Introduction758
Hamartomas and tumors of hair germ758
Desmoplastic trichoepithelioma
761
Cutaneous lymphadenoma (trichoblastoma variant)
763
Trichoblastic carcinoma/sarcoma/carcinosarcoma
763
Follicular hamartoma syndromes
764
Generalized hair follicle hamartoma
764
Basaloid follicular hamartoma
764
Linear unilateral basal cell nevus with comedones
764
Infundibular and isthmic tumors764
Tumor of the follicular infundibulum
764
Pilar sheath acanthoma
765
Inverted follicular keratosis
765
Tricholemmal (external sheath) tumors766
Outer root sheath acanthoma
767
Tricholemmal carcinoma
768
Tumors with matrical differentiation768
Tumors with prominent perifollicular mesenchyme770
Fibrofolliculoma/trichodiscoma
771
Birt–Hogg–Dubé syndrome
771
Neurofollicular hamartoma
772
Ectopic sebaceous glands772
Fordyce’s spots and related ectopias
772
Hamartomas and hyperplasias773
Folliculosebaceous cystic hamartoma
773
Benign sebaceous tumors774
Reticulated acanthoma with sebaceous differentiation
776
Malignant sebaceous tumors777
Tumors with focal sebaceous differentiation778
Cysts and hamartomas779
Apocrine hidrocystoma (apocrine gland cyst)
780
Syringocystadenoma papilliferum
780
Benign apocrine tumors781
Hidradenoma papilliferum
781
Tubular adenoma (apocrine adenoma)
781
Apocrine mixed tumor (apocrine chondroid syringoma)
783
Malignant apocrine tumors787
Apocrine adenocarcinoma
787
Extramammary Paget’s disease
788
Adenoid cystic carcinoma
790
Endocrine mucin-producing sweat gland carcinoma
791
Hidradenocarcinoma (nodular)
791
Malignant spiradenoma (spiradenocarcinoma)
792
Tumors of modified apocrine glands793
Adenocarcinoma of Moll’s glands
793
Erosive adenomatosis of the nipple
793
Ceruminous adenoma and adenocarcinoma
793
Tumors of anogenital mammary-like glands
793
Hamartomas and benign tumors794
Papillary eccrine adenoma
795
Poroma group797
Hidroacanthoma simplex
799
Hidradenoma800
Hidradenoma (poroid hidradenoma)
800
Malignant eccrine tumors800
Microcystic adnexal carcinoma
801
Eccrine carcinoma (syringoid carcinoma)
802
Squamoid eccrine ductal carcinoma
803
Polymorphous sweat gland carcinoma
803
Digital papillary adenocarcinoma
804
Miscellaneous sweat gland carcinomas
805
Signet-ring cell carcinoma of the eyelid
805
Small-cell sweat gland carcinoma of childhood
805
Adnexal clear cell carcinoma with comedonecrosis
805
COMPLEX ADNEXAL TUMORS805
Organoid nevus (nevus sebaceus)
806
Adnexal polyp of neonatal skin
807
Combined adnexal tumor
807
Hemifacial mixed appendageal tumor
807
INTRODUCTION
Appendageal tumors will be considered under the traditional headings:
• complex adnexal tumors.
A case could be made on embryological grounds to have only two categories – folliculosebaceous–apocrine and eccrine.
3 More and more tumors are being recognized in the first of these two categories with divergent differentiation, although monophasic differentiation is the usual characteristic.
Occasional tumors defy classification such as the extraparotid Warthin’s tumor and the
ocular adnexal oncocytoma.
4.5. and 6. The oncocytoma is a benign neoplasm composed of polygonal cells with abundant finely granular eosinophilic cytoplasm. The cells are arranged as sheets with some tubular and cystic spaces.
7 The cells express pancytokeratin but not S100 or smooth muscle markers.
7. and 8. Cytoplasmic immunoreactivity for androgen receptors has been reported.
8 None of the lesions arising in the caruncle has behaved aggressively, in contrast to occasional tumors in other oculocutaneous sites.
