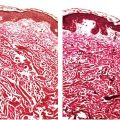
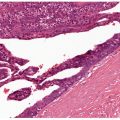
Figure 35-1 Pacinian corpuscle. This pacinian body has a distorted outline. At the periphery, there is a well-formed dense, fibrous membrane. Elongated spindle cells are compressed among the compactly arranged collagen fibers. At the center, an acidophilic punctum is representative of a single axon. Cytoplasmic processes of sheath cells are clustered about the axon. There is an intermediate zone in which spindle cells are loosely spaced in a clear matrix. Regardless of the results of immunoreactions, the loosely spaced spindle cells have the morphologic features of perineurial cells. There is a morphologic continuum from periphery to, but not including, axon.
ONTOGENY AND REACTIONS TO INJURY AS RELATED TO THE INTERPRETATION OF PHENOTYPIC EXPRESSIONS OR PATTERNS IN NEOPLASIA
Neurosustentacular cells of peripheral nerves, related cells of the peripheral ganglia, melanocytes, and even cells of some cranial mesenchyme are all of neurocristic origin.
In reactions of peripheral nerves to injury, reserve cells of neural origin proliferate; they subsequently express either schwannian or perineurial phenotypes. Expressions of phenotype are fortuitous; the environs, in which uncommitted reserve cells find themselves, likely influence the final phenotypic expression. The available options probably include a capacity to function facultatively as endoneurial fibroblasts (6).
Histologic morphologies of neural tumors may be classified according to the similarities to either normal structures, or to the reactions of normal nerves to injury. Thus, the structure of Meissner corpuscles is recapitulated in the tactile corpuscle–like structures (tactoid bodies) of diffuse (extraneural) neurofibromas (4). Some of the features of neural wallerian degeneration (Fig. 35-2) are recapitulated in granular cell nerve sheath tumors (GCNST). The intravaginal hyaline nodules of Renaut (4) may provide a model for the patterns manifested in nerve sheath myxomas (NSMs). When damaged, axons form multiple, sproutlike extensions (6). In the ensuing reparative process, newly formed Schwann cells accompany newly formed axons (Fig. 35-3). These patterns are very similar to those seen in spontaneous, intraneural neuromas.
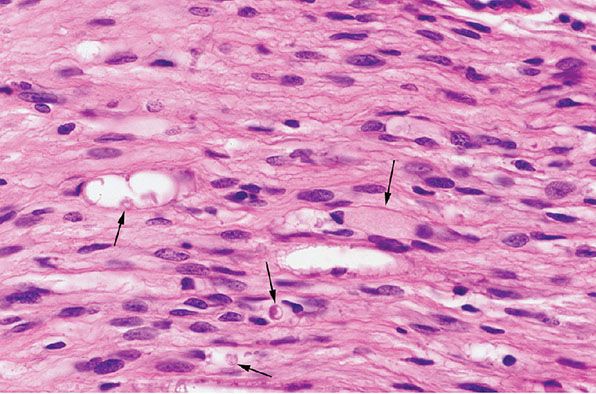
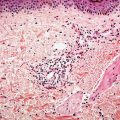
Figure 35-2 Wallerian degeneration. This peripheral nerve, bordering the site of an injury, shows poorly defined myelin sheaths; as a result, reticular fibers (basement membranes) of individual Schwann cells are more closely spaced. Scattered microcystic areas are representative of sites in which myelin sheaths have been disrupted. In some of the defects, there are globular deposits, some of which are concentrically laminated ( black arrows); these “myelin figures” are collections of lipid membranes. Some of the Schwann cells are swollen and have pale, faintly granular cytoplasm ( green arrow).
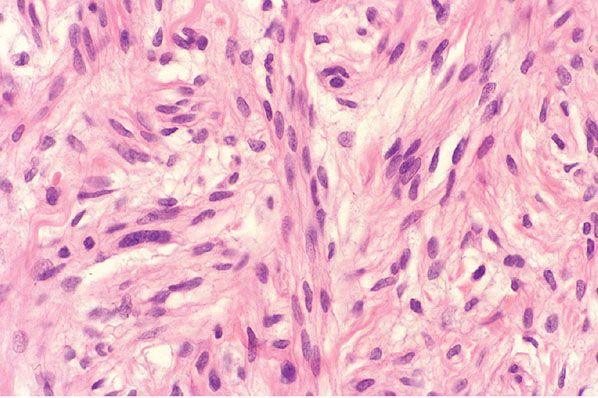
Figure 35-3 Extraneural neuroma. In extraneural neuroma, Schwann cells cluster to form thin fascicles among collagen bundles of the preexisting soft tissue. Proliferating axons, as they extend in the process of repair into the fibrous tissue, provide a scaffold for proliferating Schwann cells.
PHENOTYPES AND HISTOCHEMICAL AND IMMUNOHISTOCHEMICAL REACTIONS
Neurosustentacular cells, melanocytes, and even some mesenchymal cells, being of neurocristic origin, share a common progenitor. The cells of tumors of peripheral nerves commonly express the ultrastructural qualities of either Schwann cells or perineurial cells, but there may be conflicting findings; for example, those cells of tactile corpuscles that have the ultrastructural features of perineurial cells mark with antibodies for S-100 protein (as do Schwann cells).
S-100 protein is a lightly acidic, calcium-binding protein of glial cells but also is expressed in the skin in a variety of other cells, including melanocytes, adipocytes, chondrocytes, myoepithelial cells of sweat glands, and Schwann cells. Neuron-specific enolase (NSE), neurofilaments, and peripherin are cytoplasmic proteins common to a variety of cells including neurons and their axons. In addition, peripherin is also expressed by a variety of melanocytes. An immunoreaction for the demonstration of neural filaments or peripherin provides a pure demonstration of the reaction of axons. On a histochemical level, silver impregnation techniques demonstrate axons (Fig. 35-4) but are less sensitive than immunohistochemistry. The luxol fast blue stain, and antibodies for either myelin basic protein (MBP) or CD57 (Leu-7) antigen demonstrate myelin products. An immunoreaction for glial fibrillary acidic protein (GFAP) is positive in glial cells; it is also positive in some of the cells in some tumors of salivary glands. In some large schwannomas of soft tissue, neoplastic Schwann cells also express GFAP. Epithelial membrane antigen (EMA) is a cytoplasmic antigen commonly expressed in a variety of normal and neoplastic tissue, including perineurial, eccrine/apocrine, and sebaceous cells.
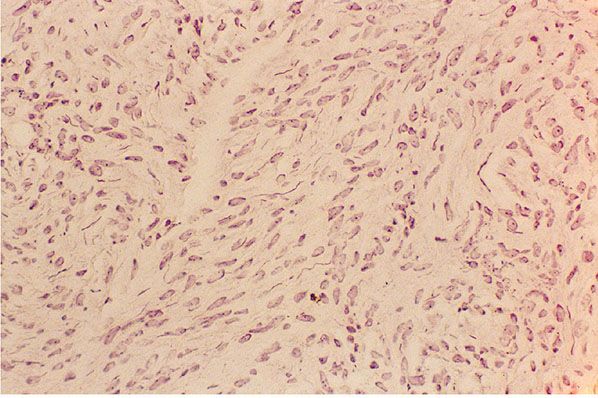
Figure 35-4 Extraneural neuroma (Bodian stain). The loosely spaced, delicate fibers are axons; axons are argyrophilic.
NEOPLASMS
Neurofibromas (Hamartomas with Phenotypic Diversity)
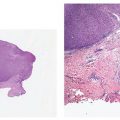
Clinical Summary. Cutaneous neurofibromas (CNFs) (the common, sporadic neurofibromas) are soft, polypoid, and skin-colored or slightly tan; they are small (rarely larger than a centimeter in diameter) (Fig. 35-5A). They usually arise in adulthood. The identification of CNFs, in the absence of other stigmata, does not establish a diagnosis of neurofibromatosis.
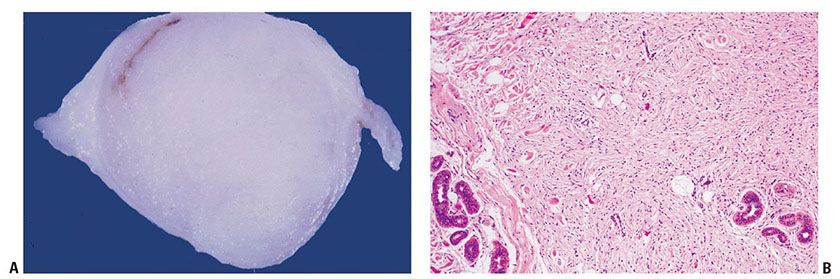
Figure 35-5 A: Extraneural sporadic CNF. This symmetrically rounded, circumscribed tumor bulges into the subcutaneous fat. The cut surface is white and homogeneous. B: Extraneural sporadic CNF. A circumscribed, nonencapsulated tumor of the dermis is composed of loosely spaced spindle cells and wavy, collagenous strands. Near the margin, collagen bundles of the dermis are entrapped in the lesion. A cluster of sweat glands also is present within the tumor; in this relationship, a defect in dermal mesenchyme has been inlaid with a neurocristic derivative.
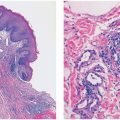
Histopathology. Under low magnification, most examples of CNF are faintly eosinophilic. They are extraneural, small dermal tumors, relatively circumscribed, but not encapsulated (Fig. 35-5B) (3,4). Thin spindle cells with elongated, wavy nuclei are regularly spaced among thin, wavy collagenous strands (Fig. 35-6). The strands are either closely spaced (homogeneous pattern), or loosely spaced in a clear matrix (loose pattern); the two patterns often are intermixed in a single lesion. Rarely are CNFs hypocellular lesions composed of widely spaced spindle and stellate cells in a myxoid matrix. The demonstration of preserved cutaneous adnexa admixed with the cells of a CNF is a combination that qualifies the lesion as hamartomatous (Fig. 35-5B). Small nerves are easily identified around CNFs. In rare occasions, there are tactoid (tactile corpuscle–like) bodies and pigmented, dendritic melanocytes (4,7). Also exceptional is a signet ring cell–like morphology resembling lipoblasts (8).
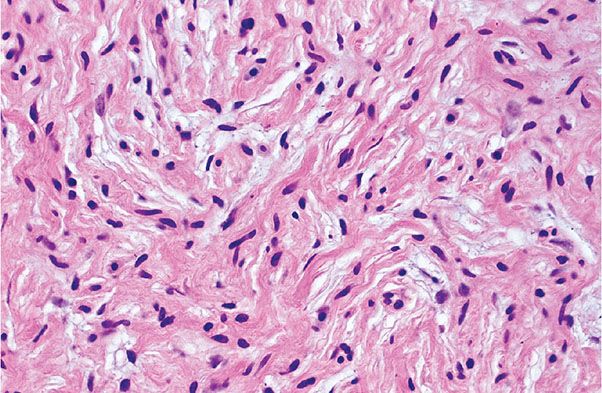
Figure 35-6 Extraneural sporadic CNF. Thin spindle cells are associated with thin, wavy collagen bundles. The cells and collagen bundles are loosely spaced in a clear or mucinous matrix.
Histogenesis. Silver impregnations (Bodian or Bielschowsky stain) reveal scattered axons, mostly in small, embedded nerves, throughout the lesion. Immunohistochemically, axons appear more numerous and more uniformly distributed. Anti-peripherin and anti-neurofilaments label the axons in either punctate (when cut tangentially) or fiberlike (when cut parallel to their long axis) patterns. Anti-NSE labels the axons; the cytoplasm of Schwann cells is faintly, but diffusely, labeled.
