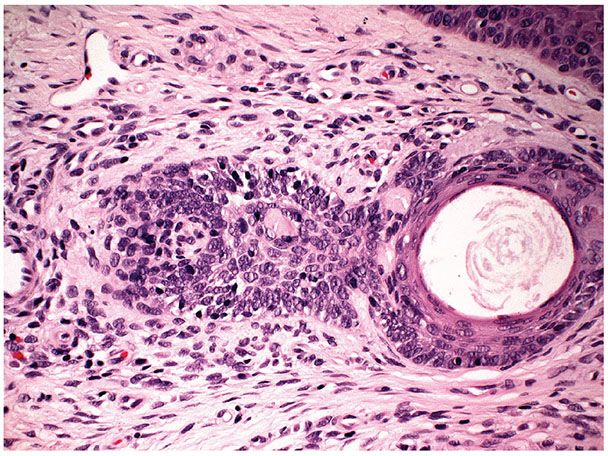
Figure 30-14 Trichoepithelioma. Some of the basophilic islands form papillary mesenchymal bodies resembling follicular papillae.
Additional findings, observed in some but not all trichoepitheliomas, are the presence of a foreign-body giant cell reaction in the vicinity of ruptured horn cysts, calcium deposits either within the foci of the foreign-body reaction or within intact horn cysts, and amyloid (78,93). Occasionally, some lesions in patients with multiple trichoepitheliomas show relatively little differentiation toward hair structures. They contain only a few horn cysts, with many areas resembling basal cell carcinoma (78). Therefore, on a histologic basis, it may be difficult to distinguish definitively between multiple trichoepitheliomas and basal cell carcinoma (see Differential Diagnosis).
Solitary trichoepithelioma often has a high degree of differentiation toward hair structures. Solitary lesions with relatively little differentiation toward hair structures could be classified as keratotic basal cell carcinoma. If a lesion is to qualify for the diagnosis of solitary trichoepithelioma, it should contain numerous horn cysts and abortive hair papillae and show only few areas with the appearance of basal cell carcinoma (86). Mitotic figures should be rare or absent, and the lesion should not be unduly large, asymmetric, or infiltrative.
Additional Studies. It is assumed that the basophilic cells surrounding horn cysts are similar to hair matrix cells and that the horn cysts represent attempts at hair shaft formation. The eosinophilic cells seen occasionally around horn cysts probably represent cells with initial keratinization and are similar to the nucleated cells seen in normal hair shafts at the keratogenous zone. An immunohistochemical analysis using a panel of monoclonal antikeratin antibodies suggests that trichoepitheliomas differentiate toward the outer root sheath (94). The expression of CK15 in a subset of trichoepitheliomas suggests that these tumors contain cells that are related to hair follicle stem cells in the follicular bulge (95).
Numerous mutations in the CYLD gene, an ubiquitin-specific protease, have been identified in up to 88% of patients with Brooke–Spiegler syndrome (77). Brooke–Spiegler is characterized by the development of multiple adnexal cutaneous neoplasms including spiradenoma, cylindroma, spiradenocylindroma, and trichoepithelioma. Patients with multiple familial trichoepitheliomatosis, a phenotypic subtype of Brooke–Spiegler, also have CYLD mutations in 44% to 72% of cases (77,96–101).
The close relationship between trichoepithelioma and basal cell carcinoma has been explained on the theory that they have a common genesis from pluripotent cells, which, like primary epithelial germ cells, may develop toward hair structures (102). This hypothesis is supported by the fact that PTCH gene mutations are seen in both tumors (103). Additionally, basal cell carcinoma has been reported arising in a trichoepithelioma in a patient with known Brooke–Spiegler syndrome (82).
Differential Diagnosis. The difficulty of differentiating multiple trichoepitheliomas from keratotic basal cell carcinoma on histologic grounds has been pointed out. Diagnosis may be assisted by clinical data, such as the number and distribution of the lesions and the presence of hereditary transmission. The presence of well-formed horn cysts, papillary mesenchymal bodies, and lack of high-grade atypia and mitoses favor the diagnosis of trichoepithelioma (92). Other histologic features that favor basal cell carcinoma include the presence of myxoid stroma and stromal retraction or clefting around the basaloid islands (93).
Several immunohistochemical stains may be used to differentiate trichoepithelioma from basal cell carcinoma. A study of trichoepitheliomas and basal cell carincomas using a microarray found that the most reliable stains for distinguishing the two are CD10, CK15, CK20, and D2-40 (102). CD10 stains the stromal cells but not the epithelial cells in trichoepitheliomas, whereas in basal cell carcinomas, CD10 is present in the epithelial cells and rarely in the stromal cells (104,105). Staining tumors for CK20 to identify Merkel cells, which are more common in trichoepithelomas, may be also used (106). PHLDA1 (pleckstrin homology-like domain, family A, member 1) staining typically highlights trichoepitheliomas and is either negative or stains less than 25% of the cells in basal cell carcinomas (107,108).
The differentiation of multiple trichoepitheliomas from the nevoid basal cell carcinoma syndrome on histologic grounds can be just as difficult. This diagnosis also requires clinical information. Although both diseases are dominantly inherited and have multiple lesions, the lesions in multiple trichoepitheliomas present mainly in the nasolabial fold, remain small, and hardly ever ulcerate, whereas the lesions in the nevoid basal cell carcinoma syndrome are haphazardly distributed and, especially in the late, “neoplastic” phase, can grow to considerable size, ulcerate deeply, and show severely destructive growth. In addition, patients with the nevoid basal cell carcinoma syndrome almost invariably show multiple skeletal and central nervous system anomalies and frequently show multiple palmar and plantar pits (109).
Principles of Management. Solitary lesions are typically excised. Mutiple lesions can be treated with cryotherapy, laser ablation, or dermabrasion.
Desmoplastic Trichoepithelioma
Clinical Summary. Desmoplastic trichoepithelioma, typically a sporadic lesion, has sufficient clinical and histologic characteristics to be regarded as a distinct variant of trichoepithelioma. The term desmoplastic trichoepithelioma appears preferable to the designation sclerosing epithelial hamartoma because it stresses the relationship of the lesion to solitary trichoepithelioma (110,111).
Clinically, the tumor almost always is located on the face, measures from 3 to 8 mm in diameter, and is markedly indurated. In many instances, there is a raised, annular border and a depressed nonulcerated center, causing the lesion to resemble granuloma annulare (110). Most commonly, the lesion appears in early adulthood, but it can appear in the second decade of life (111). It is much more common in females than in males. Familial desmoplastic trichoepitheliomas have been reported infrequently (112,113).
Histopathology. The three characteristic histologic features are narrow strands of tumor cells, horn cysts, and a desmoplastic stroma (Figs. 30-15 and 30-16) (110). The tumor strands range from one to three cells thick and are composed of small basaloid cells with prominent oval nuclei and scant cytoplasm. Usually there are numerous horn cysts, which in some cases are large (114). Considerable amounts of densely collagenous and hypocellular stroma are present. Large aggregates of tumor cells are not seen. Foreign-body granulomas at the site of ruptured horn cysts and areas of calcification within some of the horn cysts are common (114).

Figure 30-15 Desmoplastic trichoepithelioma. Horn cysts and narrow strands of epithelial cells are embedded in a fibrous stroma.

Figure 30-16 Desmoplastic trichoepithelioma. The strands of tumor cells are composed of small basaloid cells and are usually one to three cells thick.
Differential Diagnosis. The resemblance to microcystic adnexal (eccrine) carcinoma and syringoma may be considerable, especially if a superficial specimen is taken for biopsy. Like desmoplastic trichoepithelioma, microcystic adnexal carcinoma has horn cysts, strands of basaloid cells, and a dense desmoplastic stroma. Histologic features such as skeletal muscle and subcutaneous tissue invasion, perineural invasion, ductal differentiation, and the presence of mitotic figures are significantly more frequent in microcystic adnexal carcinoma. CK19 is also more commonly positive in microcystic adnexal carcinoma (114). Features such as keratocysts, keratin granuloma, and calcification are more frequent in desmoplastic trichoepithelioma. It should be noted however that desmoplastic trichoepithelioma can rarely demonstrate perineural invasion (115,116).
The diagnosis of microcystic adnexal carcinoma should be considered in any tumor resembling a trichoepithelioma or a syringoma in which the lesional cells extend to the base of the specimen. Syringomas, which usually are periorbital, rarely have horn cysts, foreign-body granulomas, or calcification (110).
Desmoplastic trichoepithelioma may also resemble morpheaform (fibrosing) basal cell carcinoma. However, in morphea-like basal cell carcinoma, horn cysts are absent. Of value in ruling out basal cell carcinoma, especially the morphea-like form, are the absence of mitoses, individual cell necrosis, and mucinous stroma, and the lack of foci of separation artifact of lesional epithelium and stroma (110). Signs of infundibular, follicular, or sebaceous differentiation, calcification, osteoma, association with a melanocytic nevus, and absence of solar elastosis below the lesion favor a diagnosis of demsoplastic trichoepithelioma over a morpheaform basal cell carcinoma (117).
Many immunohistochemical markers have been described, which may help differentiate desmoplastic trichoepithelioma and morpheaform basal cell carcinoma. Desmoplastic trichoepithelioma is currently considered a follicular hamartoma mimicking the outer root sheath, based on strong p75NTR expression as seen in later stages of hair follicle development. In contrast, morpheaform basal cell carcinoma generally lacks or is weakly positive for p75NTR expression and is therefore more likely a tumor of a more primitive follicular lesion (118). Matrix metalloproteinase stromelysin-3 (ST-3) expression has been demonstrated in morphea-like basal carcinomas but not desmoplastic trichoepitheliomas (119).
The presence of Merkel cells identified by CK20 expression has been shown in desmoplastic trichoepitheliomas and not morpheaform basal cell carcinomas (120). Combining androgen receptor (AR) and CK20 staining can significantly aid in diagnosis. The AR-negative, CK20-positive immunophenotype is sensitive (87%) and specific (100%) for desmoplastic trichoepithelioma, whereas the AR-positive, CK20-negative immunophenotype is specific (100%) and moderately sensitive (61%) for morpheaform basal cell carcinoma (106,121). PHLDA1 is most commonly positive in desmoplastic trichoepithelioma and negative in morpheaform basal cell carcinoma, which can also aid in diagnosis (122). Finally, fibroblast activation protein is expressed in peritumoral fibroblasts of morpheaform/infiltrative basal cell carcinoma but not in desmoplastic trichoepithelioma (123).
Principles of Management. While desmoplastic trichoepitheliomas are considered to be indolent, their overlapping features with microcystic adnexal carcinoma, similarity to morpheaform basal cell carcinoma, and their own rare potential for malignant degeneration favor treatment with Mohs micrographic surgery upon diagnosis (124).
Trichoblastoma
Clinical Summary. Trichoblastomas are benign skin lesions that recapitulate in some form the developing hair follicle. They differ from trichoepitheliomas in size, location, and lack of keratinizing cysts (125). Trichoblastomas are usually about 1.0 cm in size and most commonly occur on the scalp (Fig. 30-17) (126). While these tumors are usually solitary, multiple lesions have been described (127,128). As mentioned, these lesions are benign, though aggressive forms, termed trichoblastic carcinomas, may represent a type of basal cell carcinoma (129,130). Trichoblastomas and rare trichoblastic carcinomas have been reported to arise decades following radiation therapy (131).
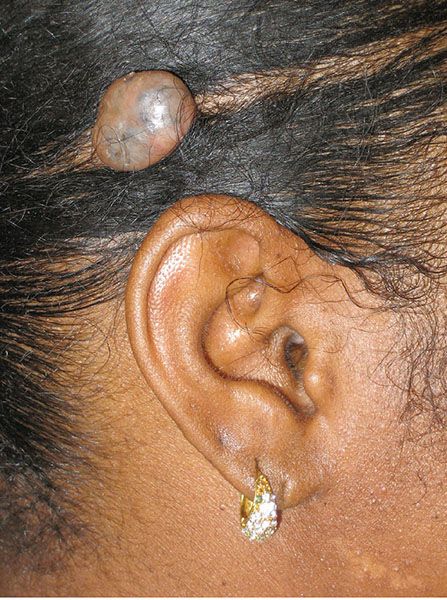
Figure 30-17 Trichoblastoma. A circumscribed, pigmented nodule is noted on the scalp.
Histopathology. Trichoblastomas consist of a proliferation of follicular germ cells manifested by a combination of various proportions of mesenchymal and epithelial cells. A spectrum of lesions is seen, depending on the proportions of mesenchymal and epithelial components. At one end of the spectrum is the predominantly mesenchymal variant, termed the trichoblastic fibroma, and at the other end is the classic trichoblastoma composed predominantly of basaloid epithelial islands (126).
The classic large lesion is composed of islands of basaloid cells occupying the dermis with occasional extension into the subcutaneous fat (Fig. 30-18). These basaloid islands demonstrate peripheral palisading and a fibrocellular stroma similar to that surrounding follicles, and mitotic activity can be seen (Fig. 30-19). There is no overlying epidermal connection. Less common variants have been described, including giant, clear cell, pigmented, cystic, giant melanotrichoblastoma, rippled-pattern trichoblastomas, and cutaneous lymphadenoma (128,132–139). Both neuroendocrine carcinoma and melanoma have been reported to occur within a trichoblastoma (140,141).