8 The cell of origin is not known.
Organ transplant recipients have a high frequency and diversity of appendageal tumors.
9 Their tumors are more likely to be malignant and of sebaceous origin.
9
HAIR FOLLICLE TUMORS
There is as yet no universally acceptable classification of hair follicle tumors. Headington, in his comprehensive review in 1976, proposed a detailed histogenetic classification,
10 whereas Mehregan in 1985 used a much simpler classification
11 with three subgroups: hyperplasias (nevi), adenomas and epitheliomas. Rosen has published a classification in which the benign tumors are divided into seven categories, depending on which part or parts of the hair follicle the lesion differentiates towards or most closely resembles.
12In their book on follicular neoplasms, Ackerman, Reddy, and Soyer have published, with a critique, the classifications used in eight textbooks of dermatopathology.
13 No two classifications are the same, although there is some unanimity as to the entities that ought to be regarded as follicular tumors. The new classification of Ackerman and colleagues differs in categorization and nomenclature from the one used here.
13 For example, they now classify trichoepitheliomas as trichoblastomas while conceding that what is known as desmoplastic trichoepithelioma is clinically distinctive. Tricholemmomas and inverted follicular keratoses are warts (still), and basal cell carcinomas have been renamed trichoblastic carcinoma. It is not proposed to modify substantially the classification previously used for follicular tumors.
The distinction between malignant and benign follicular tumors is usually quite easy on histological examination. In difficult cases, DNA image cytometry may be helpful.
14
HAMARTOMAS AND TUMORS OF HAIR GERM
The hamartomas and tumors of hair germ constitute the largest group of pilar tumors. They have in common the formation of nests, strands, and cords of basaloid cells with varying degrees of differentiation towards a hair follicle. At one end of the spectrum there are highly structured and differentiated tumors, such as the hair follicle nevus and trichofolliculoma, whereas at the other end the epithelial proliferations show only limited follicular differentiation and may resemble basal cell carcinomas. Included in this group are the rare trichogenic tumors, which are somewhat analogous to odontogenic tumors in having variants that may contain both epithelial and mesenchymal components. The better differentiated variants will be considered first.
HAIR FOLLICLE NEVUS
These hamartomas are exceedingly rare lesions of the head and neck, presenting as a small nodule or area of hypertrichosis.
11.15.16. and 17. A linear variant has been reported.
18 Such cases may follow Blaschko’s lines.
19 They are composed of closely arranged mature vellus follicles.
20 Headington has suggested the term ‘congenital vellus hamartoma’ for these lesions.
10 They must be distinguished from accessory tragi, in which many vellus follicles can also be seen.
21 The hair follicle nevus has been confused with the trichofolliculoma, but they are histologically distinct.
22Also included in this group is the entity known as ‘faun-tail’, a patch of hairs over the lower sacral area.
11 It is relevant to mention here the rare occurrence of hair follicles on the palms and soles.
23 Becker’s nevus (see
p. 294) is sometimes included in the general category of hair follicle hamartomas.
10The term
congenital panfollicular nevus has been proposed by Finn and Argenyi for a rare lesion of abortive hair follicles arranged in multiple dermal nodules.
24 There was some resemblance to folliculosebaceous cystic hamartoma.
24 The nodules were surrounded by fibrous sheaths.
25
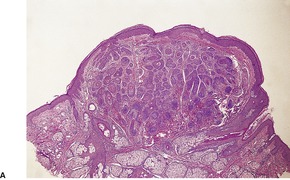
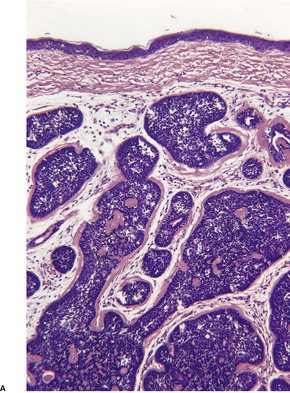
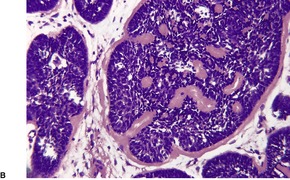
TRICHOFOLLICULOMA
Trichofolliculoma is a rare pilar tumor, intermediate in differentiation between a hair follicle nevus and a trichoepithelioma (see below). It usually presents as a solitary tumor, approximately 0.5 cm in diameter, on the head and neck, usually the face.