Differential Diagnosis. Because schwannomas are intraneural lesions, they are well circumscribed/encapsulated. In contrast with neurofibromas, schwannomas contain very few axons, and they are usually located at the periphery of the lesion, in a subcapsular location between the tumor and the perineurium (tumor capsule). In contrast with neurofibromas, there is no loose intermingling of irregularly dispersed remnants of the axial bundle of the nerve of origin in the tumoral component.
Neurotized nevi show areas indistinguishable from neurofibromas. However, they contain at least a few, superficial nests and fascicles of nevus cells. They usually contain Wagner–Meissner corpuscles and they are rare in CNFs.
Some sporadic neurofibromas, including subcutaneous variants, are confined, at least in part, by the perineurium of the nerve of origin, and thus are intraneural and circumscribed. This architecture thus may raise the possibility of plexiform neurofibroma.
Dermatofibromas may also be confused with neurofibromas, particularly those lesions with markedly fibrous stroma. However, dermatofibromas only have rare, scattered dendritic cells expressing S-100 protein. Medallion-like dermal dendrocyte hamartoma is another fibrohistiocytic lesion that may be confused with neurofibroma. This is a benign, rare congenital lesion, comprising a benign dermal proliferation of fusiform cells, presenting as a well-circumscribed atrophic and wrinkled patch located on the upper trunk or neck. Tumor cells express CD34 and factor XIIIa, but not S-100 (9,10).
Principles of Management. With the exception of malignant transformation (usually associated with rapid growth in a long-standing lesion; see also below), CNF are removed only when they result in cosmetic or functional problems.
Neurofibromatosis
Clinical Summary. Neurofibromatosis (NF) is a genetic disease that involves the nervous system. It has three major subtypes: neurofibromatosis type 1 (NF1) (the most common subtype, also known as von Recklinghausen disease), neurofibromatosis type 2 (NF2), and schwannomatosis.
NF1 (von Recklinghausen Disease)
Clinical Summary. Multiple CNFs are characteristic of most, but not all, examples of NF1. Other important stigmata include plexiform (intraneural), and deep, diffuse (extraneural) neurofibromas; multiple periareolar neurofibromas (Fig. 35-7); intraocular Lisch nodules; and macular, cutaneous hyperpigmentation (e.g., café-au-lait spots and bilateral axillary freckling) (11,12).
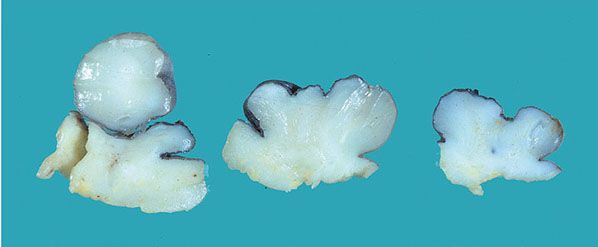
Figure 35-7 Periareolar neurofibroma. The lesion has been breadloafed. Multiple polypoid components project above the skin surface. The cut surface is white and glistening. The lesion diffusely involves the dermis and protrudes irregularly into the subcutaneous fat.
Pathogenesis. NF1 may arise as an autosomal dominant condition but it is secondary to spontaneous mutation in nearly half of the patients (13). CNFs usually appear first in late childhood or in adolescence; the disease progresses by showing gradual increases in the size and number of neurofibromas.
For NF1, its gene is located to the pericentromeric region of the long arm of chromosome 17 (14). The large gene’s size may account for the high incidence of sporadic cases. The gene shows functional and structural homology with the guanosine triphosphatase–activating protein, which controls the ras oncogene. As a result, the role of the ras oncogene in growth, development, and differentiation may be aberrantly expressed in the NF1 phenotype (14). The protein product of the gene is called neurofibromin (15).
In NF1, all sorts of nerves may be affected, including superficial peripheral nerves, deep peripheral nerves, nerve roots, and the autonomic nerves of viscera and blood vessels. Large, pendulous plexiform neurofibromas have the flabby texture of a “bag of worms.” Deep, diffuse neurofibromas are poorly defined at their limits with the adjacent soft tissue (4). Because of the diffuse nature of the lesions and their size, the limbs may have an elephantiasic quality. Spinal nerve root tumors may cause compression of the spinal cord (13). Tumors of the central nervous system occur in 5% to 10% of the patients (11). Neurofibromas adjacent to bone may erode it. Lesions that are variably associated with NF include kyphoscoliosis and an increase in the length of long bones. NF1 may be associated with diffuse intestinal ganglioneuromatosis and multiple endocrine neoplasia (MEN), type 2b (16,17). Gastrointestinal stromal tumors are more common in NF1 than in the general population (18,19), as well as xanthogranulomas and juvenile chronic myelogenous leukemias (19).
In localized or segmental NF, CNFs are few in number and there is no family history (20–22).
Neurofibromatosis Type 2
Clinical Summary. NF2 has also been called “MISME Syndrome,” for “Multiple Inherited Schwannomas, Meningiomas, and Ependymomas.” Manifestations include café-au-lait spots, neurofibromas, schwannomas, and a variety of intracranial tumors, such as bilateral schwannomas of the acoustic nerve (15) (Fig. 35-8) and meningioangiomatosis (23). The gene locus has been linked to chromosome 22q11-q13. The protein gene product, called merlin, is similar to a group of cytoskeleton-linked proteins (15) and appears to work as a tumor suppressor. Its loss results in de-differentiation and abnormal growth of Schwann cells. A recent study has indicated that loss of SOX-10 function, a marker present in both neural and melanocytic cells, may contribute to Schwann cell proliferation (24). Interestingly, schwannoma cell lines have receptors for curcumin, which might be useful as a therapeutic tool (25).
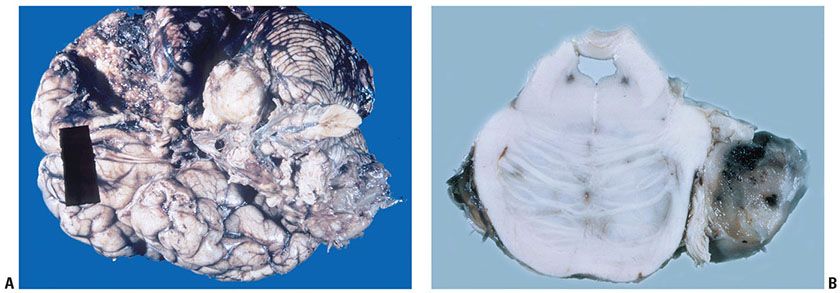
Figure 35-8 A: Acoustic neuroma (schwannoma involving the VIII cranial nerve) and meningiomas). The schwannoma is expansile and white. Portions of a meningioma are also represented along the base of the brain; other portions had been surgically removed. The patient had NF. B: Acoustic nerve schwannoma (cut surface of brain stem at level of pons). On the right, an acoustic neuroma (schwannoma) involves the VIII cranial nerve. The cut surface focally is hemorrhagic. In other areas, the cut surface is tan, a coloration often seen in association with hemosiderin deposits.
Schwannomatosis
Clinical Summary. Schwannomatosis is characterized by multiple and plexiform schwannomas, as a clinical entity distinct from NF2 (26,27). It accounts for about 15% of patients with NF. Lesions appear in any anatomic location outside the acoustic nerve and are typically associated with significant pain. Mutations of the SMARCB 1 (also known as INI 1 and BAF 47) tumor suppressor gene occur in 40% to 50% of familial cases and in 8% to 10% of sporadic cases of schwannomatosis (28). This gene is also involved in rhabdoid tumors characterized by the presence of malignant rhabdoid tumors of the kidney, atypical teratoid/rhabdoid tumors (AT/RT), AT/RT of CNS, and extrarenal rhabdoid tumors that develop in childhood. Café-au-lait spots occur in nearly all patients with NF, and they usually precede cutaneous tumors (12). The presence of more than six spots, each exceeding 1.5 cm in diameter, is indicative of NF (11,13).
Histopathology of Neurofibromatosis. (For a complete description of the histologic features of schwannomas, please see below.) The histologic spectrum of neurofibromas in NF1 and NF2 is broad. Some CNF are small; others are large, cutaneous, or deep, circumscribed or plexiform (intraneural) variants (Fig. 35-9) and still others are diffuse (extraneural).
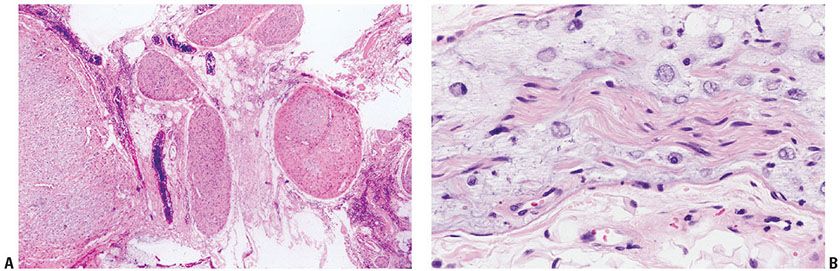
Figure 35-9 A: Components of a cutaneous, plexiform, intraneural neurofibroma. For the most part, components of the plexus in this field are cut in cross section. The tumor is intraneural; it sits in fibrofat without an extraneural component. Each component of the plexus is confined by a thickened perineurium. B: Plexiform intraneural neurofibroma; mucinous endoneurial type. This small branch of a large, cutaneous, plexiform neurofibroma shows a well-defined, central bundle of Schwann cells (an axial bundle). In this central bundle, Schwann cell fascicles of the preexisting nerve are symmetrically arranged along the long axis of the nerve. The endoneurial space is widened and mucinous. In it, distinctive, rounded cells (endoneurial mucocytes) are loosely spaced. The expanding mucinous matrix has displaced some of the Schwann fascicles of the axial bundle away from their neighbors. The muciparous cells resemble the “cellules godronne” as seen in the “intravaginal hyaline system” of Renaut. The perineurium is delicate; it is a single layer of flattened cells.
In many deep, circumscribed and in most plexiform neurofibromas, there are axial bundles composed of symmetrically arranged nerve fibers, remnants of the axial bundle of the nerve of origin (Figs. 35-9 and 35-10) (3,4,29). Their role in the histogenesis of NF is uncertain. The axial bundle retains its symmetry in a field of distorted and asymmetrical patterns (Fig. 35-10). In some axial bundles, Schwann cells are hyperplastic and tightly packed. In areas, these remnants of the nerve of origin focally evolve into micronodules composed of interlacing fascicles of Schwann cells; such micronodules may qualify as microscopic schwannomatosis (4). Many of these cellular micronodules may be independent of a true axial bundle, but they might function as growth centers (Fig. 35-11). Cells and collagenous strands stream from their periphery and extend asymmetrically into the expanded, mucinous, endoneurial matrix. In some intraneural variants, there are similar, streaming cells blending with the perineurium and thus suggesting that the perineurium contributes to the tumor. Those cells actually show features of perineurial cells, such as EMA expression.
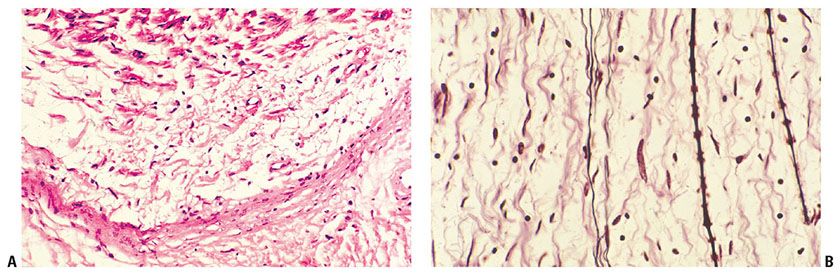
Figure 35-10 A: Intraneural neurofibroma; schwannian pattern of differentiation. A thickened perineurium forms a concave band near the bottom of the field; it defines the limits of the tumorous component at the interface with the adjacent soft tissue. At the top of the field, a distorted axial bundle (a remnant of the nerve of origin) is represented. The tumor is represented in the space between the perineurium and the distorted axial bundle; thin, pale collagen bundles are loosely and randomly spaced in a stringy, myxoid matrix. Thin spindle cells with elongated, wavy nuclei are closely associated with some of the collagen bundles. The fibrous component of the lesion is asymmetrical in its relationships with both the long axis of the involved nerve and the central axial bundle. B: Intraneural neurofibroma; remnant of axial bundle (Bodian stain). Argyrophilic, myelinated, and nonmyelinated axons are parallel; they have axial symmetry and are a remnant of the axial bundle. Wavy collagen bundles of the tumor are more asymmetrical in orientation and distribution.