Figure 30-18 Trichoblastoma. Large islands of basaloid tumor cells are present in the dermis exhibiting peripheral palisading associated with a fibrous stroma.
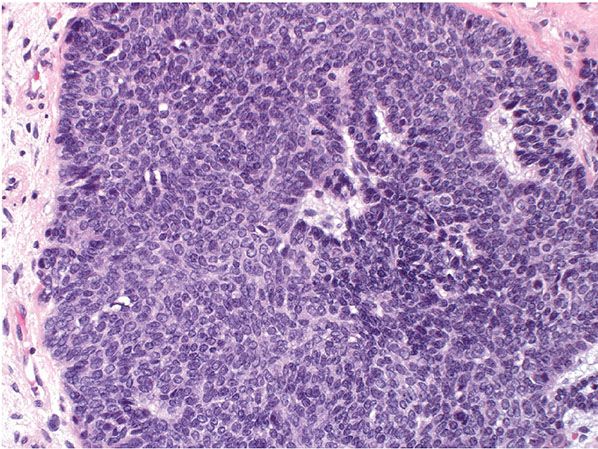
Figure 30-19 Trichoblastoma. Higher power showing epithelial structures reminiscent of follicular germs and occasional mitotic activity.
The trichoblastic fibroma variant displays small basaloid epithelial islands, exhibiting follicular differentiation, dispersed in a fibrotic stroma (Figs. 30-20 and 30-21). A rare trichoblastic sarcoma has been described as arising from a trichoblastic fibroma (142).

Figure 30-20 Trichogenic fibroma. The lesion manifests small basaloid islands in a prominent fibrotic stroma.
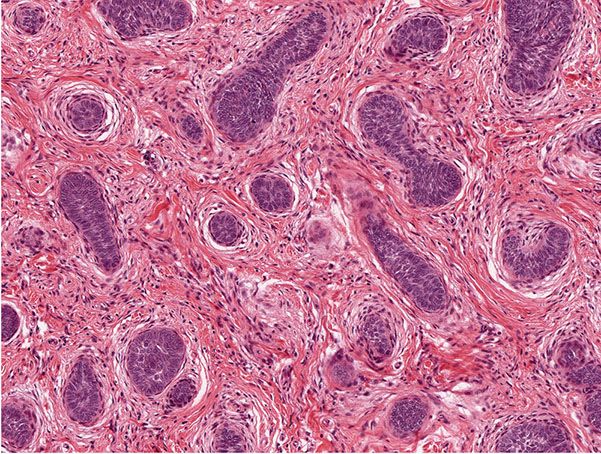
Figure 30-21 Trichogenic fibroma. The epithelial–stromal structures mimic those of hair follicles.
Lesions that are infiltrative, exhibit cytologic atypia, or mitotic activity may be termed trichoblastic carcinoma, but these lesions may overlap with basal cell carcinoma (Figs. 30-22 and 30-23). Such problematic lesions may be signed out descriptively as malignant adnexal neoplasm with a description of the apparent differentiation. Useful criteria to distinguish trichoblastomas from basal cell carcinomas include the following: the presence in the former of symmetry, circumscription with smooth margins and “shelling out” of the normal tissue, follicular and “racemiform” patterns of lesional cells, the lack of a clefting artifact between stroma and epithelium, the lack of stromal edema and lymphocytes, the formation of a delicate stroma reminiscent of that formed around immature hair follicles, and the lack of ulceration (143).
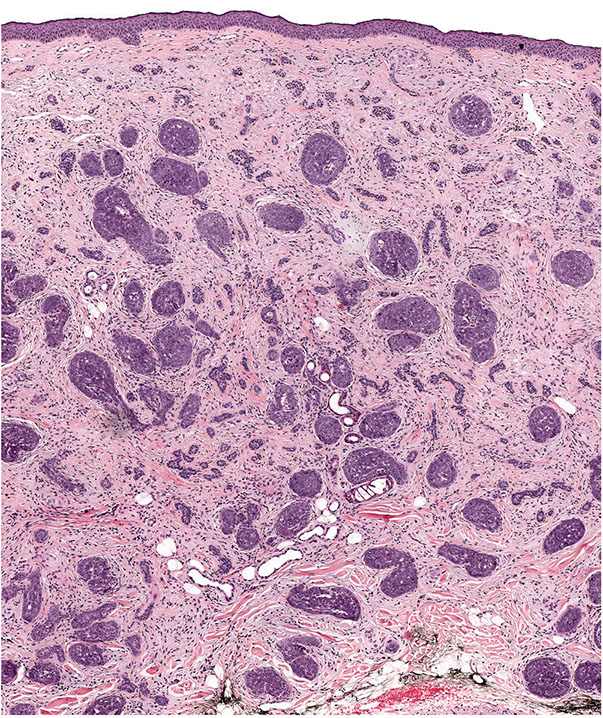
Figure 30-22 Trichoblastic carcinoma. A large dermal tumor composed of irregular epithelial islands.

Figure 30-23 Trichoblastic carcinoma. Higher power demonstrates cytologic atypia, numerous mitoses, and a fibrocellular stroma.
Immunohistochemical stains may help differentiate trichoblastoma from basal cell carcinoma. Nestin is typically positive in the stromal cells of trichoblastomas, including those seen in nevus sebaceus, whereas the stromal cells in basal cell carcinoma are negative for nestin (144). PHLDA1 positivity can be used to distinguish clear cell trichoblastoma from clear cell basal cell carcinoma (145). Additionally, laminin-332 is most commonly negative in trichoblastoma, and is consistently positive in all forms of basal cell carcinoma (146).
Additional Studies. The genetic basis for trichoblastomas is unclear. Genetic sequencing of sporadic trichoblastomas showed that classical PTCH mutations are not involved in their pathogenesis unlike trichoepitheliomas and basal cell carcinomas (147). Rarely, trichoblastomas may demonstrate mutations in the CTNNB1 gene (148).
Principles of Management. Trichoblastomas are benign and may be reexcised or removed by Mohs technique for cosmetic reasons or for concern that any remaining lesion may transform into trichoblastic carcinoma.
Trichoadenoma
Clinical Summary. A rare, solitary tumor first described in 1958, trichoadenoma usually occurs on the face or buttocks as a nodular lesion. It varies in size from 3 to 15 mm in diameter (149,150). Other rare presentations of trichoadenoma occur in the nail bed and in the bony external auditory canal (151,152). It is more common in males, and typically arises anytime during adult life (153). However, trichoadenoma can be congenital or appear in infancy (154,155). Trichoadenoma may clinically mimic basal cell carcinoma or sebaceous carcinoma (156).
Histopathology. Numerous horn cysts are present throughout the dermis (Fig. 30-24). They are surrounded by eosinophilic cells, which greatly resemble the eosinophilic cells that are often seen in trichoepithelioma located between the basophilic cells and the central horn cysts (Fig. 30-25). In some instances, a single layer of flattened granular cells is interpolated between the horn cysts and the surrounding eosinophilic cells (150,157). Some islands consist only of eosinophilic epithelial cells without central keratinization. Sparse intercellular bridges have been observed between the eosinophilic cells. Foci of foreign-body granuloma are present at the sites of ruptured horn cysts (149).
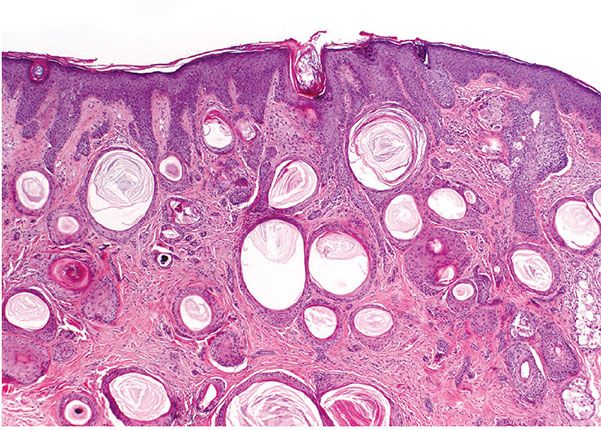
Figure 30-24 Trichoadenoma. Numerous horn cysts are present in the dermis.
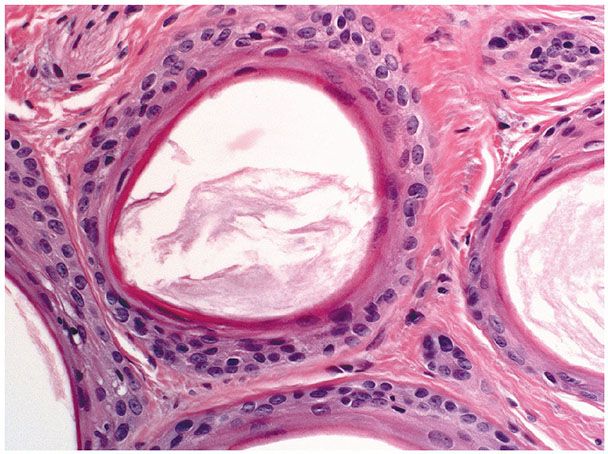
Figure 30-25 Trichoadenoma. The horn cysts are lined by eosinophilic cells and contain keratin.
A variant has been described, termed verrucous trichoadenoma, which clinically resembles a seborrheic keratosis or a deep fungal granuloma (158,159). Trichoadenoma can arise within a melanocytic dermal nevus, mimicking malignant degeneration (160).
Additional Studies. In terms of morphologic differentiation, the trichoadenoma is situated between a trichofolliculoma and a trichoepithelioma (161). The general architecture of trichoadenoma greatly resembles that of trichoepithelioma and thus suggests the development of immature hair structures. However, because the cyst wall consists of epidermoid cells and the keratinization may take place with formation of keratohyaline granules, it has been suggested that the tumor differentiates largely toward the infundibular portion of the pilosebaceous unit (150). In fact, the keratin expression pattern of trichoadenomas resembles that seen in follicular infundibula and the follicular bulge (162). Trichoadenoma and desmoplastic trichoepithelioma share some morphologic features; however, trichoadenoma is immunohistochemically distinct from desmoplastic trichoepithelioma in that it generally lacks positivity for Ber EP4 (153).
Principles of Management. No treatment is necessary; however, these lesions can be excised for cosmetic reasons or if they become irritated.
Basaloid Follicular Hamartoma
Clinical Summary. The lesions of basaloid follicular hamartoma were originally described in generalized hair follicle hamartoma. This distinctive condition is characterized by progressive alopecia starting in adulthood, diffuse papules and plaques of the face, and an association with alopecia, myasthenia gravis, or systemic lupus erythematosus (163). Since the original description, generalized hair follicle hamartoma has been reclassified as one of several clinical settings in which basaloid follicular hamartoma occurs (164). Basaloid follicular hamartoma has a variable clinical appearance and can present as macules, patches, papules, or nodules, all with variable amounts of pigmentation (165).
Basaloid follicular hamartomas can be either solitary or multiple and can be congenital or acquired. The congenital forms include an autosomal dominantly inherited generalized type associated with cystic fibrosis and diffuse alopecia, a localized linear and unilateral type, and a solitary plaque or nodule type (164,166,167). A form of congenital linear and unilateral basaloid follicular hamartoma associated with cerebral and ocular developmental abnormalities has been termed Happle–Tinschert syndrome (165,168,169). The acquired form is typically generalized and may have systemic associations such as myasthenia gravis and diffuse alopecia (163).
Histopathology. There is slight histologic variation in the basaloid follicular hamartomas found in different clinical contexts. In the context of generalized hair follicle hamartoma, the lesions within areas of alopecia that lack papules or plaques reveal more or less advanced replacement of the hair follicles by a lacelike network of basaloid cells with follicular differentiation resembling trichoepithelioma (170). The papules and plaques of the face show a complete lack of hair structures and extensive proliferations of basaloid cells embedded in a cellular stroma with occasional formation of horn cysts. The histologic appearance can be indistinguishable from that of trichoepithelioma (171).
Basaloid follicular hamartomas seen in other clinical contexts tend to demonstrate strands and cords of small, basaloid cells emanating from the infundibular portion of the hair follicle (Figs. 30-26 and 30-27) (170). The tumor stroma is mildly fibrocellular. There is no significant clefting between tumor and stroma, and mitotic activity is rare (Fig. 30-27). These lesions may have a similar appearance to infundibulocystic basal cell carcinoma, but usually they can be distinguished on clinical and histologic grounds (172,173).