26 A tuft of fine hairs may protrude from a central umbilication. A collision tumor of trichofolliculoma and basal cell carcinoma has been reported.
27Schulz and Hartschuh have published their detailed observations on the evolution of a trichofolliculoma.
28 They suggest that the trichofolliculoma undergoes changes corresponding to the regressing hair follicle in its well-known cycle.
28
Histopathology10.11. and 26.
Scattered vacuolated cells representing sebaceous differentiation may be present within the follicles or the rudimentary structures. Sebocytes are seen more often in late stage lesions. Epidermolytic hyperkeratosis has been found as an incidental change.
30 A variant of trichofolliculoma in which large sebaceous follicles connect to a central cavity or sinus has been reported as a
sebaceous trichofolliculoma.
31 It has some similarities to the folliculosebaceous cystic hamartoma (see
p. 773), which appears to be a trichofolliculoma at a late stage in its evolution.
28.32. and 33.The secondary follicles in trichofolliculoma express CK14 but not CK1 and CK10. CK15 is present in the basal layers.
34
TRICHOADENOMA
Trichoadenoma (of Nikolowski)
35 is a rare tumor with hair follicle-like differentiation; it is found as a nodular lesion, particularly on the face and buttocks.
36. and 37. A combined trichoadenoma and intradermal nevus has been reported.
38A lesion reported as a congenital trichoadenoma
39 was regarded as a nevus sebaceus in subsequent correspondence from Ackerman.
40
Histopathology36
Trichoadenomas contain cytokeratin 20-positive Merkel cells but lack Ber-EP4 and androgen receptor expression.
42Verrucous trichoadenoma is a variant of trichoadenoma which clinically resembles a seborrheic keratosis.
43 It is composed of small epidermoid cysts, some of which contain vellus hairs. There is abundant keratin on the epidermal surface.
43
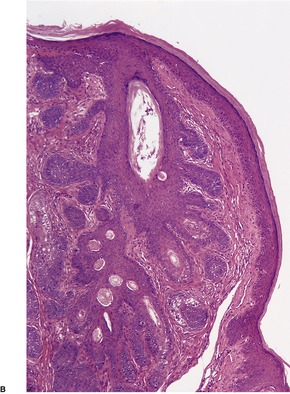
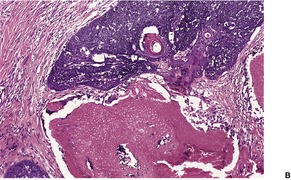
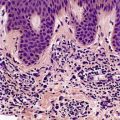
TRICHOEPITHELIOMA
Trichoepitheliomas (trichoblastomas in the classification of Ackerman et al) are regarded as poorly differentiated hamartomas of hair germ.
10. and 13. There are three variants of trichoepithelioma: solitary, multiple, and desmoplastic. The histological features of the solitary and multiple types are identical and they will be considered together. Desmoplastic trichoepithelioma is a distinct clinicopathological entity and it will be discussed separately below.
Solitary trichoepitheliomas are found as skin-colored papules, with a predilection for the nose, upper lip, and cheeks. They measure up to 0.5 cm in diameter. Most of the lesions reported as giant solitary trichoepitheliomas are trichoblastomas.
44.45.46. and 47. Rare presentations include a linear form and as a large, hemifacial plaque.
48. and 49. Vulvar trichogenic tumors can be misdiagnosed as basal cell carcinomas, leading to overtreatment.
50
Multiple familial trichoepitheliomas (epithelioma adenoides cysticum – OMIM 601606) have an autosomal dominant mode of inheritance, with lessened expressivity and penetrance in the male. It is due to a mutation in the
CYLD (cylindromatosis) gene on chromosome 16q12–q13.