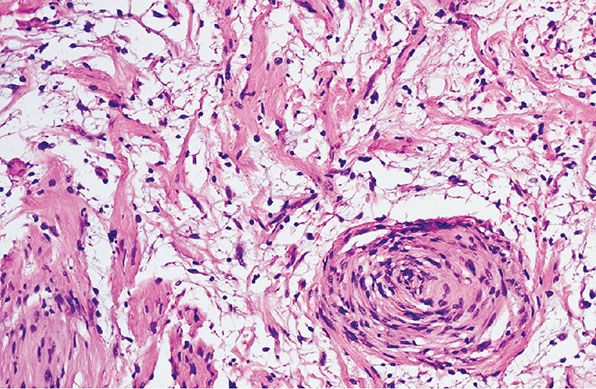
Figure 35-11 Intraneural neurofibroma; focal, microscopic schwannomatosis. Coarse collagen bundles are asymmetrically arranged in a loosely cellular, myxoid matrix. Near the right lower corner, collagen bundles and cells form a whorl; the whorl, as a nidus, is an example of microscopic schwannomatosis.
The cells of perineurial variants are bipolar or tripolar with rigid processes; they are loosely spaced in either a myxoid or fibrous matrix (Figs. 35-12 and 35-13). Endoneurial (schwannian) variants are characterized by a thickened perineurium, one or several axial bundles, and isolated cells in an expanded, mucinous, endoneurial component (Fig. 35-14). Some of the cells in the mucinous matrix resemble the “cellules godronné” of Renaut (round cells with portions of the mucinous matrix that are either enclosed within the cytoplasm or focally partitioned—encircled and sequestered—by cytoplasmic processes) (Fig. 35-9B) (1,3,4,30). One or several axial bundles may be found among asymmetrical collagen bundles of the tumor (Figs. 35-14 and 35-15). In the expanded endoneurial component of both perineurial and endoneurial (schwannian) variants, the matrix is myxoid, rich in hyaluronic acid (4).
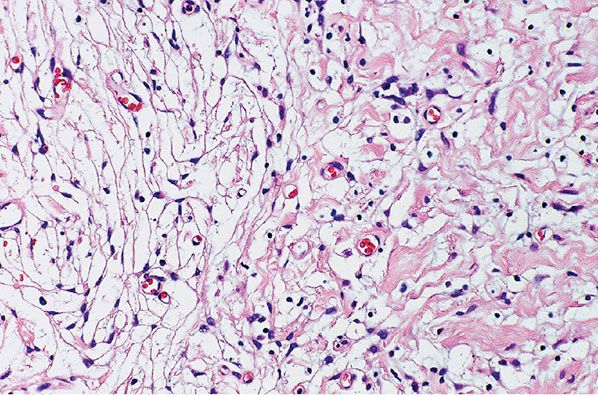
Figure 35-12 Intraneural neurofibroma; biphasic (perineurial and schwannian) patterns. To the right, wavy collagen bundles are loosely spaced; spindle cells are closely associated with the collagen bundles (schwannian pattern). To the left, distinctive, bipolar and tripolar cells are loosely spaced in a clear or myxoid matrix; these distinctive cells have perineurial qualities.
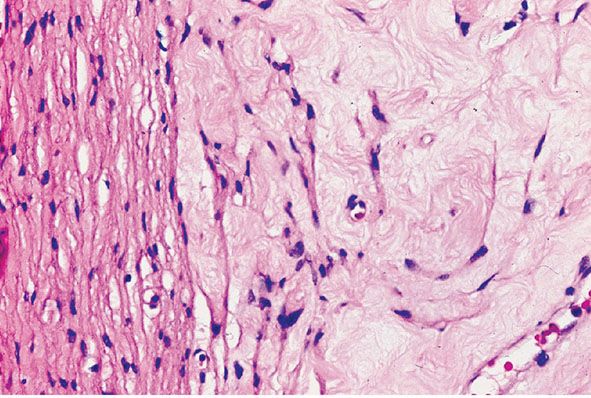
Figure 35-13 Intraneural neurofibroma; perineurial cell differentiation near the capsule of the tumor. The membrane to the left of the field is a thickened perineurium (capsule). In the fibrous matrix to the right of the perineurium, distinctive cells are loosely spaced; some blend with the inner surface of the perineurium. The loosely spaced cells have the morphologic features of perineurial cells.
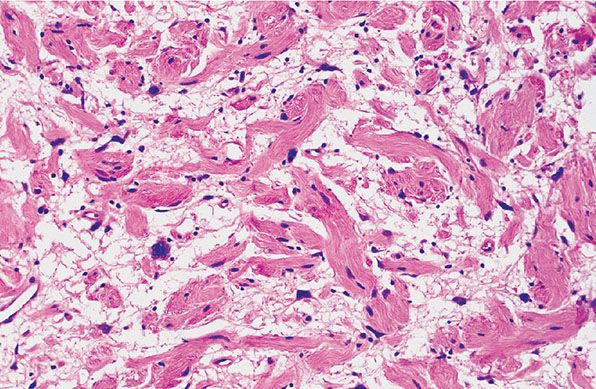
Figure 35-14 Intraneural neurofibroma; cytologic atypia. Coarse collagen bundles are loosely, and randomly, spaced in a myxoid matrix. Spindle cells are closely associated with the collagen bundles. Spindle and stellate cells are present among the collagen bundles in the myxoid matrix. Some of the cells in the myxoid matrix show nuclear atypia; they show variations in nuclear size and outline.
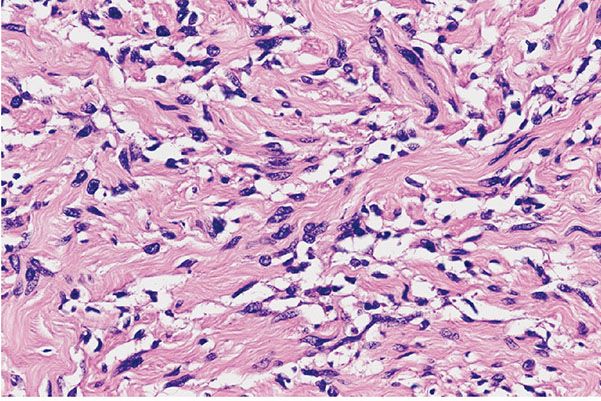
Figure 35-15 Intraneural neurofibroma; cellular pattern. Spindle cells are increased in number among the collagen bundles; focally, they are present within the collagen bundles. The cells have enlarged, wavy nuclei and scanty cytoplasm.
Focal cytologic atypia (“ancient” change), defined as scattered, enlarged, atypical nuclei (Figs. 35-14 and 35-16) is not associated with malignant change, as long as the lesion does not have hypercellularity and mitotic figures (3,4).
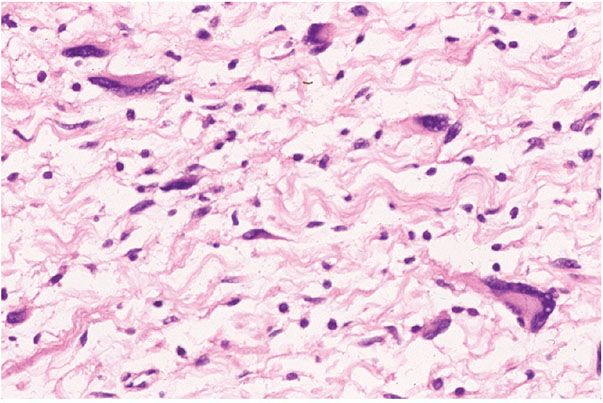
Figure 35-16 Intraneural neurofibroma; cellular atypia. Some of the cells among the collagen bundles are cytologically atypical; some of the atypical cells are multinucleated.
At the peripheral edge of the extraneural (diffuse) neurofibroma, margins are poorly defined (Fig. 35-17), typically involving adipocytes (3,31). The matrix of extraneural neurofibroma is either delicately fibrous and faintly acidophilic, or more coarsely fibrous and brightly acidophilic. In the absence of plexiform components, nerves within diffuse neurofibromas usually are small, internally symmetrical, and hypercellular; perineuria of such nerves often are hyperplastic. In cross section, these nerves resemble pacinian corpuscles (32). Patterns of both plexiform and diffuse neurofibroma are combined in so-called paraneurofibroma (Figs. 35-18 and 35-19) (4). Focal collections of structures resembling tactile corpuscles (tactoid bodies) (Fig. 35-20), and pigmented, dendritic melanocytes (a relatively common feature of deep, extraneural [diffuse] neurofibromas) are rare in the intraneural components of plexiform, or circumscribed, intraneural neurofibromas (4,7).
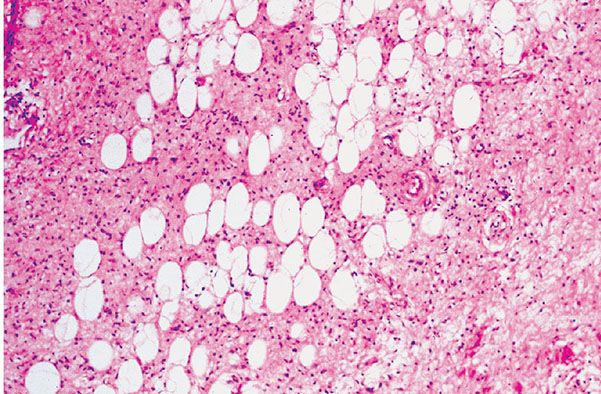
Figure 35-17 Extraneural (diffuse) neurofibroma. A delicately fibrous matrix is loosely cellular. The cells have scanty cytoplasm; they appear as naked nuclei. Adipocytes are entrapped.
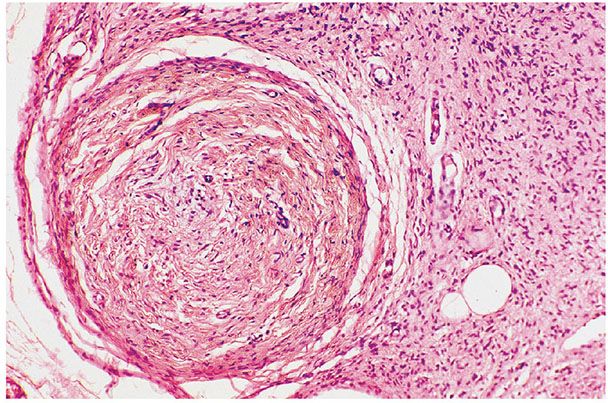
Figure 35-18 Diffuse and plexiform neurofibroma (paraneurofibroma). The round nodule to the left is a portion of an intraneural plexiform neurofibroma; it has been cut in cross section. The delicate fibrous matrix to the right is a portion of a diffuse (extraneural) component.