Figure 30-26 Basaloid follicular hamartoma. Present is a small, well-circumscribed lesion composed of strands of epithelial cells with scattered cystic structures.
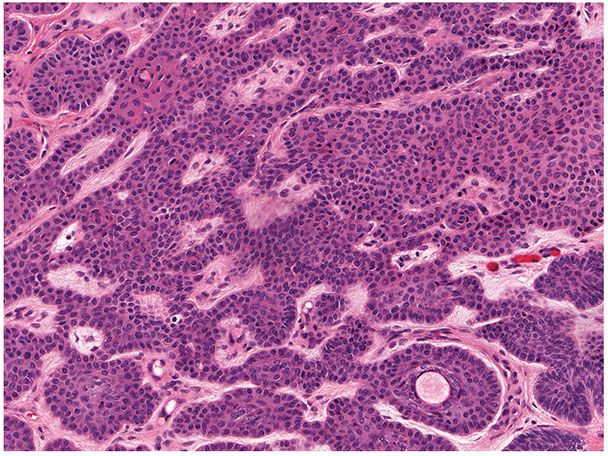
Figure 30-27 Basaloid follicular hamartoma. The lesion exhibits small basaloid cells lacking necrosis or mitotic activity. A mildly fibrocellular stroma is seen.
Differential Diagnosis. Mosaic forms of basal cell nevus syndrome (Gorlin) that are due to PTCH mutations may display basaloid follicular hamartomas, but typically also contain adnexal tumors and basal cell carcinoma (168). PTCH mutations are typically absent in linear unilateral basaloid follicular hamartoma syndrome (169). Basaloid follicular hamartoma can be distinguished from basal cell carcinoma on the basis of immunohistochemical studies. Basaloid follicular hamartoma is typically positive for CK20 highlighting Merkel cells, positive for CD34 in the stromal cells, and negative for the androgen receptor and Ber EP4 (174).
Additional Studies. Transgenic mice with increased epidermal expression of smoothened demonstrate activation of the Shh signaling pathway and exhibit lesions similar to basaloid follicular hamartomas (175). Abnormalities in PTCH signaling are associated with basaloid follicular hamartomas; however, they differ in intensity and distribution when compared with basal cell carcinoma (172).
Principles of Management. Typically, these lesions do not require excision except for cosmetic reasons. Lesions demonstrating recent growth may merit excision as basal cell carcinoma can occasionally develop in these lesions. Diffuse lesions have been treated with topical 5-aminolevulinic acid coupled with a filtered tungsten–halogen lamp (590 to 700 nm) or argon dye-pumped laser (176).
Pilomatricoma
Clinical Summary. Pilomatricoma (pilomatrixoma), or calcifying epithelioma of Malherbe, is a tumor with differentiation toward hair cells, particularly hair cortex cells. Pilomatricoma occurs usually as a solitary lesion; however, 5% of children with pilomatricomas will have multiple lesions (177). The head, neck, and upper extremities are the most common sites. Generally, the tumor varies in diameter from 0.5 to 3.0 cm, but it may be as large as 5 cm (178). The tumors may arise in persons of any age, but about 40% of them arise in children younger than 16 years of age, with a mean age of 32 years (179).
Pilomatricoma frequently presents as a firm, deep-seated nodule that is covered by normal skin. Occasionally the tumor is more superficially located, causing a blue-red discoloration of the overlying skin, and can rarely protrude as a sharply demarcated, dark red nodule.
Although solitary pilomatricoma is not hereditary, there are cases of familial occurrence with multiple lesions, most commonly associated with myotonic dystrophy. Additionally, multiple pilomatricomas and pilomatricoma-type changes can be seen in the epidermal cysts of Gardner syndrome (180). Less commonly, multiple pilomatricomas have been associated with sarcoidosis, skull dysostosis, Rubinstein–Taybi syndrome, Churg–Strauss syndrome, Turner syndrome, Soto syndrome, frontoparietal baldness, gliomatosis cerebri, and trisomy 9 (177). Unusual clinical variants include large, extruding, bullous or perforating lesions, multiple eruptive cases, and familial cases (181,182).
Histopathology. The tumor is sharply demarcated and often surrounded by a connective tissue capsule (Fig. 30-28). It is usually located in the lower dermis and extends into the subcutaneous fat. Embedded in a rather cellular stroma are irregularly shaped islands of epithelial cells. As a rule, two types of cells—basophilic cells and shadow cells—compose the islands (Fig. 30-29) (183). In some tumors, however, basophilic cells are absent. The basophilic cells possess round or elongated, deeply basophilic nuclei and scanty cytoplasm so that the nuclei lie close together (Fig. 30-29). The cellular borders of the basophilic cells often are indistinct so that it appears as if the nuclei were embedded in a contiguous mass. The basophilic cells are arranged either on one side or along the periphery of the tumor islands. In some areas, the transition of basophilic cells into shadow cells is abrupt, whereas in others the transition is gradual. In areas of gradual transition, there are cells showing a gradual loss of nuclei and ultimately appearing as faintly eosinophilic, keratinized shadow cells. The shadow cells have a distinct border and possess a central unstained area as a shadow of the lost nucleus. In tumors of recent origin, numerous areas of basophilic cells are usually present. As the lesion ages, the number of basophilic cells decreases because of the development into shadow cells, and in long-standing tumors, few or no basophilic cells remain (183).
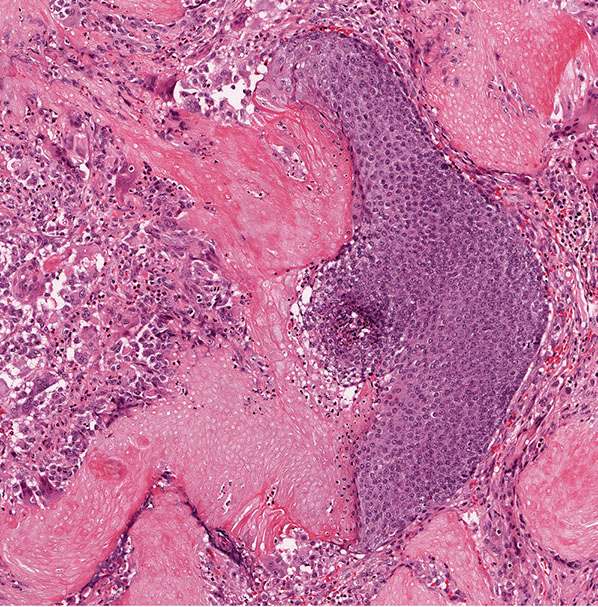
Figure 30-28 Pilomatricoma. The tumor is often well-circumscribed and composed of epithelial islands embedded in a cellular stroma. Two types of cells comprise the islands: basophilic cells and shadow cells. The basophilic cells resemble hair matrix cells. The shadow cells show a central unstained shadow at the site of the lost nucleus. In the center of the field, one can see transformation of the basophilic cells into shadow cells.
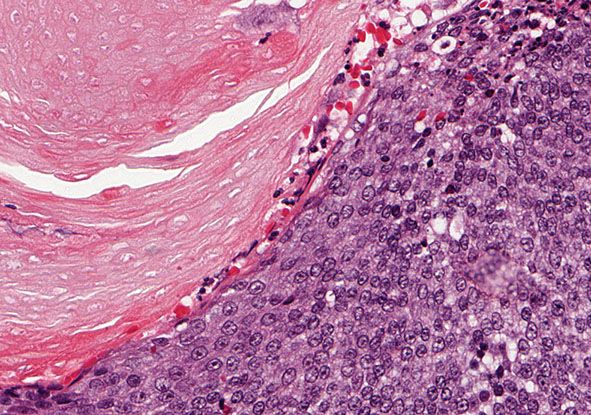
Figure 30-29 Pilomatricoma. The transformation of basaloid cells into shadow cells is associated with loss of nuclei.
In many tumors, small, round, eosinophilic centers of keratinization are seen within the areas of basophilic cells or within the aggregates of shadow cells. The keratinization within these centers is abrupt and complete (184). In some tumors, melanin is present, as can be expected in tumors with differentiation toward hair bulbs. It is found most commonly in shadow cells or within melanophages of the stroma. A variant of pilomatricoma with numerous dendritic melanocytes located in the islands of basophilic cells has been described and termed melanocytic matricoma (185).
Calcium deposits are typically apparent as deeply basophilic collections in sections stained with hematoxylin–eosin. Using the von Kossa stain, these deposits are found in approximately 75% of tumors (186). Most of the tumors containing calcium are composed largely of shadow cells. The calcium is seen either as fine basophilic granules within the cytoplasm of the shadow cells or as large sheets of amorphous, basophilic material replacing the shadow cells. Occasionally, foci of calcification are seen in the stroma of the tumors. Areas of ossification are seen in 15% to 20% of the cases (187). Ossification takes place in the stroma next to areas of shadow cells, probably through metaplasia of fibroblasts into osteoblasts. Bone morphogenic protein-2 has been shown in the cytoplasm of shadow cells but not in basophilic cells, indicating a possible role in bone formation (188). The stroma of pilomatricomas often shows a considerable foreign-body reaction containing many giant cells adjacent to the shadow cells (Fig. 30-30).
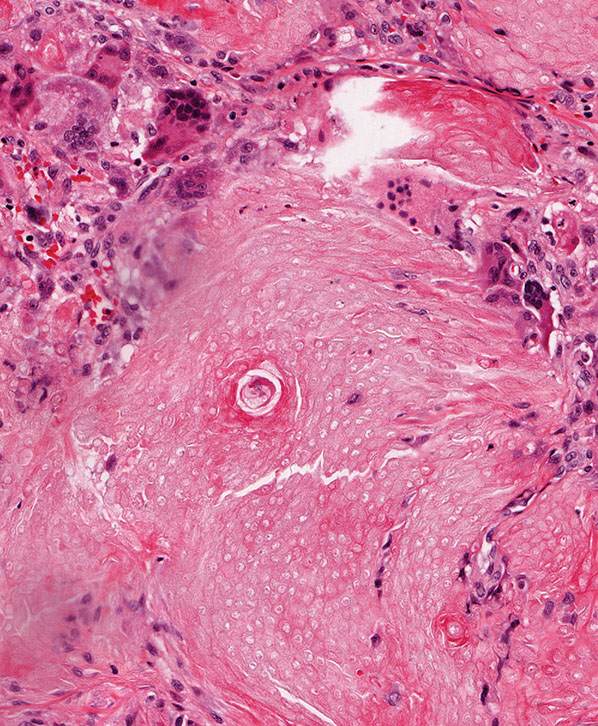
Figure 30-30 Pilomatricoma. The stroma often contains numerous multinucleated giant cells reacting to tumoral keratin.
Additional Studies. Pilomatricoma was originally described in 1880 as calcified epithelioma of sebaceous glands; however, it was recognized in 1942 that the cells of the tumor differentiate in the direction of hair cortex cells, a finding that was subsequently confirmed by electron microscopic studies (189,190). On this basis, the designation pilomatricoma was suggested (187).
Several studies using immunohistochemistry and in situ hybridization with various cytokeratins and hair-specific keratins support the notion that pilomatricomas differentiate toward components of the normal hair shaft during their maturation process (191,192). Further investigations of keratin and filaggrin expression in pilomatricomas indicate that the tumor can differentiate not only toward hair matrix and hair cortex, but also toward follicular infundibulum, outer root sheath, and hair bulge. In pilomatricoma, basophilic cells, transitional cells, and shadow cells were negative for epithelial keratin and filaggrin antibodies as well as hair matrix and hair cortex keratins. However, the infundibular-like epithelium showed positivity for K1, K10, and filaggrin, and epithelium showing trichilemmal keratinization was positive for K14 and K16. Finally, K19 is distinctly positive in the hair bulge–like structure. This heterogeneity of keratin and filaggrin expression indicates that the differentiation of pilomatricoma is diversified (193).
Mutations in the gene for β-catenin (CTNNB1) have been described in pilomatricomas (148). The resulting mutation stabilizes the β-catenin protein, which translocates into the nucleus, where it activates gene transcription through members of the Lef/Tcf family (194).
Differential Diagnosis. The wall of trichilemmal cysts also contains basophilic cells, which gradually lose their nuclei and often undergo calcification as they keratinize. The peripheral layer of basophilic cells in trichilemmal cysts, however, shows a palisading pattern, whereas the basophilic cells of pilomatricoma do not. Furthermore, shadow cells characterized by a central unstained area at the site of the disintegrated nucleus are seen only in pilomatricomas and, exceptionally, in basal cell carcinomas with foci of matrical differentiation (195). Pilomatricoma must also be differentiated from proliferating pilomatricoma (see Proliferating Pilomatricoma section).
Principles of Management. Treatment is surgical excision.
Proliferating Pilomatricoma
Clinical Summary. Proliferating pilomatricoma is a rarely reported lesion, which usually presents as a solitary nodule measuring 1.5 to 5.5 cm on the head and neck. It may present with alopecia (196). It is most common in the elderly, with a mean age at presentation of 66 years (197). The tumors can recur locally after excision, but have not been reported to metastasize (196,197).
Histology. Proliferating pilomatricoma demonstrates a large lobular proliferation of basaloid cells in association with small foci of eosinophilic shadow cells. The basaloid cells may show nuclear atypia and demonstrate a mitotic rate up to 15 per high-power field (196,197).
Differential Diagnosis. Proliferating pilomatricoma must be differentiated from pilomatricoma, which classically demonstrates larger areas of shadow cells and fewer basaloid cells. Proliferating pilomatricoma also has a high number of mitotic figures and atypia compared to pilomatricoma. Proliferating pilomatricoma can also be differentiated from pilomatrix carcinoma on the basis of symmetry, relatively small size, circumscription, and lack of numerous basaloid aggregates. It also lacks lymphatic and perineural invasion seen in pilomatrix carcinoma. Basal cell carcinoma with matrical differentiation typically is attached to the epidermis and demonstrates retraction between lobules and the stroma (197). Whether these lesions represent a transition to pilomatrix carcinoma from a preexisting pilomatricoma is currently unclear.
Principles of Management. The treatment is surgical excision (198).
Pilomatrix Carcinoma
Clinical Summary. Pilomatrix carcinoma is the rare malignant counterpart of pilomatricoma. The lesions have a male predominance and occur in older patients. Some pilomatricomas show apparent transformation into carcinomas. Other cases are malignant from the onset. Pilomatrix carcinomas are not necessarily larger than pilomatricomas, and may be mistaken clinically for an epidermoid cyst (199). The lesions recur locally in more than 60% of cases if not widely excised. The tumor may be more aggressive in the immunosuppressed (200). Pulmonary and bone metastases may occur in over 10% of cases, and life expectancy ranges from 3 months to 2 years from the diagnosis of metastatic disease (201).
Histopathology. Pilomatrix carcinomas are asymmetric, cellular, infiltrative neoplasms (Fig. 30-31). Many areas, especially at the periphery of the tumor, show proliferations of large, anaplastic, hyperchromatic basophilic cells with numerous mitoses (Fig. 30-32). Toward the center of the tumor, there may be transformation of basophilic cells into eosinophilic shadow cells of the type seen in benign pilomatricomas, or there may be large cystic centers containing necrotic debris (199). Features that are helpful in making the diagnosis include asymmetry and poor circumscription, presence of several markedly sized and variably shaped basaloid aggregations of tumor cells, continuity of basaloid cells with the epidermis, extensive areas of necrosis en masse, infiltrative growth pattern, and presence of ulceration (202).

Figure 30-31 Pilomatrix carcinoma. There is a cellular, asymmetric, ulcerated tumor that infiltrates the dermis.

Figure 30-32 Pilomatrix carcinoma. Hyperchromatic, anaplastic cells exhibit mitotic activity and transition into shadow cells.
Differential Diagnosis. See the text above regarding proliferating pilomatricoma and pilomatricoma.
Additional Studies. Pilomatrix carcinoma has been found to express K5, K14, and K17, whereas pilomatricomas typically lack these keratins (203). In pilomatrix carcinomas, basaloid tumor cells demonstrate nuclear positivity with β-catenin, and mutations in exon 3 of CTNNB1 have been detected (148,204).
Principles of Management. Recommended treatment is surgical excision with margins of 5 mm to 2 cm (202,205). External beam radiotherapy may be useful as adjuvant treatment (206).
Proliferating Trichilemmal Tumor
Clinical Summary. The proliferating trichilemmal tumor, also referred to as proliferating trichilemmal cyst, or proliferating pilar cyst/tumor is typically a single lesion; rarely, there are multiple lesions (207). About 85% of the cases occur on the scalp, with the remainder occurring mainly on the back. Lesions have been described in the vulva and other hair-bearing sites off the scalp (208). More than 80% of the patients are women, with a mean age of 62 years (range: 21 to 88 years) (209).
Starting as a subcutaneous nodule suggestive of a wen, the tumor may grow into a large, elevated, lobulated mass that may undergo ulceration and thus greatly resembles a squamous cell carcinoma (210). The tumor may occur in association with one or even several trichilemmal cysts of the scalp (211). There is evidence that a proliferating trichilemmal tumor may develop from an ordinary trichilemmal cyst. However, the tumor may also give rise to one or several trichilemmal cysts, which can ultimately separate from the tumor (212). Rapid enlargement of nodular scalp lesions often indicates malignant transformation (213,214).
Histopathology. The proliferating trichilemmal tumor is usually well-demarcated from the surrounding tissue. It is composed of multiple, variably sized lobules of squamous epithelium, and the epithelial lining is typically thicker than that seen in pilar cysts (Fig. 30-33). Some of the lobules are surrounded by a vitreous layer and show palisading of their peripheral cell layer (212). Characteristically, the epithelium in the center of the lobules abruptly changes into eosinophilic amorphous keratin (Fig. 30-34). This amorphous keratin is of the same type as that seen in the cavity of ordinary trichilemmal cysts. In addition to showing trichilemmal keratinization, some proliferating trichilemmal tumors exhibit changes resembling the keratinization of the follicular infundibulum. These changes consist of epidermoid keratinization resulting in horn pearls, some of which resemble “squamous eddies” (209,215).