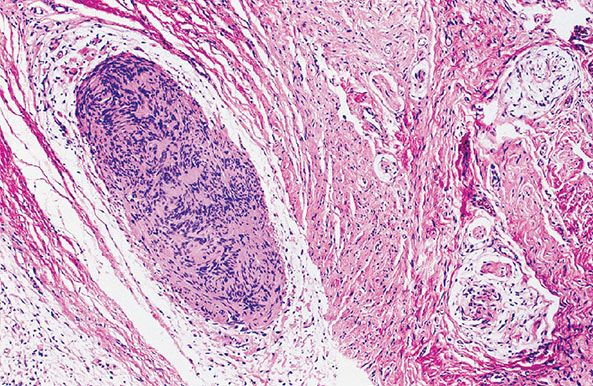
Figure 35-19 Diffuse and plexiform neurofibroma (paraneurofibroma) with focal microscopic schwannomatosis. The intraneural portions are pale, rounded, and faintly basophilic; in these components, wavy collagen bundles are loosely spaced. The extraneural component is a uniformly cellular fibrous matrix. Near the center of the field, a cellular nodule is confined within the intraneural matrix; in this cellular nodule, nuclei are arranged in palisades. The nodule is an example of microscopic schwannomatosis. The patterns of Verocay bodies are represented.
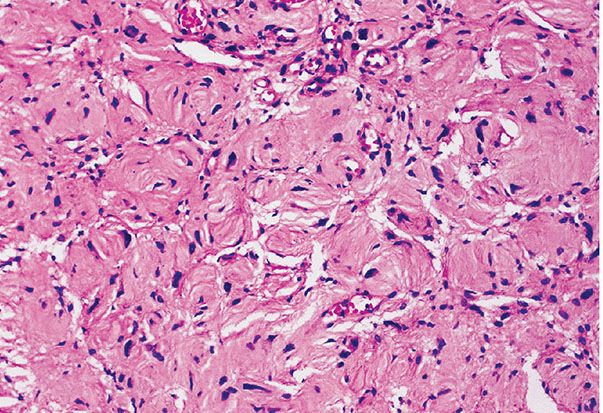
Figure 35-20 Extraneural neurofibroma; tactile corpuscle–like patterns. As a regional variation in this extraneural neurofibroma, spindle cells, in stacked lamellae, form tactile corpuscle–like structures.
Ultrastructurally, the cells of neurofibromas (including those of tactoid bodies) mostly resemble those of perineurial cells (33), but there are also axons and cells with the characteristics of Schwann cells and endoneurial fibroblasts (34). Endoneurial fibroblasts, like common mesenchymal fibroblasts, do not have basal lamina. The average diameter of their collagen fibrils lies well below that of dermal collagen fibrils, which is about 100 nm (35).
Histogenesis. The immunohistochemical profile of neurofibroma is as follows: S-100 protein (+), with variable expression of CD57 (Leu-7) antigen (+), and MBP (+). In extraneural CNFs, axons, as demonstrated with NSE, or with antibodies to neurofilaments, are fairly uniform in distribution among collagen bundles. This finding is less consistent in deep, intraneural neurofibromas. Perineural cells express EMA, while endoneurial fibroblasts express CD34+ (36). Furthermore, the pattern of CD34 reaction named “fingerprinting” (slender, CD34-positive cells forming whorls around dermal vessels) (37) has been described as characteristic of neurofibromas and not in melanoma (38), although rare cases of spindle cell melanoma may have similar immunohistochemical features (39).
Immunohistochemical and ultrastructural analysis reveals that Schwann and perineural cells with enclosed axons are variably distributed in CNFs (40,41). Their presence has been interpreted as either being an integral part of the tumor or entrapped nerve fibers (33). Nonplexiform neurofibromas of von Recklinghausen disease contain abundant, peptide-rich nerve fibers (40). Similar associations undoubtedly exist in plexiform variants. Thus, the innervation of neurofibromas of NF1 appears to be much richer than appreciated in the past.
The extraneural distribution of the common dermal neurofibroma may be a manifestation of the extension of tumor through the open-ended terminal of perineurial sheaths, or through the thin perineurium of small, dermal nerves (29). Deep, extraneural neurofibromas may represent a diffuse, neurocristic dysplasia (42). In this approach, the presence of adipocytes within such a tumor may be evidence of mesenchymal differentiation rather than simple entrapment.
There are hybrid peripheral nerve sheath tumors (43), including neurofibroma-schwannoma (44), schwannoma-perineurioma (45), neurofibroma-perineurioma (46), and congenital nevi with features of schwannoma-perineurioma (47). That study emphasized histopathologic and immunohistochemical findings, with one case having been analyzed ultrastructurally. Furthermore, one case had a point mutation in exon 15 of the NF2 gene (43).
A recent study in mice has suggested that mast cells and macrophages may be important in the maintenance of nerve sheath tumors (48,49). That study has shown that depletion of macrophages with targeted therapy (PLX3397) resulted in cell death and tumor volume regression.
Differential Diagnosis. Plexiform lesions of the skin and subcutaneous tissue include plexiform schwannoma (4), plexiform neuroma (50), neuromas of the mucosal neuroma syndrome (MNS) (51), NSM (52), plexiform fibrous histiocytoma (53), and possibly perineurioma (54). Also, some fasciculated melanocytic tumors are described as “plexiform” (55–57).
Dendritic cell neurofibroma with pseudorosettes is a poorly documented, benign, “plexiform” lesion of the skin (58,59). These lesions have two cell types: large dendritic and small, dark cells. The dark cells tend to cluster in palisades about the large dendritic cells. The tumor cells are immunoreactive for S-100 protein. The large cells are not neurons. Michal et al. (60) described a tumor that was both diffuse and plexiform. As photographically documented, the dendritic cells were intraneural and sheltered from the small-cell component by a complex of cell processes resembling neuropil. The histologic features of the population of small cells can be compared to those of the small cells in some examples of “epithelioid” small-cell populations in some variants of schwannoma. Such schwannomas are of the type associated with zones of radial sclerosis (29). In neither category of nerve sheath tumor do the small cells have the cytologic features of a population of “blast” (immature) cells. To borrow an old designation, but in the process to alter its definition, the patterns of differentiation in the small-cell population might qualify as “neuroglial” differentiation. However the similarities, we believe that, on the basis of their distinct clinical behavior, the term “neuroblastoma-like” should probably not be used to describe these dendritic cell neurofibromas.
Subcutaneous, diffuse neurofibromas may be confused with the deep component of dermatofibrosarcoma protuberans (DFSP). Some authors have mislabeled the pigmented form of dermatofibosarcoma (Bednar tumor) as pigmented storiform neurofibroma (32,61).
Fibrous hamartoma (of infancy) has fascicles of spindle cells with a myxoid background, thus resembling diffuse neurofibroma.
The nodular myxoid components of a low-grade malignant myxoid fibrous histiocytoma might be misinterpreted as the pattern of a plexiform neurofibroma.
Mucinous and proliferative endarteritis, and aneurysms are occasional features in the setting of NF1; they affect mainly muscular renal vessels, but similar changes can be manifested in other sites (4,62,63). Aneurysmal changes have been a prominent feature of muscular vessels in some examples of extraneural neurofibromas of the skin and soft tissue, particularly in the distribution of the trigeminal nerve (4). Such lesions clinically may be mistaken for hemangioma.
Principles of Management. As mentioned above, with the exception of malignant transformation (usually associated with rapid growth in a long-standing lesion; see also below), CNF are removed only when they result in cosmetic or functional problems.
TRUE NEOPLASMS OF SCHWANN CELLS
Schwann cells are specialized sustentacular cells, normally distributed as a single layer about, and along, one or more axons. In a sense, the insulation that they provide along normal nerve fibers favors proper propagation of nerve impulses.
Schwannomas
Schwannomas (neurilemmomas; anaxonal, intraneural, Schwann cell tumors; “peripheral neurogliomas”) are benign, circumscribed, expansile neoplasms (1,29,33). The cells of schwannomas mostly have the characteristics of Schwann cells.
Clinical Summary. In schwannomas, the population of Schwann cells exists independent of a plexus of axons. As solitary, skin-colored tumors along the course of peripheral or cranial nerves, their usual size is between 2 and 4 cm, usually on the head or the flexor aspect of the extremities (Fig. 35-21A). Schwannomas are rare in the subcutaneous tissue and even less common in the dermis. In deep soft tissue, they may be large (64). In the central nervous system, the VIII cranial is the favorite site. Internal viscera (65) and bones may be involved. When small, most schwannomas are asymptomatic but may develop pain, localized to the tumor or radiating along the nerve of origin.
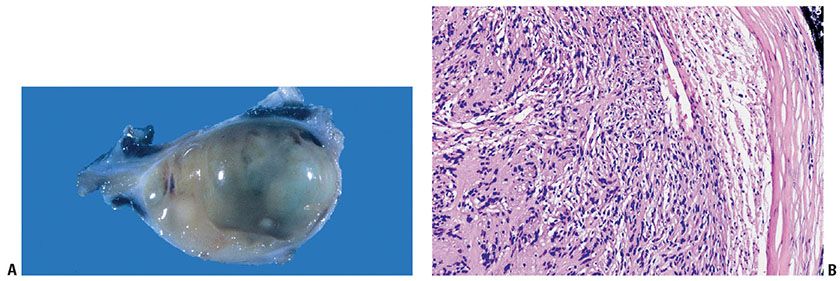
Figure 35-21 A: Schwannoma. The lesion is circumscribed and encapsulated. The lesion bulges over a portion of the cut surface. B: Schwannoma and nerve of origin. This schwannoma is uniformly cellular; the Schwann cells focally form nuclear palisades and Verocay bodies. To the right, a thickened perineurium, at its interface with the neighboring soft tissue, defines the limits of the tumor. The loosely cellular zone between the perineurium and the tumor is a compressed nerve of origin; the nerve maintains its identity at the periphery of the tumor; it is not enclosed within the substance of the tumor.
Histopathology. Schwannomas, with the exception of infiltrating, fascicular variants, are intraneural and symmetrically expansile; they are confined by the perineurium of the nerve of origin (Fig. 35-21B). Most of the symmetrically bundled nerve fibers of the nerve of origin are displaced eccentrically; if identified in a histologic section, they usually are to be found between the tumor and the perineurium (see Fig. 35-21B) (4,29). At the interface with the nerve of origin, some of the nerve fibers may extend from the compressed, eccentric axial bundle into the tumor. The observations of Russell and Rubinstein (66) contradict the dictum that schwannomas are universally devoid of axons. Rather, loosely, and randomly, spaced, nonmyelinated axons have been observed in small peripheral schwannomas, and in small and medium-sized acoustic nerve schwannomas (66).
The tumor cells are arranged in two patterns, Antoni A and Antoni B (3,65). In the Antoni type A areas, uniform spindle cells are arranged back-to-back (Fig. 35-22); each cell is outlined by delicate, rigid, reticular fibers (basement membranes). The cells tend to cluster in stacks, and the respective nuclei often form palisades. Two neighboring palisades, the intervening cytoplasm of Schwann cells, and associated reticular fibers, all in combination, constitute a Verocay body (see Figs. 35-22 and 35-23).
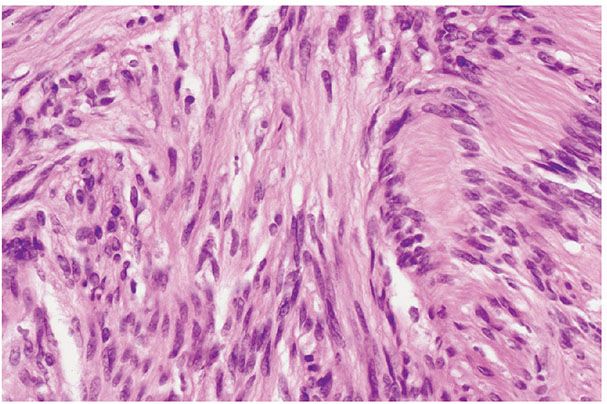
Figure 35-22 Schwannoma; Antoni A patterns. Pale spindle cells with elongated, wavy nuclei are arranged in fascicular patterns. To the right of the field, nuclear palisades are regularly spaced; two nuclear palisades and the enclosed cytoplasmic processes comprise a Verocay body. Cellular components of this type qualify as Antoni A tissue.