Figure 30-33 Proliferating trichilemmal cyst. The tumor is composed of irregularly shaped lobules of squamous epithelium undergoing an abrupt change into amorphous keratin. Central portions of the lobules demonstrate amorphous keratin.
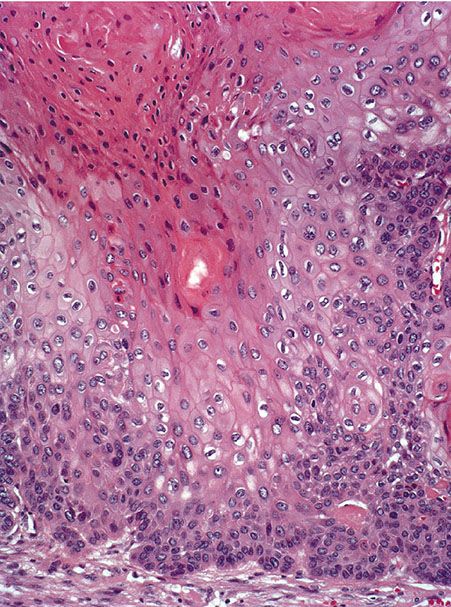
Figure 30-34 Proliferating trichilemmal cyst. Large, pale keratinocytes undergo abrupt keratinization without keratohyaline granules.
The tumor cells in many areas show some degree of nuclear atypia, as well as individual cell keratinization, which at first glance suggests a squamous cell carcinoma (Fig. 30-34). The tumor differs from a squamous cell carcinoma by a rather sharp demarcation from the surrounding stroma, as well as an abrupt mode of keratinization. Foci of calcification, although generally small, are often present in the areas of amorphous keratin (216). Some tumors show vacuolization or clear cell formation within the tumor cells as a result of glycogen storage (215).
Differential Diagnosis. In addition to its well-circumscribed nature, the presence of numerous sharply demarcated areas of amorphous eosinophilic keratin in the center of the tumor strands and lobules and the absence of severe atypia and invasion of the surrounding tissue permit differentiation from forms of malignant proliferating trichilemmal tumor and squamous cell carcinoma (209). Regardless of atypia or invasive properties, most proliferating trichilemmal tumors show positive staining with AE13 and AE14. These monoclonal antibodies are directed at pilar-type keratin polypeptides, and squamous cell carcinoma is consistently negative for these markers (209).
Additional Studies. Keratinization in proliferating trichilemmal tumors is of the same type as in ordinary trichilemmal tumors. Focal calcification is also seen in both lesions. Analogous to the outer root sheath, proliferating trichilemmal tumors show (a) an abrupt change of squamous epithelium into amorphous keratin, (b) vacuolated cells containing glycogen, like the cells of the outer root sheath, and (c) a prominent glassy layer of collagen surrounding some tumor formations (217). Owing to its close relationship with and potential transformation to a malignant proliferating trichilemmal tumor, treatment with wide local excision and close clinical follow-up is recommended (209,215,218).
Principles of Management. Complete excision with a margin of normal tissue is recommended; however, no standard has been determined regarding the margin size of uninvolved tissue.
Malignant Proliferating Trichilemmal Tumor
Clinical Summary. Malignant transformation of proliferating trichilemmal tumors takes place more commonly than previously thought. They have recently been subclassified as low-grade or high-grade malignant proliferating trichilemmal tumors, on the basis of clinical and histologic features. Low-grade lesions tend to display local recurrence, whereas high-grade lesions are more likely to display regional metastases (209). Generalized metastases do occur and can be fatal (209,219). Overall, the local recurrence rate is reported at approximately 3% to 6%. Lymph node metastases occur in 1% to 2% of patients (209). Penetration of the tumor into cerebral sinuses has occurred, resulting in death (218,220).
Histopathology. Low-grade proliferating trichilemmal tumor demonstrates irregular infiltration of the surrounding dermis, a finding that should be absent in proliferating trichilemmal tumor. This growth pattern likely results in discontinuous foci in the dermis that may be the root cause of frequent local recurrence after excision. Low-grade proliferating trichilemmal tumors lack the marked anaplasia seen in high-grade forms. High-grade proliferating trichilemmal tumors demonstrate marked cytologic atypia and significant anaplasia, likely giving them the ability to embolically metastasize. Owing to these aggressive features, it may only be possible to distinguish high-grade proliferating trichilemmal tumors from squamous cell carcinoma by positive AE13 and AE14 staining (209).
Additional Studies. Unlike proliferating trichilemmal tumors, there is loss of CD34 expression in malignant proliferating trichilemmal tumors (221). DNA aneuploidy and a higher proliferation index have also been observed (221). Malignant proliferating trichilemmal tumors are on a spectrum with proliferating trichilemmal tumors (209,222). In addition, similar to squamous cell carcinomas, loss of heterozygosity at chromosome 17p and increased p53 immunoreactivity have been observed in malignant proliferating trichilemmal tumors (223,224).
Principles of Management. Owing to the chance of malignant transformation, all proliferating trichilemmal tumors should be treated with wide local excision and adequate follow-up (222). Radiotherapy and/or chemotherapy may be indicated for lesions with high metastatic potential.
Trichilemmoma
Trichilemmoma is a fairly common solitary tumor. Multiple facial trichilemmomas are specifically associated with Cowden disease.
Solitary Trichilemmoma
Clinical Summary. Solitary trichilemmoma, first recognized as an entity in 1962, generally is a small tumor, 3 to 8 mm in diameter, which normally occurs on the face (225). Occasionally, it measures several centimeters in diameter (226). It has no characteristic clinical appearance. In some instances, it is found at the base of a cutaneous horn (227).
Histopathology. One or several lobules are seen descending from the surface epidermis into the dermis. In some instances, the lobules are oriented about a central hair-containing follicle. A variable number of tumor cells have the appearance of clear cells because of their glycogen content (Figs. 30-35 and 30-36). The periphery of the tumor lobules usually shows palisading of columnar cells and a distinct, often thickened basement membrane zone resembling the vitreous layer surrounding the lower portion of normal hair follicles (Fig. 30-36) (225). Paradoxically, trichilemmomas do not show the trichilemmal type of keratinization seen in trichilemmal cysts and proliferating trichilemmal cysts. Instead, at the surface, trichilemmomas display epidermoid keratinization, which is frequently pronounced, often associated with verrucous hyperplasia, and may even lead to the formation of an overlying cutaneous horn (228).
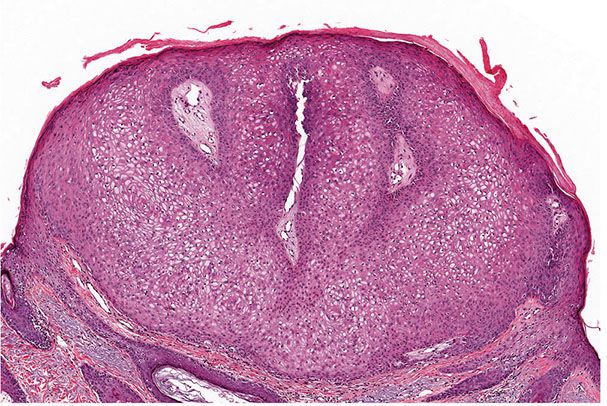
Figure 30-35 Trichilemmoma. The tumor shows verrucous hyperplasia with lobular formations extending into the superficial dermis. As a result of their differentiation toward outer root sheath cells, many cells appear clear.
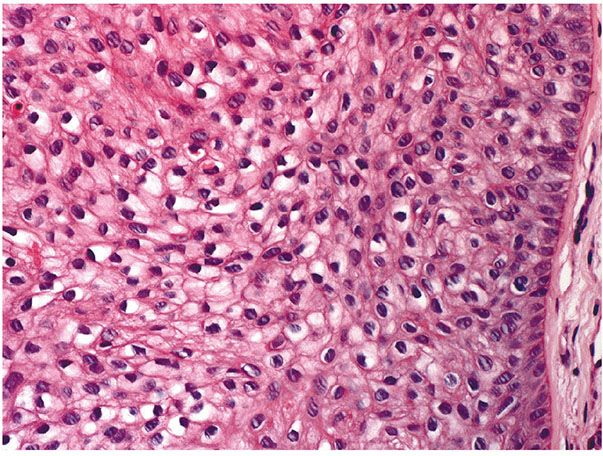
Figure 30-36 Trichilemmoma. Many cells demonstrate clear cytoplasm. The peripheral epithelial cells demonstrate palisading.
In desmoplastic trichilemmoma, there are irregular extensions of cells of the outer root sheath type that project into sclerotic collagen bundles and mimic invasive carcinoma (Figs. 30-37 and 30-38) (229). Superficially, the lesion shows changes of a trichilemmoma, a finding that aids in the distinction from an invasive carcinoma. In addition, unlike basal cell carcinomas, desmoplastic trichilemmomas stain with CD34 (230). Although desmoplastic trichilemmoma is a benign tumor, it may coexist with basal cell carcinoma, and therefore complete surgical excision with histologic confirmation of clear margins is recommended (231). As with most adnexal tumors, and in contrast with metastatic carcinomas to the skin, desmoplastic trichilemmoma is immunohistochemically positive for p63 and D2-40 (232).
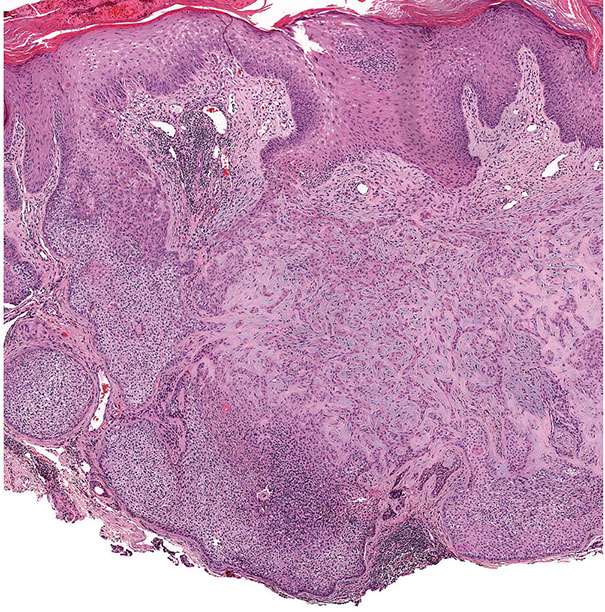
Figure 30-37 Desmoplastic trichilemmoma. The lesion exhibits features of a trichilemmoma with focal infiltrative growth in the dermis.
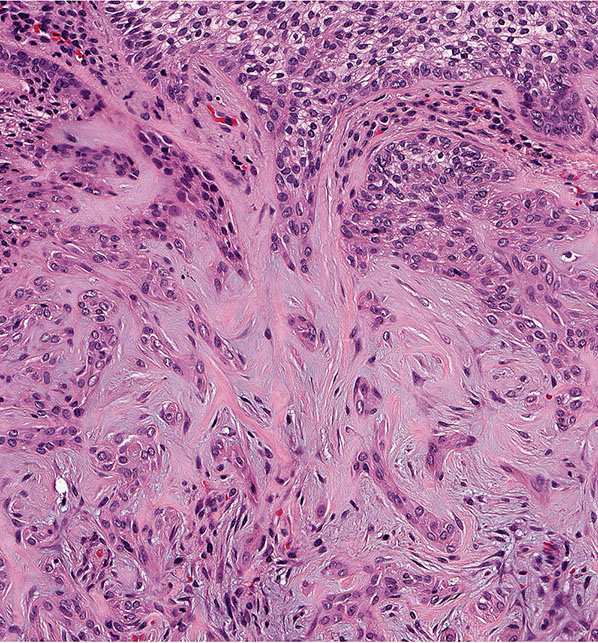
Figure 30-38 Desmoplastic trichilemmoma. Cords of bland epithelial cells infiltrate the dermis mimicking invasive carcinoma.
Differential Diagnosis. In instances with relatively few clear cells and marked hypergranulosis and hyperkeratosis, differentiation from a verruca vulgaris may be difficult. A periodic acid–Schiff (PAS) stain for the demonstration of glycogen may aid in differentiating these two lesions. However, old verrucae, which often lack viral cytopathic changes, may acquire cytoplasmic glycogen and mimic many features of trichilemmoma. Trichilemmomas differ from ordinary verrucae by their lobulated rather than papillary (verrucous) configuration, their localization about follicular infundibula, the presence of basaloid palisading at the periphery of tumor lobules, and the presence of a thickened, hyalinized basement membrane that can be highlighted by a PAS stain. Molecular studies have not supported the notion that all trichilemmomas result from human papillomavirus (HPV) infection (233,234).
Principles of Management. No treatment is required. For cosmetic reasons, lesions have been completely excised or ablated with carbon dioxide lasers.
Multiple Trichilemmomas in Cowden Syndrome
Clinical Summary. Cowden syndrome, or multiple hamartoma syndrome, is an autosomal dominant genodermatosis with distinctive mucocutaneous and systemic findings. The syndrome is characterized by multiple hamartomas in several organ systems including the skin, breast, thyroid, gastrointestinal tract, endometrium, and brain (235). The mucocutaneous lesions are a near-constant manifestation and include trichilemmomas, fibromas, oral papillomatoses, and acral and palmoplantar keratoses. The most frequent systemic findings include fibrocystic breast disease, breast fibroadenomas, thyroid adenomas, goiter, and intestinal polyposis. Individuals are at highest risk for the development of breast cancer, but other visceral malignancies, including thyroid and endometrial carcinomas, may occur (236). Rarely, certain Cowden patients develop immunosuppression with recurrent pyogenic and fungal skin infections, decreased total T- and B-cell counts, and an abnormal T-cell helper:suppressor ratio (235).
Multiple trichilemmomas are found in nearly all patients with Cowden disease, with most manifesting by the second or third decade of life. The trichilemmomas are mostly limited to the face, where they are found mainly about the mouth, nose, and ears, but can also occur in flexural sites (235). They consist of flesh-colored, pink or brown papules that may resemble verruca vulgaris (234). Multiple trichilemmomas precede the development of breast cancer, and thus can aid in identifying women with a high risk of developing this cancer. Thus, recognition of Cowden disease is important because of the high incidence and early development of breast cancer in women with this disease (237). In addition, there may be closely set oral papules, giving the lips, gingiva, and tongue a characteristic “cobblestone” appearance, as well as multiple small acral keratoses. Small hyperkeratotic papillomas are commonly found on the distal portions of the extremities (236).
The gene for Cowden disease was localized to chromosome 10q22-23 and subsequently identified as PTEN (phosphatase and tensin homolog deleted on chromosome 10) (238). Cowden syndrome is one of the PTEN hamartoma syndromes along with Lhermitte–Duclos disease (dysplastic cerebellar gangliocytoma) and Bannayan–Riley–Ruvalcaba syndrome (macrocephaly, lipomatosis, hemangiomatosis, and speckled penis) 239–241). PTEN encodes a dual-specificity phosphatase and is the major 3-phosphatase in the phosphoinositol-3-kinase pathway, which inactivates signals from (PI-3K)/AKT kinases, an anti-apoptotic pathway. Therefore, loss of PTEN function results in increased anti-apoptotic signaling in the PI-3K/AKT pathway (242). Mutational analysis has revealed germline mutations in the gene in multiple members of Cowden families (243,244).
Immunohistochemistry studies investigating trichilemmomas in Cowden syndrome show a loss of PTEN expression in most tumors. While most solitary or spontaneous trichilemmomas in patients who do not meet clinical criteria for Cowden syndrome show normal PTEN expression, roughly 13% of these cases can show decreased PTEN expression. Therefore, lack of PTEN staining in a trichilemmoma is not diagnostic for Cowden syndrome (245).
Histopathology. Multiple biopsy specimens may be needed to find the diagnostic histologic picture of trichilemmoma in the facial lesions. Thus, in one series, only 29 of 53 facial lesions showed findings diagnostic of trichilemmoma (228).
The oral lesions may show a fibromatous nodule composed of relatively acellular fibers patterned in whorls (sclerotic fibroma) or fibrovascular tissue with acanthosis (228). The extrafacial cutaneous lesions may resemble verruca vulgaris or acrokeratosis verruciformis (see Chapters 6 and 25). Some fibroma specimens show mild follicular hyperplasia as seen in trichilemmoma (235).
Differential Diagnosis. See Differential Diagnosis under Solitary Trichilemmoma section.
Principles of Management. The management of Cowden syndrome is complex, and patients should ideally be referred for genetic counseling and should be managed by experienced physicians and appropriate specialists as necessary. Principles include screening for early diagnosis of breast cancer, and consideration of prophylactic mastectomy on an individual basis. Annual dermatologic exam should be considered, and the risk of endometrial cancer should be addressed by appropriate follow-up testing, which may include blind endometrial biopsy. Individual skin lesions are managed as necessary.
Tumor of the Follicular Infundibulum
Clinical Summary. The tumor of the follicular infundibulum most commonly occurs as a solitary, flat, keratotic papule on the head and neck. It commonly presents as a plaque, or more rarely as a hyper- or hypopigmented macule. Tumor of the follicular infundibulum may rarely present as multiple lesions. Eruptive and widespread reticulated variants have also been described (246–248). There is a slight male predominance, and the average age at presentation is 67 years (range 24 to 92 years) (246). Tumor of the follicular infundibulum presents in association with other primary lesions approximately 25% of the time. These lesions include basal cell carcinoma, actinic keratosis, squamous cell carcioma, desmoplastic malignant melanoma, junctional melanocytic nevus, tricholemmoma, nevus sebaceus, and epidermal inclusion cyst (246,249). These associations have raised the possibility that tumor of the follicular infundibulum represents a reactive process.
Histopathology. There is a platelike growth of epithelial cells in the upper dermis extending parallel to the epidermis and showing multiple connections with the lower margin of the epidermis (Fig. 30-39). The peripheral cell layer of the tumor plate shows palisading, and the centrally located cells show a pale-staining cytoplasm as a result of their glycogen content; small cysts resembling the follicular infundibulum are seen often (Fig. 30-40). Small hair follicles enter the tumor plate from below and lose their identity and then are no longer recognizable (250). Other common histologic features seen in tumor of the follicular infundibulum include an eosinophilic cuticle, ductal differentiation, cornoid lamellae, and desmoplasia (246).
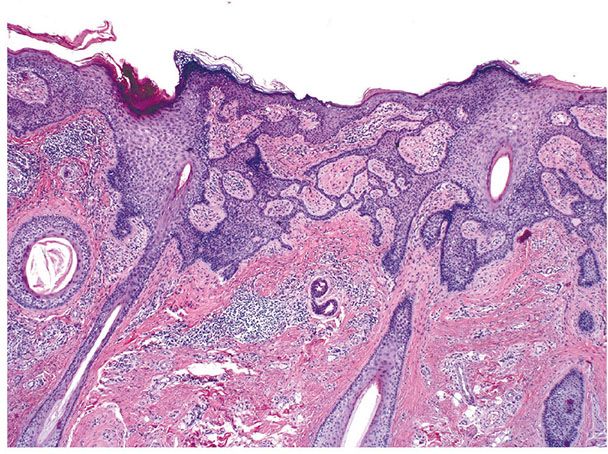
Figure 30-39 Tumor of the follicular infundibulum. A platelike growth of anastomosing epithelial cords extends parallel to the epidermis in the upper dermis and shows multiple connections with the epidermis.

Figure 30-40 Tumor of the follicular infundibulum. Lesional cells demonstrate focal cytoplasmic clearing and horn cyst formation.
Differential Diagnosis. The platelike growth with multiple connections to the epidermis resembles superficial basal cell carcinoma, which may also show peripheral palisading. Certain authors have suggested that tumor of the follicular infundibulum represents a form of basal cell carcinoma (251). However, atypia, necrosis, and mitotic activity are generally lacking in tumor of the follicular infundibulum (246). It is also negative for Ber EP4 staining, in contrast to basal cell carcinoma (252). The key histologic finding in tumor of the follicular infundibulum, as mentioned previously, is the presence of a platelike growth of epithelium with multiple connections to the epidermis.
Principles of Management. Solitary lesions are treated by simple excision.
Trichilemmal Horn and Trichilemmomal Horn
Clinical Summary. Trichilemmal and trichilemmomal horns clinically have the appearance of a cutaneous horn, as seen in several other conditions (see Chapter 29). Both growths are solitary. The more common trichilemmal horn may be seen in many different areas; the trichilemmomal horn occurs on the face or scalp (227,253).
Histopathology. In a trichilemmal horn, trichilemmal keratinization occurs. At the base of the lesion, there is a prominent basement membrane zone, with palisading of the basal cell layer and a tendency of the viable epithelial cells to become large and pale-staining. As trichilemmal cells, they keratinize without a granular layer (254). A seborrheic keratosis or verruca vulgaris with exuberant trichilemmal keratinization may mimic a trichilemmal horn (255). Immunohistochemical staining with CD34 has been shown, confirming an outer root sheath origin (256). A trichilemmal horn has been reported to arise within a burn scar (257).
In a trichilemmomal horn, a trichilemmoma is seen at the base of the lesion. At the surface, the trichilemmoma shows a cutaneous horn displaying epidermoid keratinization that is frequently pronounced, with a thick granular layer and massive hyperkeratosis (Fig. 30-37, top) (227).
Principles of Management. Solitary lesions can be treated by simple excision.
Trichilemmal Carcinoma
Clinical Summary. Trichilemmal carcinoma occurs largely on the face or ears as a slow-growing epidermal papule, indurated plaque, or nodule that may ulcerate (258). The pathogenesis is unclear; however, sun exposure has been proposed as a trigger. It can arise within other skin neoplasms, such as seborrheic keratosis (259). Unlike trichilemmomas, trichilemmal carcinoma is an unusual finding in Cowden disease (260). A case with neuroendocrine differentiation has been reported (261). Recurrence and metastases are uncommon, and the lesions are rarely locally aggressive.
Histopathology. This tumor is histologically invasive and consists of cytologically atypical clear cells resembling those of the outer root sheath (21). The lesional cells have abundant glycogenated clear cytoplasm. The lesional cells form solid, lobular, or trabecular growth patterns with foci of pilar-type keratinization. They demonstrate peripheral palisading of cells with subnuclear vacuolization. Cytologic atypia is prominent, and there may be pagetoid spread of lesional cells into the epidermis, mimicking melanoma. The cells have hyperchromatic, pleomorphic, very large nuclei. The cytoplasm contains glycogen, which is PAS-positive and diastase-sensitive (262). Trichlemmal carcinoma may rarely develop perineural invasion. Immunohistochemical positivity with CK17 distinguishes trichilemmal carcinoma from invasive squamous cell carcinoma (263).
Differential Diagnosis. A trichilemmal carcinoma may resemble a trichilemmoma by showing a lobular architecture or may resemble a “tumor of the follicular infundibulum” by replacing the surface epidermis (262). Differentiation from other carcinomas with clear cell features such as squamous cell carcinoma, basal cell carcinoma, and sebaceous carcinoma may be difficult. Typical features of these carcinomas are usually seen adjacent to the clear cell component.
Additional Studies. The pathogenesis of trichilemmal carcinomas is unclear. The distribution of lesions suggests that sunlight may play a role. A few cases have been reported in association with a burn scar, radiation, and xeroderma pigmentosa (258,264,265). Loss of p53 has been shown in a trichilemmal carcinoma arising from a proliferating trichilemmal cyst (266). As with many skin adnexal carcinomas, trichilemmal carcinoma stains positively with D2-40, in contrast with metastatic adenocarcinomas (267).
Principles of Management. Conservative surgical excision with clear margins is normally curative; however, any recurrent tumor should be managed with wide excision or Mohs surgery (268).
TUMORS WITH SEBACEOUS DIFFERENTIATION
Nevus Sebaceus
Clinical Summary. Nevus sebaceus of Jadassohn is a single lesion located commonly on the scalp or the face and is present often at birth. In childhood, this lesion consists of a circumscribed, slightly raised, hairless plaque that is often linear in configuration, but it may be round or irregularly shaped. In puberty, the lesion becomes verrucous and nodular. Less commonly, nevus sebaceus consists of multiple extensive lesions found on the face, neck, or trunk. Usually, at least some of the lesions have a linear or Blaschkoid configuration (269). Giant forms of nevus sebaceus have also been described (270).
Certain patients with extensive nevus sebaceus may also develop epilepsy, mental retardation, other neurologic defects, or skeletal deformities, causing a neuroectodermal syndrome termed Shimmelpenning or nevus sebaceus syndrome (271). There is significant overlap with keratinocytic epidermal nevi and the epidermal nevus syndrome, and it is likely that these syndromes represent different phenotypes arising from the same underlying mutations in HRAS and KRAS (see Chapter 6) (271,272).
The constellation of nevus sebaceus, central nervous system malformations, aplasia cutis congenita, limbal dermoid, and pigmented nevus together constitute the SCALP syndrome (273). Occasionally, nevus sebaceus syndrome is associated with hypophosphatemic rickets. The pathogenesis of hypophosphatemia is unclear and has been attributed to increased levels of fibroblast growth factor-23 (274,275). There is also a rare hemimegalencephalic variant of the nevus sebaceus syndrome/epidermal nevus syndrome defined by severe epilepsy, mental retardation/developmental delay, ocular/visual involvement, and facial abnormalities (276).
Nevus sebaceus can also present with a speckled lentiginous nevus, skeletal and neurologic abnormalities as part of a defined epidermal nevus syndrome termed phacomatosis pigmentokeratotica. Rarely, these cutaneous lesions may coexist without associated systemic abnormalities (277).
Histopathology. The sebaceous glands in nevus sebaceus follow the pattern of normal sebaceous glands during infancy, childhood, and adolescence. In the first few months of life, they are well-developed. During the childhood or first stage, the sebaceous glands in nevus sebaceus may be underdeveloped and greatly reduced in size and number (192,278). Thus, the diagnosis of nevus sebaceus may be missed. However, the presence of incompletely differentiated hair structures is typical of nevus sebaceus. There often are cords of undifferentiated cells resembling the embryonic stage of hair follicles (279). Some hair structures consist of dilated, keratin-filled infundibula showing multiple buds of undifferentiated cells (Fig. 30-41).
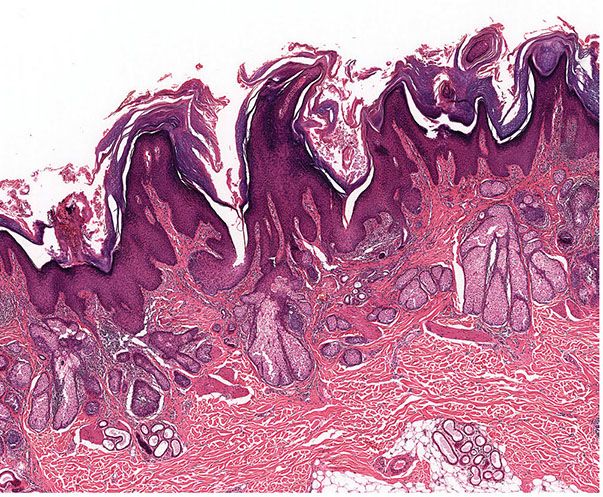
Figure 30-41 Nevus sebaceus. There is epidermal hyperplasia and papillomatosis. Dilated apocrine glands are present in the dermis, and anagen follicles are lacking.
At puberty, or second stage, the lesion assumes its diagnostic histologic appearance. Large numbers of mature or nearly mature sebaceous glands and overlying papillomatous hyperplasia of the epidermis are seen. The hair structures remain small, except for occasional dilated infundibula. Ectopic apocrine glands develop in about two-thirds of the patients at puberty and sometimes at a younger age (Fig. 30-41). There are often buds of undifferentiated cells that resemble foci of basal cell carcinoma and represent malformed hair germs (Fig. 30-42). The apocrine glands are located deep in the dermis beneath the masses of sebaceous gland lobules (192,278).