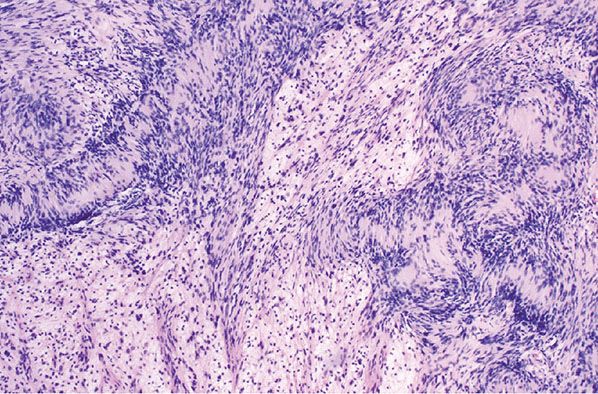
Figure 35-23 Schwannoma; mixed Antoni A and Antoni B patterns. The Antoni A component is cellular; Verocay bodies are a prominent feature. The loosely cellular, pale zones are Antoni B patterns. In the Antoni B component, there is some variation in nuclear size and staining.
In Antoni type B areas, files of elongated cells, arranged end-to-end are loosely spaced in a clear, watery, or myxoid matrix (Figs. 35-23 to 35-25) (1); in some examples, the cells somewhat resemble glial cells; in other examples, they resemble perineurial cells.
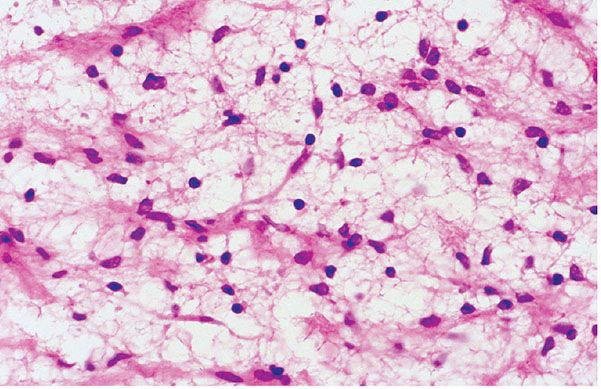
Figure 35-24 Schwannoma; Antoni B patterns. In this example, the cells of an Antoni B component are small with complex, delicate cytoplasmic processes. The cytologic features have a glial cell–like quality.
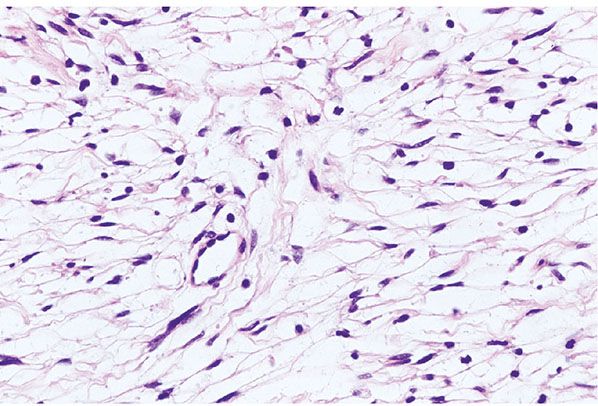
Figure 35-25 Schwannoma; Antoni B patterns. In this example, the cells of an Antoni B component are bi- or tripolar; the cytoplasmic processes are elongated and straight; the cells cytologically resemble perineurial cells.
Typically there are clusters of dilated, thick, congested blood vessels (3), occasionally with fibrinoid necrosis or hyalinization, thrombi, or subendothelial collections of foam cells (Fig. 35-26) (67). The vascular changes can be found in both Antoni A and Antoni B tissue. Some lesions may have cystic changes (Fig. 35-27), extravasated erythrocytes, or hemosiderin (4,67).
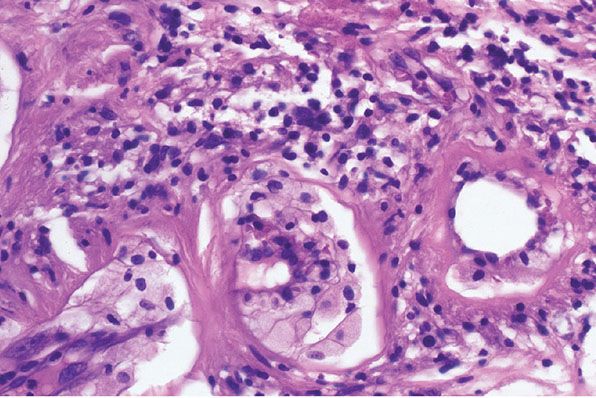
Figure 35-26 Schwannoma; Antoni B component, vascular changes. In this Antoni B component, several vessels are loosely clustered. They have thickened, hyalinized walls. There are prominent, subintimal collections of foam cells. Loose infiltrates of lymphoid cells are adjacent to the vessels, and among the foamy histiocytes. In part, the hyalin represents a stage in the organization of fibrinoid; fibrinoid degeneration and thrombosis are common vascular changes in some schwannomas.
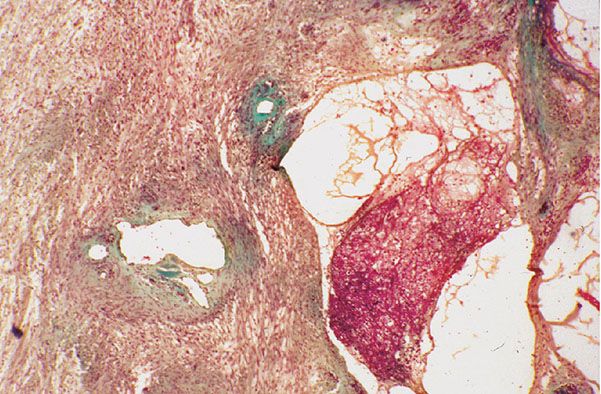
Figure 35-27 Schwannoma; microcystic and vascular changes. With this connective tissue stain, cells have a reddish tinge and connective tissue is green. Vessels are dilated; their walls are fibrous and irregularly thickened. To the right, multiple, small cysts contain a loose meshwork of fibrin.
Ultrastructurally, most tumor cells manifest the features of Schwann cells (long cytoplasmic processes and myelin with long-spacing collagen) (33,65).
The patterns in Antoni type B areas are interpreted to be of a “degenerative” nature (67). “Ancient change” characterizes the nuclear atypia as well as the large, thick, dilated vessels with fibrin often seen in some schwannomas (4,67) (see also below). Ultrastructurally, tumor cells are widely separated by an electron-lucid, homogeneous matrix containing strands of fibrin and detached segments of basement membrane. Autophagic lysosomes, some containing myelin figures, are seen in many cells (i.e., a “histiocytic” quality). The cellular changes include extensive loss of basement membrane, disruption of cell membrane, and nuclear degenerative changes (68).
Schwannomas associated with NF2 or schwannomatosis may display prominent myxoid stroma or “mixed” features, resembling a combination of schwannoma, neurofibroma, and perineurioma. Regarding immunohistochemistry, there may be a mosaic pattern of expression of SMARCB1/INI1. Sporadic schwannomas rarely show such pattern (69).
Histogenesis. The immunohistochemical profile is S-100 protein (+), and two antigens characteristic of basement membrane—collagen type IV and laminin (+). In the capsule, cells are EMA (+). A Bodian stain and an immunoreaction for the demonstration of neural filaments reveal few or no axons, except in and near the peripherally displaced axial bundle.
Differential Diagnosis. If a nerve sheath tumor involves a significant sensory or motor nerve, the histologic distinction between intraneural neurofibroma and schwannoma becomes an important consideration for a surgeon. The intact, but displaced, symmetrical axial bundle (Fig. 35-21) of a schwannoma often can be preserved during surgical dissections. On the other hand, excision of an intraneural neurofibroma requires sacrifice of the nerve of origin. Most neurofibromas are poorly circumscribed and show a “hamartomatous” appearance owing to the combination of Schwann cells, axons, perineural cells, and fibroblasts. Axons are usually distributed through the lesion.
Palisaded and encapsulated neuroma (PEN) (see also below) should be considered in the differential diagnosis of schwannoma. They are mostly located on the face. Although they are circumscribed, as are most schwannomas, they contain numerous axons within the lesion.
Variant Types of Schwannomas
Cellular Schwannoma
In cellular schwannoma (CS), as defined by Harkin and Reed (3), most cells are arranged with an Antoni A–type pattern; Schwann cells are closely aggregated with scanty intercellular matrix. Some scattered cells may have large, hyperchromatic nuclei (Fig. 35-28). Increased cellularity per unit area, a preponderance of type A patterns, and a low mitotic rate are requisites for the diagnosis of CS (Fig. 35-29). Other authors have expanded the definition of CS (1) to include some features that overlap with malignant peripheral nerve sheath tumor (MPNST). The dichotomy, in the approaches to CS is given recognition herein by the definition of two classes: CS1 and CS2 (70).
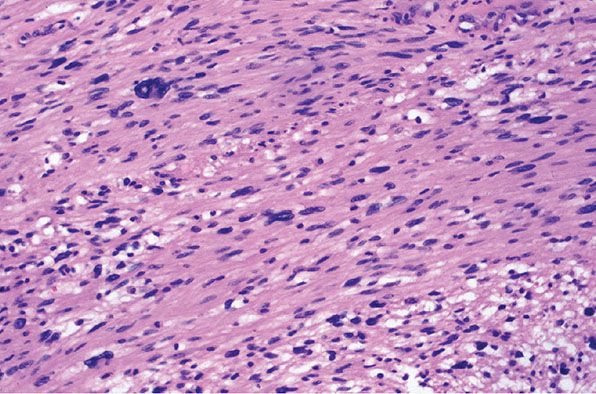
Figure 35-28 Schwannoma; cytologic atypia (ancient change of cytologic type). Spindle cells form interlacing fascicles. The cells show variations in nuclear size, staining, and outlines; in many of the nuclei, chromatin is dense and smudgy. Mitoses are not a feature of “ancient change.”
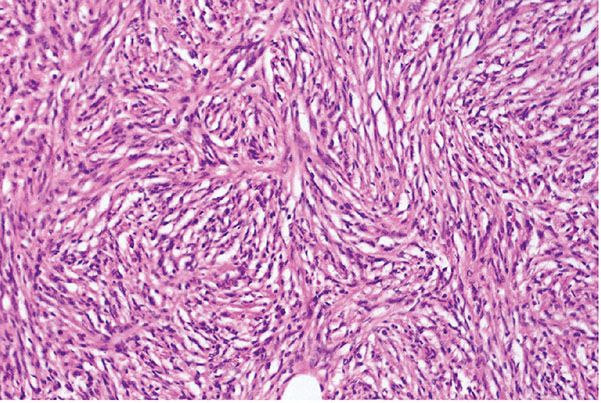
Figure 35-29 Schwannoma; cellular variant (CS1). In this schwannoma, patterns are uniformly cellular (Antoni A type). Uniform spindle cells are arranged in the pattern of interlacing fascicles. In this example, mitotic activity is not a significant feature.
Clinical Summary. Most examples of CS are large tumors of the retroperitoneum and mediastinum and may show immunoreactivity for GFAP (71,72).