Figure 30-42 Nevus sebaceus. The basaloid epithelial proliferations often resemble basal cell carcinoma. However, the proliferations exhibit features of appendageal neoplasms as evidenced by the hyaline material within the lesion.
In approximately 15% to 20% of nevus sebaceus in the third stage or adult stage, various appendageal tumors develop. In two large case series, the most commonly associated appendageal tumors were basaloid neoplasms most consistent with trichoblastoma and syringocystadenoma papilliferum (SCAP) (Fig. 30-43) (192,278). Less commonly found benign appendageal tumors include nodular hidradenoma, syringoma, sebaceous epithelioma, chondroid syringoma, trichilemmoma, trichoadenoma, and cylindroma (279–283). Certain lesions of nevus sebaceus may contain several secondary neoplasms, and benign and malignant neoplasms may coexist (281,283).
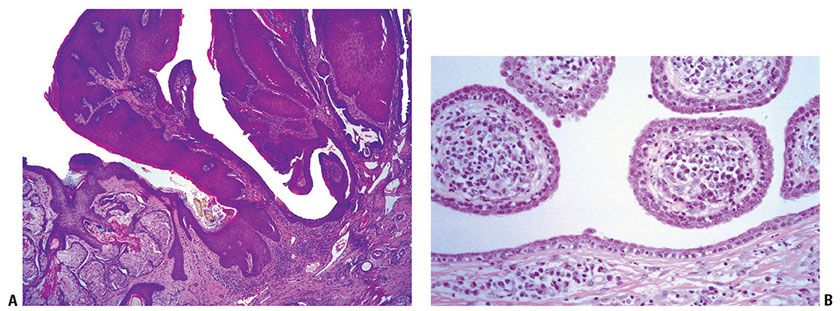
Figure 30-43 Nevus sebaceus. A syringocystadenoma papilliferum is also present.
Previously it was held that the basaloid neoplasms in nevus sebaceus represented basal cell carcinoma. However, this view was revised in several critical reviews stating that most of these basal cell carcinomas actually represented trichoblastomas (192,278). The basaloid neoplasms that arise in nevus sebaceus may represent basal cell carcinoma or trichoblastoma, and it can be extremely difficult to differentiate them on the basis of histologic grounds alone. Interestingly, the majority of basaloid tumors arising in nevus sebaceus are negative for PHLDA1, a follicular stem cell marker. PHLDA1 is generally positive in trichoepithelioma and trichoblastoma and negative in primary basal cell carcinoma. These findings indicate that many of the basaloid neoplasms may represent basal cell carcinoma rather than trichoblastoma (284). Further study of these lesions is necessary to fully define whether these represent basal cell carcinoma or trichoblastoma.
Basaloid epithelial proliferations resembling basal cell carcinoma are clinically evident in 5% to 7% of the cases of nevus sebaceus (285–287). In many instances, however, basal cell carcinomas are found that are small, clinically not apparent, and show no aggressive growth pattern (287). It is not always possible to histologically differentiate between a basal cell carcinoma and the “basaloid proliferations” that arise in as many as half of all cases of nevus sebaceus (285). As original evidence that the basal cell carcinoma–like proliferations in many instances are not true basal cell carcinomas, some proliferations contain Sudan-positive granules as an indication of sebaceous differentiation or glycogen as an indication of pilar differentiation (288). Other proliferations show follicular differentiation with formation of hair papillae and hair bulbs (289). Immunohistochemical studies with Ber EP4, p53, Ki-67, bcl-2, CD34, and factor XIIIa support the concept that these basaloid proliferations represent areas of follicular differentiation rather than true basal cell carcinoma (286).
Rarely, malignant tumors other than basal cell carcinoma may arise within nevus sebaceus, including squamous cell carcinoma (290). Squamous cell carcinoma arising within a nevus sebaceus may be associated either with regional lymph node metastasis or with generalized metastases. Spindled cell squamous cell carcinoma within a nevus sebaceus has also been reported (283,291). Other malignant tumors include apocrine carcinomas, malignant eccrine poroma, keratoacanthoma, proliferating trichilemmal tumor, leiomyosarcoma, microcystic adnexal carcinoma, sebaceous carcinoma, and mucoepidermoid carcinoma (291–299). Additionally, primary skin adenocarcinoma arising in a nevus sebaceus with resulting metastases has been reported (300).
Additional Studies. On the basis of whole exome sequencing, keratinocytic epidermal nevi and sebaceus nevi are both associated with activating HRAS p.Gly13Arg and KRAS p.Gly12Asp mutations (269,271). The mutations are likely to occur postzygotically (272,301,302). Interestingly, both genital-type and epidermodysplasia verruciformis–type HPV DNA have been detected within nevus sebaceus, though the pathogenesis and implication of such infections is not clear (303).
Nevus sebaceus demonstrates differential cytokeratin expression on the basis of the age of the patient. In the infant stage, CK1 and CK10 are reduced, with increased CK14. In the adolescent stage, CK17 is additionally strongly expressed. In the adult stage, CK14, CK17, and CK19 were all expressed. On the basis of these findings, nevus sebaceus likely qualifies as a hamartomatous growth rather than a hyperproliferative one (304).
The frequent association of nevus sebaceus with other appendageal tumors and with apocrine glands suggests that nevus sebaceus is derived from the primary epithelial germ (305). Moreover, as has been reported in basal cell carcinomas, loss of heterozygosity of the human homolog of the Drosophila patched gene (Ptch) has been demonstrated in nevus sebaceus (306–308). This could partly explain the association of basal cell carcinomas and other basaloid appendageal tumors with nevus sebaceus. However, a separate study of 11 lesions of nevus sebaceus found no demonstrable loss of heterozygosity. The reason for the discrepancy is uncertain; however, differences such as sampling during microdissection of other cell populations may be responsible (309).
Nevus sebaceus generally presents with marked alopecia because it lacks terminal hairs. Sebocytes within nevus sebaceus show an increase in expression of hair growth–suppressing bioactive factors, including FGF-5, and a decrease in expression of hair growth–stimulating factors, which may result in the decrease of terminal hairs within the lesion (310).
Principles of Management. Nonsurgical approaches include photodynamic therapy with topical aminolevulinic acid. Given their low malignant potential, strict guidelines for excision of nevus sebaceus have not been established. However, critical review has shown that most should be removed at some time in childhood, ideally before the onset of the expansion phase of the nevus sebaceus. Though there is no recommended time point for this procedure, ideally it is performed when the parent, child, and physician are in agreement with the treatment plan (311,312).
Sebaceous Hyperplasia
Clinical Summary. The lesions of sebaceous hyperplasia nearly always occur on the face, chiefly on the forehead and cheeks, in elderly persons. It has also been reported in the vulva, penis, and aerola (313–318). It clinically presents as one or, more commonly, several elevated, small, soft, yellow, slightly umbilicated papules. The usual size is 2 to 3 mm in diameter. Rarely, sebaceous hyperplasia occurs in adolescence or young adulthood, and even more rarely in prepubescent children (319,320). A neonatal transient form of sebaceous hyperplasia is very common and lasts the first few weeks of life (321). Apparent familial forms have also been reported (322). Medications, specifically cyclosporine, may induce sebaceous hyperplasia (323).
Histopathology. Most lesions consist of a single, enlarged sebaceous gland composed of numerous lobules grouped around a centrally located, wide sebaceous duct (Fig. 30-44). Its opening to the surface corresponds to the central umbilication of the lesion. Serial sections show that all sebaceous lobules grouped around the central duct are connected with that duct. Large lesions may consist of several enlarged sebaceous glands and contain several ducts, with sebaceous lobules grouped around each of them. Although some sebaceous gland lobules appear fully mature, others show more than one peripheral row of undifferentiated, germinative cells in which there are few or no lipid droplets (324).
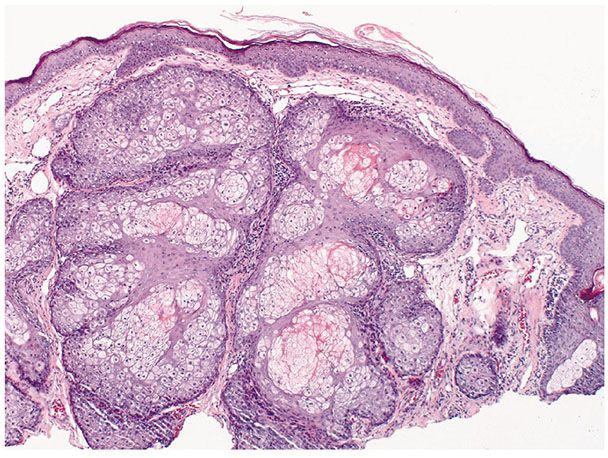
Figure 30-44 Sebaceous hyperplasia. The lesion consists of an enlarged sebaceous gland lying in close proximity to the epidermis.
Additional Studies. Labeling with tritiated thymidine has shown that the migration of sebocytes from the basal cell area to the center of the sebaceous lobules and into the sebaceous duct is distinctly slower in cases of sebaceous hyperplasia than in normal sebaceous glands (324). Microsatellite instability has been detected in very few cases of sebaceous hyperplasia as compared to other sebaceous neoplasms (see Muir–Torre Syndrome section) (325,326).
Immunohistochemical staining for androgen receptors has been shown to be fairly specific for sebaceous lesions as compared to basal cell carcinomas, squamous cell carcinomas, and clear cell acanthomas (327). Androgens affect sebaceous gland growth and differentiation, and sebocytes stain positively with androgen receptor (328). However, no increase in circulating androgens has been observed in patients with sebaceous hyperplasia (329). Sebaceous hyperplasia can occur after trauma, such as a burn, or overlying a dermatofibroma. This proliferation of sebocytes is thought to be caused by upregulation of the EGF–EGFR (epidermal growth factor receptor) signaling pathway and the Hedgehog–PTCH signaling pathway (330,331). Of note, sebaceous glands in sebaceous hyperplasia and many sebaceous neoplasms stain positively with D2-40 (332).
Differential Diagnosis. In rhinophyma, which also shows large sebaceous glands and ducts, there is no grapelike grouping of the sebaceous lobules around the ducts, and the lesion is not sharply demarcated. In nevus sebaceus, ductal structures are less apparent than in sebaceous hyperplasia, and apocrine glands are often found beneath the sebaceous glands.
Occasionally, sebaceous hyperplasia may be difficult to distinguish from a sebaceous adenoma or other sebaceous tumor, and immunohistochemistry for α-methylacyl-CoA racemase (AMACR, P504S) may aid in diagnosis. AMACR is a protein that plays an important role in mitochondrial and peroxisomal β-oxidation of branched-chain fatty acid and bile acid intermediates. Staining for AMACR is strongly positive in normal sebocytes and in sebaceous hyperplasia. AMACR shows decreasing positivity in more poorly differentiated sebaceous neoplasms, with sebaceous carcinoma showing little to no positive staining. Therefore, AMACR staining may be used to differentiate sebaceous neoplasms that are challenging to classify, and distinguish them from routine sebaceous gland hyperplasia (333).
Principles of Management. Treatment is not required. For cosmetic purposes, treatment options include photodynamic therapy, laser ablation, cryotherapy, cauterization, topical therapy with trichloroacetic acid, or excision.
Fordyce Spots and Montgomery Tubercles
Clinical Summary. In Fordyce spots, groups of minute, yellow, globoid lesions are observed on the vermilion border of the lips or on the oral mucosa composed of ectopic sebaceous glands. The incidence of the disorder increases with age so that 70% to 84% of elderly persons show such lesions (334). Montgomery tubercles are ectopic mature sebaceous glands located on the areola of the breast. Rarely, in adolescence these glands can form cystic structures that drain nonmilky fluid from the areola. These tend to resolve without intervention (335).
Histopathology. Each lesion consists of a group of small but mature sebaceous lobules situated around a small sebaceous duct leading to the surface epithelium (Fig. 30-45) (334). Because of the small size of the sebaceous duct, serial sections may be required to demonstrate the duct.
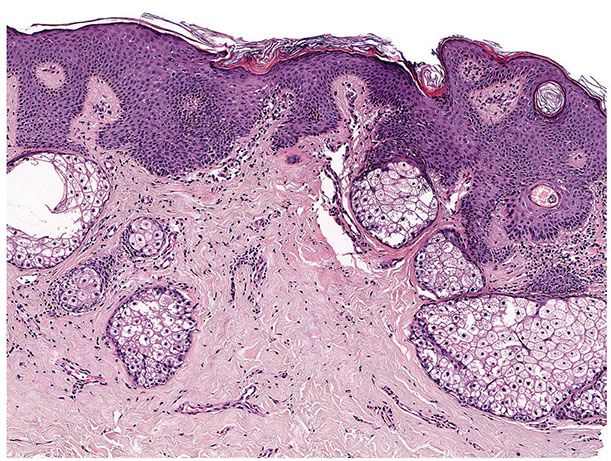
Figure 30-45 Montgomery tubercle. A mature sebaceous lobule in continuity with the epidermis is seen.
Additional Studies. There have been reports of unilateral Fordyce spots arising on the buccal mucosa in the setting of ipsilateral facial paralysis, suggesting that the sebaceous hyperplasia may be mediated by dysregulated neuroendocrine signaling (336).
Principles of Management. Treatment is not required. For cosmetic purposes, treatment options include photodynamic therapy, laser ablation, cauterization, or topical therapy with trichloroacetic acid or retinoids.
Sebaceous Adenoma
Clinical Summary. Sebaceous adenoma presents as a yellow, circumscribed nodule located on either the face or scalp. Rare intraoral and genital lesions have been described (337–339). Prior to 1968, sebaceous adenoma was regarded as a rare solitary tumor, and there were few publications about it (340). Since then, however, both solitary and multiple lesions have been well-documented in patients with Muir–Torre syndrome (see Muir–Torre Syndrome section).
Histopathology. On histologic examination, sebaceous adenoma is sharply demarcated from the surrounding tissue. It is composed of incompletely differentiated sebaceous lobules that are irregular in size and shape (Fig. 30-46). Two types of cells are present in the lobules. There are undifferentiated basaloid cells at the periphery identical to those in normal sebaceous glands (Fig. 30-47). The central cells are mature sebocytes (Fig. 30-47). Distribution of the basaloid and sebaceous cells within the lobules varies. Frequently, the proportion of mature sebaceous cells exceeds that of the surrounding basaloid cells (Fig. 30-47). Some lobules contain mainly sebaceous cells and thereby resemble mature sebaceous lobules. At most, the distribution of these two cell types occurs in approximately equal proportions. There are often transitional cells between the two types (341).

Figure 30-46 Sebaceous adenoma. The tumor is composed of enlarged sebaceous lobules of varying size and shape.
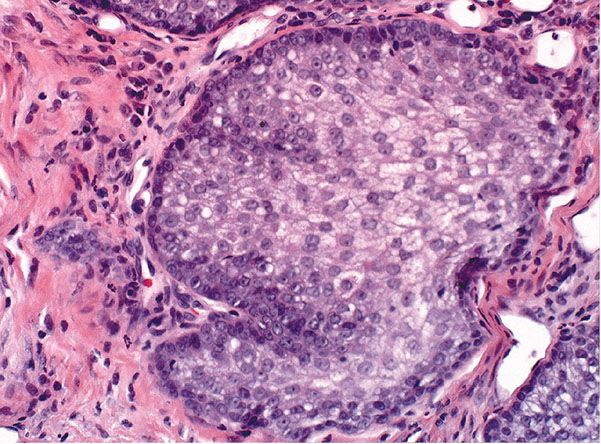
Figure 30-47 Sebaceous adenoma. In the lobules, two types of mature cells can be recognized: basaloid and sebaceous cells. However, sebaceous cells predominate. Cytologic atypia is not seen.
Fat stains, on properly preserved specimens, reveal the presence of lipid material in the sebaceous and transitional cells. Some large lobules contain cystic spaces in their center formed by the disintegration of mature sebaceous cells. In addition, there may be foci of squamous epithelium with keratinization. These foci probably represent areas with differentiation toward cells of the infundibulum. Cystic sebaceous adenomas seem to occur exclusively in patients with Muir–Torre syndrome (342).
Differential Diagnosis. In terms of differentiation, sebaceous adenoma stands between sebaceous hyperplasia and sebaceous epithelioma (see Sebaceous Epithelioma section). In sebaceous hyperplasia, the sebaceous lobules are fully or nearly fully matured and in sebaceous epithelioma, irregularly shaped cell masses predominate, and the percentage of tumor cells with mature sebaceous differentiation is less than 50%. Sebaceous adenoma and sebaceous epithelioma lack nuclear atypia and invasive, asymmetric growth patterns, which are hallmarks of sebaceous carcinoma. Considerable mitotic activity in the basaloid regions, however, may be present in both sebaceous adenoma and epithelioma.
Principles of Management. Complete excision is curative.
Sebaceous Epithelioma
Clinical Summary. Clinically, sebaceous epithelioma (formerly sebaceoma) presents as a solitary circumscribed nodule or an ill-defined plaque, often yellow in color (343). Most lesions are located on the face or scalp, though rare lesions have been described on the ear and eyelid (344–346). Sebaceous epithelioma may arise within a nevus sebaceus (281). Sebaceous epitheliomas may be multiple and can also occur in association with other sebaceous neoplasms and multiple visceral carcinomas in Muir–Torre syndrome (see Muir–Torre Syndrome section).
Histopathology. The histologic spectrum extends from that seen in sebaceous adenoma to lesions that may be difficult to distinguish from sebaceous carcinoma. Architecturally, a sebaceous epithelioma can range from a well-circumscribed nodule to irregularly shaped cell masses in the dermis. More than half of the cells are undifferentiated basaloid cells, but there are still significant aggregates of mature sebocytes and transitional cells (Figs. 30-48 and 30-49) (347). Disintegration of sebaceous cells may be present (343). Lesions verging on a sebaceous carcinoma show some degree of irregularity in the arrangement of the cell masses (348). The upper part of a sebaceous epithelioma may rarely mimic a verruca or seborrheic keratosis (349). Rarely, sebaceous epitheliomas may demonstrate sebaceous and apocrine gland differentiation (350).

Figure 30-48 Sebaceous epithelioma. The tumor is a well-circumscribed nodule and composed of basaloid epithelial cells. Scattered areas of clearer (sebaceous) cells are seen.

Figure 30-49 Sebaceous epithelioma. The majority of the lesion is composed of undifferentiated basaloid cells with islands of sebaceous cells and occasional sebaceous ducts.
Differential Diagnosis. Criteria that may be of assistance in differentiating sebaceous adenoma from sebaceous epithelioma include the often greater size and depth of the latter and the lack of structures resembling normal sebaceous lobules. In contrast to sebaceous adenoma, nuclear atypia may be seen, and mitoses are often more numerous (351).
Rippled-Pattern Sebaceoma. The rippled-pattern sebaceoma is a variant of sebaceous epithelioma. It is more common in males and presents most frequently on the scalp (352). This variant exhibits a unique arrangement of small, monomorphous, cigar-shaped basaloid cells in linear rows parallel to one another, resembling Verocay bodies; this arrangement of cells produces the rippled pattern (Fig. 30-50). Scattered sebaceous cells and ducts are also typically seen (Fig. 30-51) (352).
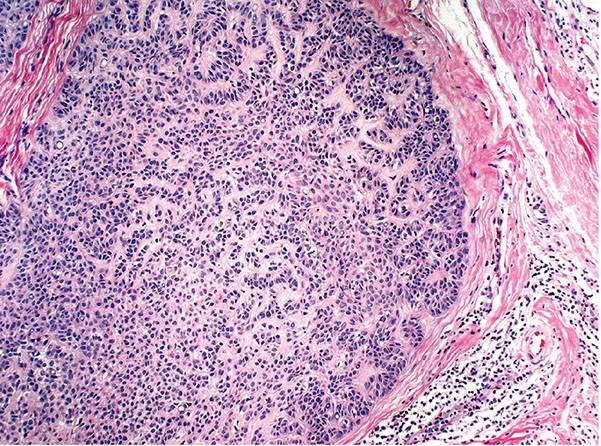
Figure 30-50 Rippled-pattern sebaceoma. Parallel arrangements of cells and nuclei are seen at low power.

Figure 30-51 Rippled-pattern sebaceoma. The lesion is composed of undifferentiated basaloid cells with occasional sebaceous cells and sebaceous ducts.
Principles of Management. Complete excision is recommended.
Muir–Torre Syndrome
Clinical Summary. The Muir–Torre syndrome is defined by the combined occurrence of at least one sebaceous skin tumor and one internal malignancy in the same patient. Since 1967, when the first publication by Muir appeared concerning the coexistence of frequently multiple sebaceous tumors and multiple visceral carcinomas, scores of cases have been reported (353). Keratoacanthomas also have occurred in these patients, in some cases without sebaceous lesions, in association with visceral malignancies (354,355). Reticulated acanthoma with sebaceous differentiation may also be associated with Muir–Torre (356). Cutaneous lesions vary in number from 1 to more than 100 lesions and can precede the first manifestation of internal malignancy (357,358).
Colon carcinoma is the most common internal malignancy in Muir–Torre syndrome. The next most common malignancy is carcinoma of the genitourinary tract (including bladder, renal, pelvis, ovary, and uterus), followed by breast, head and neck, small intestine, and lymphoma (358). Adenomatous colonic polyps are also common. Therefore, close surveillance for gastrointestinal and genitourinary malignancies in patients with sebaceous neoplasm is recommended (359). Glioblastoma multiforme also rarely occurs in Muir–Torre, overlapping with Turcot syndrome (360,361). Muir–Torre can be indolent and be unmasked by immunosuppression (362). Additionally, patients treated with localized radiation appear to be at risk for developing both further sebaceous carcinomas and soft tissue sarcomas within the radiation field (363,364).
Histopathology. Sebaceous adenomas are the most distinctive cutaneous markers of the Muir–Torre syndrome. They may be solid, cystic, or keratoacanthoma-like (341). Cystic sebaceous adenomas seem to occur exclusively in patients with Muir–Torre syndrome (342,365). Although ordinary keratoacanthomas occur in the Muir–Torre syndrome, often they have an accompanying sebaceous proliferation (366).
In addition to classic sebaceous adenomas, sebaceous epitheliomas, and basal cell carcinomas with sebaceous differentiation, there are tumors that are difficult to classify. Therefore, sebaceous proliferations that lack a definitive diagnosis should raise suspicion for Muir–Torre syndrome (366). Sebaceous carcinomas also occur in Muir–Torre, but no metastases have been reported (357,367).
Genetics. Muir–Torre syndrome, when related to microsatellite instability, is a variant of hereditary nonpolyposis colorectal carcinoma syndrome (359). Microsatellite instability is identified in many patients with Muir–Torre syndrome, and nearly 90% have a germline mutation in the gene that codes MSH-2. Approximately 10% have mutations in MLH-1 and less have MSH-6 or PMS-2 mutations (368). Mutations in MYH gene have also been associated with Muir–Torre syndrome (369). The presence of microsatellite instability in sebaceous neoplasms is helpful in the detection of an underlying inherited DNA mismatch repair defect. Loss of MLH-1, MSH-2, and MSH-6 protein expression can be demonstrated by immunohistochemical staining, and such tests are warranted to screen for microsatellite instability and possible Muir–Torre syndrome (370,371). In sebaceous neoplasms that do not demonstrate microsatellite instability, p53 overexpression may play a role (372).
Sebaceous Carcinoma
Clinical Summary. Sebaceous carcinomas have been traditionally classified into ocular and extraocular types. The ocular type most frequently occurs on the eyelids, typically originating from the meibomian glands and less commonly from the glands of Zeis. Extraocular sebaceous carcinoma has been reported most commonly on the head and neck, but also on the vulva, penis, and rarely other locations (373,374). On the eyelids, sebaceous carcinoma may be easily mistaken for chronic blepharoconjunctivitis or a chalazion (375). On extraocular sites, sebaceous carcinoma usually manifests as a nodule that may or may not be ulcerated. Giant forms of extraocular sebaceous carcinoma can grow to 20 cm in size (376). Extraocular sebaceous carcinoma is highly associated with having another primary cancer. Sebaceous carcinoma occurs predominantly in elderly Caucasian patients; however, it can occur in adolescents (377). Ocular sebaceous carcinoma occurs equally between sexes; however, extraocular sebaceous carcinoma is more common in men. For unclear reasons, the incidence of extraocular sebaceous carcinoma appears to have increased over the past several decades (378).
Ocular sebaceous carcinomas quite frequently cause regional metastases. In addition, there may be orbital invasion, and in 22% of the cases reported in one study, death resulted from visceral metastases (379). Though previously thought to be not as aggressive, extraocular sebaceous carcinomas can widely metastasize and cause death just as frequently as ocular types (380). The sebaceous carcinomas that may be found among the multiple sebaceous neoplasms occurring in association with multiple visceral carcinomas in the Muir–Torre syndrome do not metastasize, unlike the visceral malignant tumors (381). In some instances, a sebaceous carcinoma represents the only cutaneous manifestation of Muir–Torre syndrome (382).
Histopathology. The irregular lobular formations show great variations in the size of the lobules (Fig. 30-52). Although many cells are undifferentiated, distinct sebaceous cells showing a foamy cytoplasm are present in the center of most lobules (Figs. 30-52 and 30-53). Many undifferentiated cells and sebaceous cells appear atypical, showing considerable nuclear and nucleolar pleomorphism (Fig. 30-53) (383). In addition, many of the undifferentiated cells have an eosinophilic cytoplasm, and when fat stains are used on frozen sections, the cells are found to contain fine lipid globules (375). Some of the large lobules show areas composed of atypical keratinizing cells, as seen in squamous cell carcinoma (348).
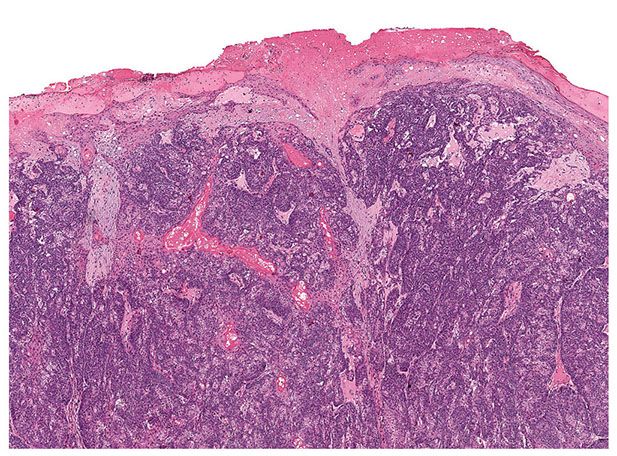
Figure 30-52 Sebaceous carcinoma. Irregular epithelial lobules are associated with epidermal ulceration and an infiltrative growth pattern in the dermis.
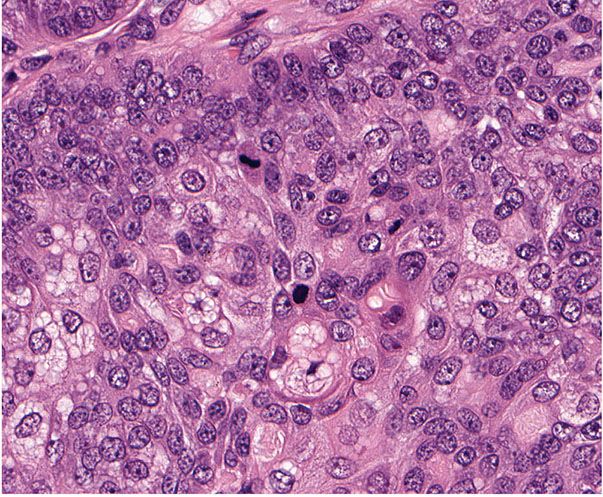
Figure 30-53 Sebaceous carcinoma. Lesional cells demonstrate marked cytologic atypia, mitotic activity, and focal sebaceous differentiation.
More than half of ocular sebaceous carcinomas show pagetoid spread of malignant cells either in the conjunctival epithelium, the epidermis of the lid skin, or both (379). These changes are seen very rarely in extraocular sebaceous carcinoma (384). The pagetoid cells contain no mucopolysaccharides, but stain positively for fat with oil red O (379). Recognition of the pagetoid growth pattern in biopsy material can be essential to recognition of the existence of an underlying sebaceous carcinoma (385). Originally, sebaceous carcinoma was thought to occur strictly de novo, but it can arise from a sebaceous adenoma, and sebaceous carcinoma in situ can progress to an invasive carcinoma (386).
Additional Studies. Certain strains of HPV have been implicated in the pathogenesis of sebaceous carcinoma (387). Mutations in the p53 gene have been seen in invasive but not in situ sebaceous carcinoma, and immunohistochemical staining for proliferating cell nuclear antigen and p53 seem to have some prognostic value (388,389). Interestingly in sebaceous carcinomas that arise in nevus sebaceus, p53 overexpression is common, and these lesions are more indolent than de novo sebaceous carcinoma (390). C-Myc may also play a role in the development of sebaceous carcinoma. C-Myc can induce sebaceous gland differentiation, and dysregulation of c-Myc signaling through loss of the androgen receptor or through aberrant p53 expression may result in lack of differentiation and progression to sebaceous carcinoma (391). Extraocular sebaceous carcinoma, when not associated with Muir–Torre, shows significantly increased EGFR expression, and may be effectively targeted with EGFR inhibitors (392).
In ocular sebaceous carcinoma, higher expression of Shh, ABCG2, and Wnt are associated with a more aggressive course and increased rate of metastatic disease (393).
Differential Diagnosis.
Related posts:

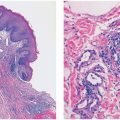
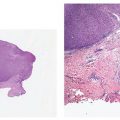
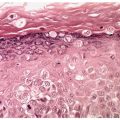
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