Histopathology. The lesions of CS1 are mostly cellular with “patternless” areas in which cells form solid sheets, alternating with areas in which intersecting fascicles of spindle cells are in a storiform pattern (Fig. 35-29). The basic patterns of more classical variants of schwannoma, such as those of a common schwannoma or an epithelioid schwannoma, are preserved, but in some examples most of the cells are cytologically atypical (Fig. 35-29). In some examples, nuclear atypia is uniform and extensive. By definition, mitotic figures are infrequent (<2/10 high-power fields [hpf]). A background population of small lymphocytes also may contribute to the cellularity. Nuclear palisades, Verocay bodies, and Antoni type B components tend to be inconspicuous; there should not be zones of necrosis (3,4).
In CS2 (CS of Woodruff [73]; transformed schwannoma of Reed and Harkin [3]), lesions may have increased cell density, storiform patterns, mitotic figures (up to 20 or more per 10 hpf), necrosis, and focal but frank cytologic atypia (1,72–77). Although histologic features might suggest a transition from benignancy to low-grade malignancy (Fig. 35-30), the large majority of these lesions behave in a benign fashion. Thus, most of these lesions are amenable to local surgery. In making a distinction between these lesions and infiltrating low-grade malignant schwannomas, confinement by the perineurium of the nerve of origin is prognostically and therapeutically important (73).
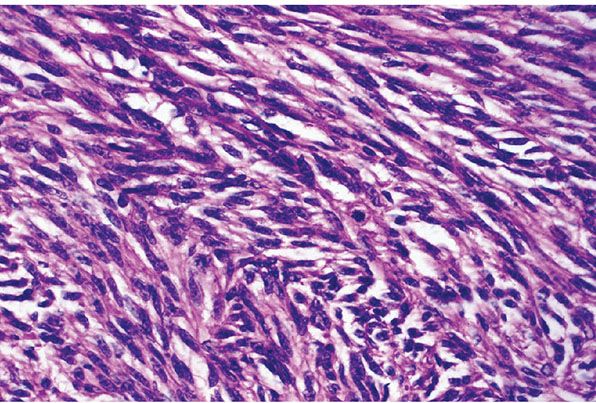
Figure 35-30 Schwannoma; cellular variant with mitotic activity (CS2). In this example of CS, the fascicles are hypercellular; nuclei are enlarged and show hyperchromatism. There are scattered mitotic figures.
Differential Diagnosis. The differential diagnosis of CS1 includes leiomyoma, low-grade leiomyosarcoma, gastrointestinal stromal tumor, and pleomorphic hyalinizing angioectoid tumor. Intense and diffuse expression of S-100 protein with negative CD34, SMA, and desmin would offer support for a diagnosis of schwannoma. Smooth muscle cells, in turn, react with antibodies to SMA and desmin. In gastrointestinal stromal tumors, divergent phenotypes may be expressed, but CD117 (c-kit) is the commonly expressed antigen. Some pleomorphic hyalinizing angioectoid tumors may express CD34 but definitely lack S-100 protein. As mentioned above, CS2 may be confused with MPNST. Circumscription and strong/diffuse S-100 expression are features that support the diagnosis of CS. MPNST may express CD117 (78), p53, and high Ki67 (79,80). Also resembling MPNST, children may develop a plexiform variant of CS (81).
Epithelioid Schwannoma
In the rare epithelioid schwannoma (with or without radial sclerosis) (4,82–84), spindle and round cells are clustered in epithelioid patterns. The nuclei often are round (Figs. 35-31 and 35-32) but they may be irregular and hyperchromatic. Typically, the cytoplasm is large and eosinophilic. In some cases, cells have round nuclei and scanty cytoplasm, and some of the cells are radially arranged in palisades around distinctive zones of sclerosis (4,85–88). In the zones of sclerosis, fibrous lamellae are radially oriented; they interdigitate with cytoplasmic projections of the surrounding tumor cells (88). Such lesions have been characterized as epithelioid schwannoma with radial sclerosis (4), “collagenous spherulosis” in a schwannoma (87), rosetoid schwannoma (88), and the so-called “neuroblastoma-like” epithelioid schwannoma (85,86). The designation “neuroblastoma-like” schwannoma gives recognition to a lymphocyte-like pattern; the nuclei of the small blue tumor cells, in size and staining, resemble those of lymphocytes. The small-cell component may be extensive, or just a focal pattern in what is otherwise a typical schwannoma. The small cells tend to cluster in rosette-like patterns about vessels and spherical zones of radial sclerosis. In our opinion, owing to the possible confusion with a malignant neoplasm, neither the histology nor the clinical behavior of these lesions justifies a characterization of “neuroblastoma-like.” Collagenous spherulosis, as originally defined for breast lesions, is distinct from the patterns of radial sclerosis seen in nerve sheath tumors (89). Some examples of epithelioid schwannoma have been characterized as cellular and others have a plexiform pattern (see Figs. 35-31 and 35-32).
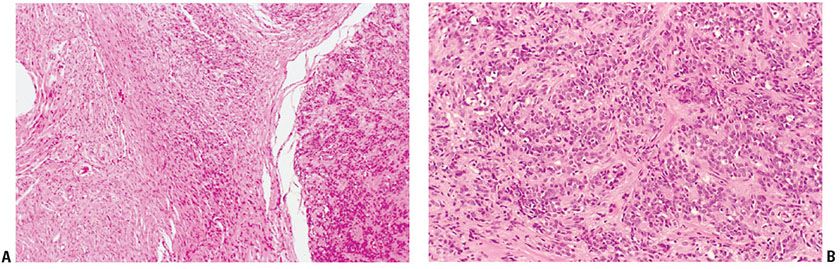
Figure 35-31 Plexiform epithelioid schwannoma, arising in extraneural neurofibroma. A: To the right, a portion of a plexiform lesion presents a sharp interface with an extraneural (diffuse) neurofibroma. Fascicles of uniform, epithelioid cells are loosely, and regularly, spaced in the plexiform component. B: At higher magnification, the fascicles of uniform, small epithelioid cells are loosely spaced in a fibrous matrix.
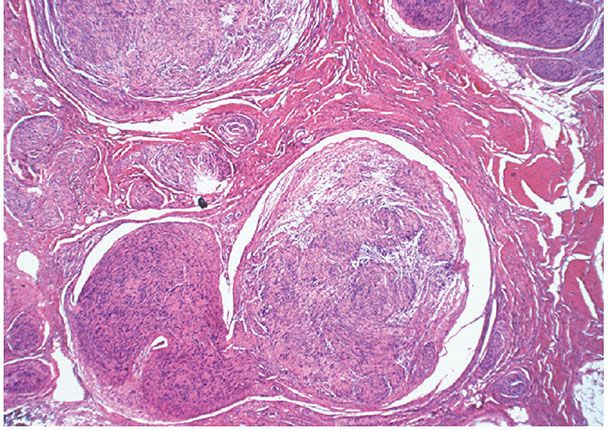
Figure 35-32 Plexiform schwannoma. Components of the plexus are mostly cut in cross section; they are supported by a dense, fibrous matrix. Within some of the components, both Antoni A and Antoni B patterns are represented.
Glandular Schwannoma
Glands, having the features of sweat glands, are occasionally found within subcutaneous schwannomas (90–93). A demonstration of a component of myoepithelial cells has fostered a proposal that the glands are nothing more than entrapped sweat glands. Yoshida and Toot (93) proposed divergent differentiation as an alternate explanation for the glandular inclusions. Glandular differentiation can also be seen in malignant schwannomas and rarely in neurofibromas (92). Some schwannomas may have cysts that contain mucoid material outlined by palisades of Schwann cells; such lesions have been classified as pseudoglandular schwannomas (94).
Plexiform Schwannoma
Clinical Summary. Plexiform schwannomas occur mostly in the subcutaneous tissue and only rarely in the dermis (4,27,95–101). They may be a feature of the syndrome of multiple schwannomas (clinical schwannomatosis or neurilemmomatosis) (98,102) or they may be solitary (99). The same gene is involved in NF2 and multiple schwannomas (26,103).
Histopathology. Plexiform schwannoma is intraneural. Spindle cells, often in solid Antoni A patterns, fill the expanded endoneurial space of a limited segment of a neural plexus; each column and nodule of the tumor is surrounded by perineurium. There may be dilated vascular spaces and Antoni type B tissue. Cystic changes are uncommon. In some examples, fascicles of Schwann cells extend for a short distance beyond the confines of the perineurium into the neighboring mesenchyme. These limited patterns, when encountered in the adjacent soft tissue, resemble those of a small, circumferential, extraneural neuroma (96).
Plexiform schwannomas may be cellular or epithelioid. Those located in the deep soft tissue frequently show histologic features of CS2 (104). In one series, lesions had hypercellularity, pleomorphism, and mitotic figures; tumor necrosis and myxoid change were less common. Females were more commonly affected than males; none of the patients had stigmata of NF. Two of six patients with follow-up had two local recurrences, and one patient had one local recurrence. At last follow-up, all six patients were free of disease (104).
The category of plexiform schwannoma may be heterogeneous. In a single case, Kao et al. (99) have documented a relationship between neurofibroma and plexiform schwannoma. One of the authors has identified cases of plexiform schwannoma that appear associated with a background of diffuse neurofibroma (70) (Figs. 35-31 and 35-32).
Principles of Management. As mentioned in neurofibromas, schwannomas are removed only when they result in cosmetic or functional problems. The aim then is to excise them while preserving neural function. In the case of CS and melanotic schwannomas, owing to their deep-seated location, they are usually excised to rule out the possibility of a sarcoma.
Schwannoma Arising in Neurofibroma (with Emphasis on Relationships with Intraneural Microscopic Schwannomatosis)
There are reports of schwannomas in continuity with neurofibromas/hybrid peripheral nerve sheath tumors (44,105,106). Intraneural microscopic schwannomatosis (see Figs. 35-11 and 35-19) (4), a proliferative change affecting axial bundles of nerve fibers in intraneural neurofibromas, may become a schwannoma ex neurofibroma (22). Rarely, remnants of an intraneural neurofibroma are found at the periphery of a CS (CS1).
Borderline Variants of Schwannnomas
These are lesions in which there is a possibility of local persistence and recurrence and perhaps rarely of metastasis.
Infiltrating (Extraneural) Fascicular Schwann Cell Tumor of Infancy and Childhood
Clinical Summary. Infiltrating fascicular Schwann cell tumor (IFSCT) (plexiform MPNST of infancy and childhood [107]; congenital hamartoma of nerve) is a rare Schwann cell neoplasm most often affecting an extremity (108). In contrast to all variants of schwannoma, IFSCT is chiefly extraneural because true plexiform patterns (intraneural by definition) are not a prominent feature. Therefore, IFSCT does not fulfill the criteria of a variant schwannoma. It also lacks the organoid qualities of a hamartoma.
Histopathology. Uniform spindle cells form tortuous, interlacing fascicles varying in size. In some of the thin fascicles, spindle cells tend to cluster in palisades; collections of cytoplasm separate the neighboring palisades. Some cases may show rapid growth, locally aggressive behavior, hypercellularity, and frequent mitotic figures. The lesion infiltrates skin and soft tissue and, in some examples, erodes bone.
Histogenesis. The most characteristic feature of IFSCT is its invasion of soft tissue in a fascicular pattern. Most tumor cells of IFSCT are clearly identifiable as Schwann cells, both immunohistochemically and ultrastructurally.
Principles of Management. In one published collection, four of the six patients with follow-up had local recurrences (107). In addition, one patient, with an invasive lesion of the orbit, died of disease. Woodruff reported six cases (81), ranging in age from 2 to 15 months. There was no association with NF1. Three of the tumors were well circumscribed but lesions were not encapsulated. Although all tumors recurred locally, children were alive and free of disease at last follow-up.
The documented behavior of reported examples has not convincingly established the nature of the lesion. The histologic patterns, if not the cytologic features, overlap with those of infiltrating and fascicular epithelioid malignant schwannoma (IFEMS); they even share some features with neurotropic melanoma. Their nature is controversial. In our opinion, IFSCT should be characterized as a borderline lesion of indeterminate malignant potential until its nature is better documented.
Psammomatous Melanotic Schwannoma
Clinical Summary. Psammomatous melanotic schwannoma (PMS) is an uncommon tumor typically seen in paraspinal areas followed by the GI tract, bone, subcutaneous tissue, and other locations. Up to two-thirds of the neoplasms cause symptoms (109). It may be either sporadic, or a stigma of the Carney complex. This complex includes some or all of the following: myxomas (including trichogenic and cardiac variants) (110); spotty pigmentation; pigmented adrenal cortical hyperplasia; large-cell calcifying Sertoli cell tumor (111); and endocrine overactivity (including Cushing syndrome) (109,112). Up to 10% of PMS develop metastasis to lungs, liver, and spleen (113).
Histologically, there usually are psammoma bodies, and pigmented, and dendritic melanocytes. Most examples of PMS are quite distinctive and only vaguely resembling schwannomas, but a few lesions show nuclear palisades and Verocay bodies as common schwannomas (109,114). Zhang studied 13 cases of non-PMSs (113). Positive immunohistochemical markers include S-100, Leu-7, HMB45 antigen, Melan-A, and vimentin. The product, in linear patterns, of immunoreactions for laminin and collagen IV correlates with the Schwann cell nature of the tumor cells. Ultrastructural features also are those of Schwann cells (see under Ultrastructure in MPNST) (113).
Principles of Management. These lesions are usually excised to rule out malignancy. The main issue is not to confuse this lesion with a metastatic melanoma. Owing to the possibility of recurrence or metastasis, the aim should be complete excision and close follow-up.
Transformed (Borderline) Schwannoma (TBS) (CS2)
This borderline lesion of uncertain malignant potential is by definition, like its benign counterpart, intraneural (i.e., confined by an expanded perineurium) (4). Its histologic features are reminiscent of those seen in low-grade MPNST of NF (a lesion that is not confined by the perineurium of the nerve of origin). Some examples of CS2 contain scattered globular deposits of melanotic pigment. As defined by Reed and Harkin (4), this lesion shares many features with CS as defined by Woodruff et al. (73), and herein we define this lesion as CS2.
Bona fide malignant transformation of benign schwannoma has been a rarely documented event (115–117). In one report, only histologically “high-grade” variants were represented (118). Such “high-grade” lesions show marked degree of cellular atypia and pleomorphism, and a high mitotic rate. They may show infiltrative growth beyond the confines of the perineurium (118), a quality that excludes CS2.
Principles of Management. As mentioned above, these lesions are usually removed to rule out sarcoma.
MALIGNANT NEOPLASMS OF PERIPHERAL NEURAL TISSUE
These lesions may be sporadic, associated with a neurofibroma, or appear in the context of NF1 (Fig. 35-33). Related malignancies may be de novo, or may be transformations in other neurocristic neoplasms, such as schwannoma or ganglioneuroma (42,119,120).
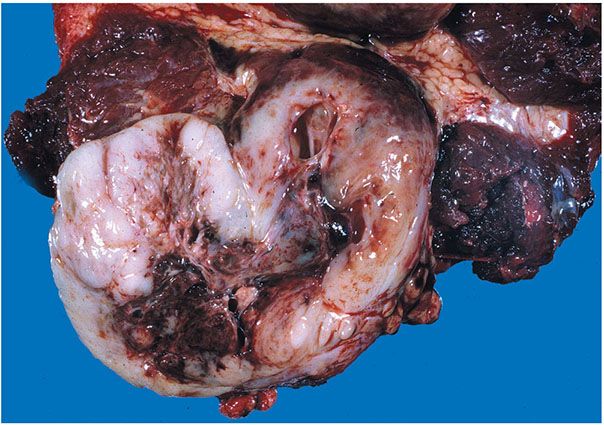
Figure 35-33 MPNST. The tumor is expansile and circumscribed. The cut surface has a fish-flesh quality. A central, degenerative cyst contains blood clot.
Malignant Peripheral Nerve Sheath Tumor of Common Type
Clinical Summary. The criteria for the diagnosis of MPNST include some of the following: origin from, or continuity with, a major nerve or neurofibroma; association with classic stigmata of NF; characteristic morphology in histologic patterns of spindled and epithelioid patterns; and demonstration of at least focal expression of S-100; or ultrastructural features of Schwann cells (and/or perineurial cells) (121).
It has been argued that because most “malignant schwannomas” arise in association with neurofibromas of NF, the designation “malignant schwannoma” is best used only for those rare cases of malignant schwannoma ex schwannoma. On the other hand, it may be argued that the designation “malignant peripheral nerve sheath tumor” provides no distinctions between the common malignancy of NF and an uncommon lesion such as peripheral neuroepithelioma (PNE) (primitive neuroectodermal tumor). It does not provide distinctions between Schwann cell variants and perineurial variants. Similarly, epithelioid variants and mesenchymal variants are not distinguished in the concept of MPNST. However, despite all these shortcomings, this designation is the most popular for malignant lesions with features of neurofibromas.
MPNST ( sheath or Schwann cell type: S-100+) is the common malignant tumor of peripheral nerves (1,3,4,122–128). Its association with NF1 is established but, even in this setting, it is uncommon. On the other hand, of the many peripheral stigmata in NF, neurofibromas are the most predisposed to malignant transformations. In a series of 678 patients with NF, an MPNST was observed in only 21 (3.1%) of the patients (125). In only 2 of the 678 patients did an MPNST primarily involve the skin and subcutaneous fat. Allison et al. (127) reviewed the clinicopathologic features of five examples of MPNST involving the skin or subcutaneous tissue. Four of the lesions occurred in women. One patient had a diagnosis of NF1. Four of the lesions were associated with a CNF; for the remaining case, the tumor involved a superficial peripheral nerve. Four of the cases were of a common type while the remaining case was an epithelioid variant. There were no local recurrences but three patients died of metastatic disease.
MPNSTs usually arise contiguous to a neurofibroma: in turn, they may be intraneural or extraneural. In the deep soft tissue, involvement of a large nerve trunk, such as a femoral, tibial, or intercostal nerve, is characteristic, but in some instances such connection is not apparent. MPNST can also arise as sporadic or de novo lesions (i.e., not associated with NF) (126). As mentioned above, there are rare reports of MPNSTs arising in a schwannoma (118). For many examples of standard MPNST, the patterns are expressive of the patterns of differentiation in embryonic mesenchyme (Fig. 35-34). The expressions can include, singly or in various combinations, fascicles of cells, often alternatingly light and dark (Figs. 35-35A,B); or elements such as chondroid matrix, osteoid (Fig. 35-36A), and rhabdomyoid cells (Fig. 35-36B) (128,129). MPNST of perineurial type has been promoted as a variant, with the predictable proposal that such a lesion is a malignant perineurioma, but the majority of such cases lack evidence that there is a relationship between a benign “perineurioma” and an MPNST (130–132). The biologic potential of such perineurial variants has not been adequately defined because of the limited number of examples.
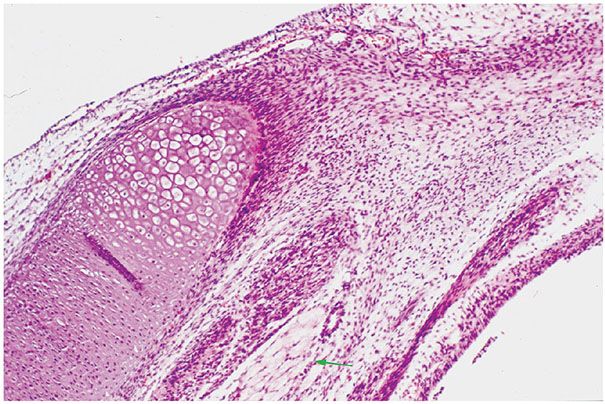
Figure 35-34 Fetal mesenchyme. Fetal mesenchyme shows varying patterns of differentiation. An island of immature cartilage is represented. In the neighboring mesenchyme, there are patterns of alternating light and dark fascicles. One fascicle of developing skeletal muscle fibers ( green arrow) is represented in the myxoid matrix at the bottom of the field. These patterns are recapitulated, in neoplastic distortions, in many examples of MPNST.
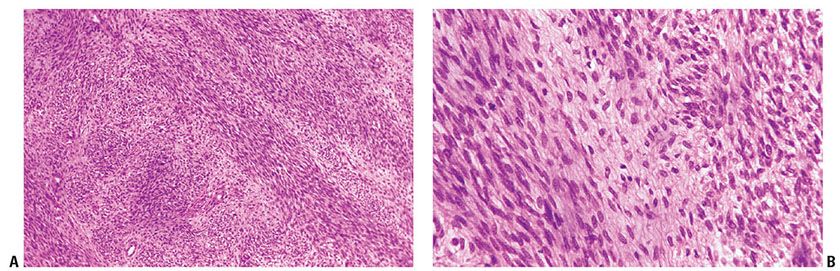
Figure 35-35 MPNST; spindle cell variant. A: Atypical spindle cells form intersecting fascicles; the fascicles are alternately light and dark. B: At higher magnification, spindle cells are cytologically atypical, but cytologic features are uniform.
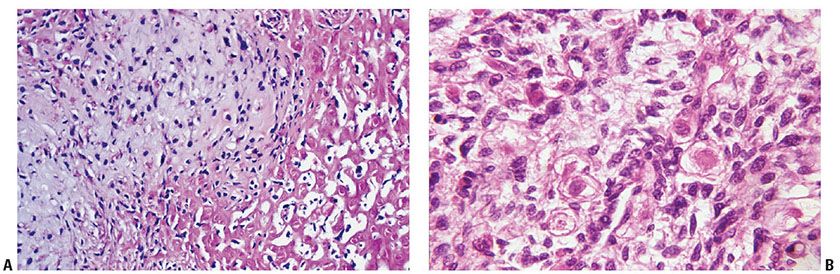
Figure 35-36 Mesenchymal differentiation in MPNST. A: Chondro-osseous differentiation; Chondroid differentiation is represented to the left of the center of the field; to the right, the patterns are those of neoplastic osteoid. B: Rhabdomyoblastic differentiation. In this field of undifferentiated small cells, there are scattered, rounded cells with plump, round nuclei; these round cells show concentrically arranged cytoplasmic myofibrils.
Histopathology. The variable patterns of differentiation in MPNST include mesenchymal (fibrosarcomatous, osteosarcomatous, and chondrosarcomatous (4); rhabdomyosarcomatous (1,118,128); and liposarcomatous) patterns; glandular (endodermal) patterns (92); epithelioid patterns in which individual spindle and round cells often have acidophilic cytoplasm (4,133,134) and are closely spaced in nests, fascicles, and sheets; and neuroepithelioma-like patterns (135).
Histologically, mesenchymal patterns are preponderant in the setting of MPNST that arises in patients with NF (3). Thin spindle cells are arranged in interlacing fascicles in a fibromyxoid matrix. The fascicles are long and straight. They are alternatingly pale and dark (more intensely stained). Often, the spindle cells are remarkably uniform (Figs. 35-35A,B). Neoplastic cells tend to be concentrically, or radially, arranged about vessels, particularly near areas of necrosis. Endothelial cells may be prominent, resembling the endothelial proliferation pattern seen in glioblastomas. As mentioned above, some lesions may show specific patterns of mesenchymal differentiation (Figs. 35-36A,B). When present, such areas usually are spotty in distribution, and there may be multiple patterns of mesenchymal differentiation in a single lesion. Detection of mitotic figures is very important for a diagnosis of MPNST, although it is not a prime determinant of biologic potential. In the setting of NF, rates may be low (<10/10 hpf) or high (>50/10 hpf), and the outcome for either category may be poor; also important are the size and location of the lesions, and the age of the patient.
Epithelioid patterns are a variant expression of MPNST in the setting of NF (3). De novo true malignant schwannomas (126) and “high-grade” MPNST arising in benign schwannomas ( malignant transformed schwannoma) (118) tend to be predominantly intraneural and show “epithelioid” morphologies (133,134). The study of Hruban et al. (135) does not support this generalization but we should take into account that it also included, as de novo variants, examples of both neuroepithelioma and MPNST arising in neurofibroma.
In these epithelioid variants, plump tumor cells, in nests and sheets, are closely spaced with scanty intercellular matrix (Figs. 35-37 and 35-38). Nuclei are large and often rounded. Giant cells are commonly seen in the pleomorphic variants. In some lesions, cells form nests and cords in a sparsely cellular, myxoid or hyaline matrix. The “purely epithelioid” (epithelial) MPNST is rare, representing a lesion without a “sarcomatous” component (136). In some “epithelioid” examples, plump, acidophilic tumor cells manifest a striking cytoplasmic argyrophilia (Fig. 35-39) (4); such examples may represent large-cell neuroendocrine carcinoma.
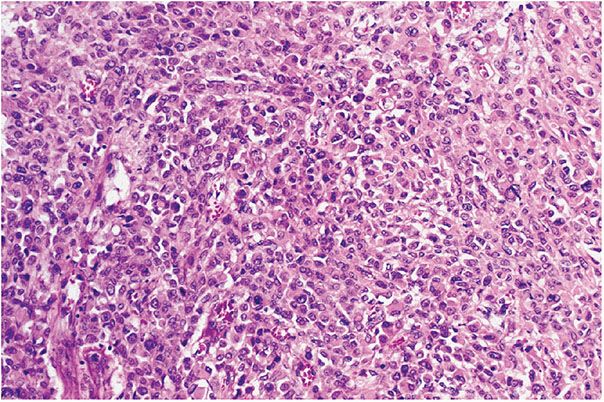
Figure 35-37 Epithelioid MPNST. Plump, atypical, spindle, and epithelioid cells form solid sheets. Intercellular matrix is scanty.
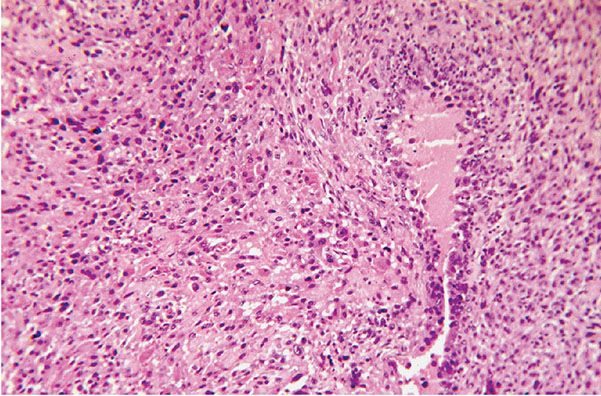
Figure 35-38 Epithelioid MPNST. Plump, rounded cells are arranged in patterns of ill-defined nests. Nuclei show variations in size and staining. A cleft to the right of the center of the field is outlined by palisades of tumor cells.
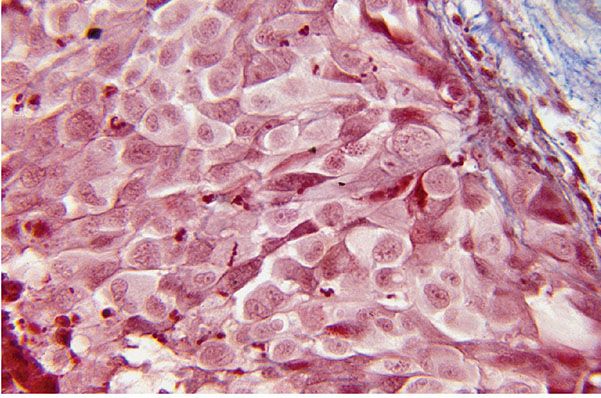
Figure 35-39 Neuroendocrine MPNST (Masson trichrome stain). Large atypical cells are closely clustered; they form distinct nests in a fibrous matrix. The preponderant cells have pale cytoplasm. Dark cells with dendritic processes are compressed among the pale cells. The dark cells are argyrophilic.
Some malignant epithelioid peripheral nerve sheath tumors, some of which appear to have had their origin from, and are in continuity with, a peripheral nerve, may manifest extraneural, infiltrating, and fascicular morphologies (32,133); they may qualify as IFEMSs (70). In the skin and subcutis, such a lesion, in recurrences, might be histologically indistinguishable from desmoplastic and neurotropic melanoma (Fig. 35-40).
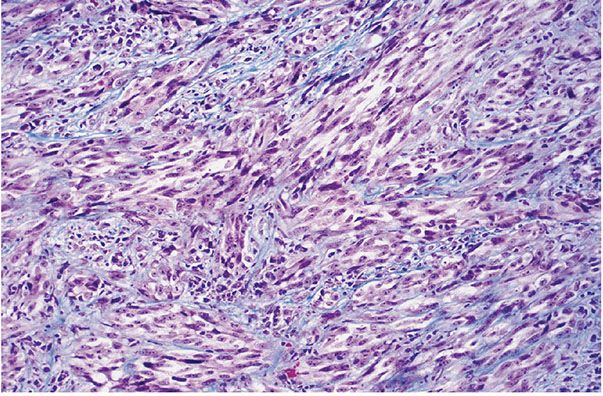
Figure 35-40 Fasciculated, malignant epithelioid peripheral nerve sheath tumor (both melanoma-like and neurotropic). Broad fascicles of atypical spindle cells infiltrate a fibrous matrix; portions of this soft tissue lesion were intraneural.
In both fusiform and epithelioid variants, there may be areas of geographic necrosis with peripheral palisades of tumor cells, reminiscent of patterns in high-grade gliomas of the central nervous system (3).
Regarding possible differences between sporadic and NF1-associated MPNST, there are no pathognomonic histologic findings, although the latter seem to portray worse prognosis.
Ultrastructure. A light microscopic diagnosis of MPNST of sheath (Schwann) cell type is supported by a variety of features. Electron microscopy studies reveal features of both Schwann and perineural cells (33). Slender, overlapping cytoplasmic processes that envelop other processes, or cell bodies, may be connected by intercellular junctions. Granular, flocculent material, preferably along and parallel to the plasma membrane, may occasionally assume the linear form of basal lamina. Fine intracytoplasmic filaments should be absent or relatively scant (121).
Purported Histogenesis. In 40% to 80% of cases of MPNST (32,136,137), the tumor cells are immunoreactive for S-100 protein. CD57 can be detected in 30% to 40% of the cases (137). MBP is less commonly detected (10%) (138). Laminin and collagen type IV are found in about 25% to 30% of cases. A high Ki67 labeling index (LI) has been correlated with a reduced survival rate; in addition, there seems to be a significant correlation between Ki67 LI and the immunohistochemical expression of p53 or MDM2 (80). It has been suggested that an MPNST that is strongly S-100+ might then be characterized as malignant schwannoma (70).
Immunohistochemistry and ultrastructural analysis have been used to highlight patterns of diverse differentiation (123), including schwannian, perineurial and endoneurial, and mesenchymal categories. In that study, some MPNSTs had a high component of cells immunoreactive for S-100 protein, and without perineurial and mesenchymal features. In a second category, less than 50% of tumor cells were reactive for S-100 protein and, in a third category, the cells did not react for S-100 protein; these latter two groups were interpreted as having patterns of heterologous (mesenchymal) and/or perineurial differentiation.
Differential Diagnosis. A moderate increase in cellularity, focal storiform patterns, mild nuclear atypia, and the presence of mitotic figures (<10/10 hpf) are helpful features that distinguish low-grade MPNST from neurofibroma (4,77,79,139). Such examples may qualify as potentially locally aggressive and will probably require only wide local excision.
In the absence of discernible stigmata of NF, differentiation of MPNST from fibrosarcoma can be difficult. Patterns of interlacing fascicles that are alternately light and dark are characteristic of MPNST. For those cases of mesenchymal variants of MPNST with patterns of specific differentiation but lacking a history or stigmata of NF, histologic features would be very similar to those of malignant mesenchymomas.
Monophasic synovial sarcoma shares many features with MPNST, namely the presence of long fascicles of monotonous, spindle cells. In contrast with MPNST, synovial sarcoma has a genetic profile with t(X;18). Common changes are a loss of NF1 locus, and a loss of the band containing gene p53, although such changes are not specific. In 50% of monophasic synovial sarcomas, the cells are focally immunoreactive for cytokeratins and EMA. With the exception of one study, MPNSTs routinely contain translocations involving chromosomes X and 18: t(X;18) (SYT-SSX) (140).
For deeply located epithelioid variants of MPNST, the main differential diagnosis is melanoma. In general, epithelioid melanocytes tend to express at least other melanocytic markers in addition to S-100, for example, MART1, HMB45 antigen, or MITF.
If confronted with infiltrating and fascicular patterns in a tumor of the skin, a distinction between neurotropic melanoma, desmoplastic melanoma, and cutaneous primary infiltrating and fascicular epithelioid MPNST may be impossible in the absence of lentiginous and junctional, melanocytic patterns in the overlying or adjacent epidermis. For those cases, the intensity and diffuseness of S-100 expression may be helpful, because melanomas frequently and strongly express this marker in the majority of tumor cells. Also helpful may be the detection of peripherin, because this is a marker that is expressed in a number of spindle cell melanomas (141) and tends to be absent in MPNST (personal observation).
IFSCT, in its histologic, but not its cytologic features, shows overlaps with the patterns of both IFEMS and neurotropic melanoma.
Glandular, entoderm-like patterns (Fig. 35-41) may be more commonly associated with epithelioid than with mesenchymal variants of MPNST. When such glandular differentiation is present, the main differential diagnosis is carcinosarcoma. Entoderm-like glandular patterns, in the absence of sarcomatous components, rarely are encountered in the soft tissue in the setting of NF; a lesion showing such patterns may represent a primary neuroendocrine carcinoma/Merkel cell carcinoma (142).
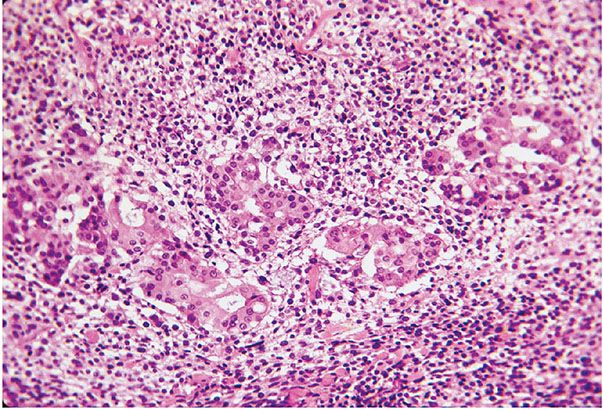
Figure 35-41 Small epithelioid cell MPNST; divergent glandular differentiation. Plump, columnar cells form solid nests and glandular patterns; the cells have small, round nuclei with uniform nuclear characteristics. The glandular spaces contain basophilic mucin. The glandular component is a random manifestation in an epithelioid MPNST.
Principles of Management. In the setting of NF, MPNST, in its many guises, generally has a poor prognosis, although a recent meta-analysis indicates that the differences between MPNST associated or not associated with NF1 are decreasing (143
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