51. and 52. Mutations in this gene may also produce familial cylindromatosis (OMIM 132700) and the Brooke–Spiegler syndrome (multiple trichoepitheliomas and cylindromas – OMIM 605041).
51.53.54.55.56. and 57. The presence also of spiradenomas may be another manifestation of Brooke–Spiegler syndrome.
58 These various manifestations have been regarded as different expressions of a folliculosebaceous–apocrine genodermatosis.
59 Multiple different mutations in this gene have been reported in familial trichoepitheliomas.
51. and 60. The gene encodes the protein CYLD, which is a deubiquitinating enzyme recently implicated in modulation of the nuclear factor (NF)-κB pathway.
51 It is down-regulated, or not expressed as a result of the gene mutations.
61 The significance of the much earlier report implicating 9p21 in multiple trichoepitheliomas is uncertain.
54 Sporadic cases of this condition also occur.
10. and 62. Multiple trichoepitheliomas present as small papules with a strong predilection for the central part of the face.
63 The trunk, neck, and scalp are sometimes involved. The papules may coalesce to give plaques.
64.65.66. and 67. The onset of lesions is usually in childhood or at the time of puberty.
68 Rare presentations include a linear and dermatomal distribution,
69 linear lesions on the face in the lines of Blaschko (a possible type 2 segmental manifestation),
70. and 71. development in an epidermal nevus,
72 an association with cylindromas (Brooke–Spiegler syndrome)
65. and 73. and sometimes also with spiradenomas (see above),
58.59. and 74. and an association with ungual fibromas,
75 dystrophia unguis congenita,
76 and the ROMBO syndrome (vermiculate atrophoderma, milia, hypotrichosis, trichoepithelioma, basal cell carcinomas, and peripheral vasodilatation).
77.78. and 79. A
PTCH gene deletion was present in the trichoepitheliomas that developed in a patient with neurofibromatosis (NF-1).
80 The simultaneous appearance of trichoepitheliomas and carcinoma of the breast has also been reported.
81
Trichoepitheliomas are benign lesions. Many of the cases of malignant transformation reported in the older literature represent cases of the nevoid basal cell carcinoma syndrome and not trichoepitheliomas.
10 Nevertheless, there are several documented cases of basal cell carcinoma developing in trichoepithelioma,
82.83. and 84. and one case of a malignant tumor, with pilar (including matrical) differentiation, arising in a trichoepithelioma.
85One study has shown deletions at 9q22.3 (the location of the
patched (PTCH) gene of the basal cell nevus syndrome) in sporadic trichoepitheliomas.
86Various treatment modalities have been successful in treating trichoepithelioma.
87 They include carbon dioxide laser vaporization,
88 cryotherapy, electrosurgery, surgical excision, and radiotherapy.
87 For multiple trichoepitheliomas additional treatment methods have included argon laser, and the combination of erbium:YAG and CO
2 laser.
89 Such therapies pose an important risk of significant scarring in the area affected.
67 Very gradual improvement has been reported following the use of aspirin and subcutaneous adalimumab to block TNF activation of NF-κB at two levels of the pathway.
90
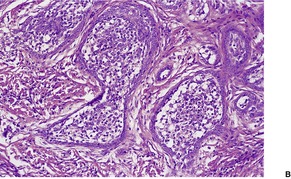
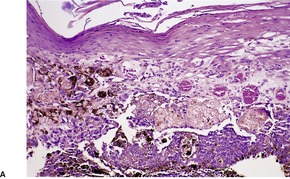
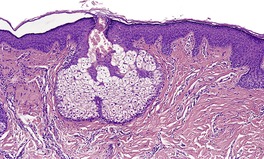
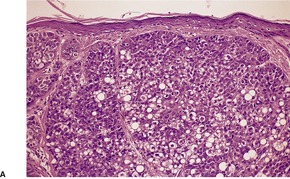
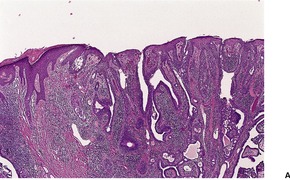
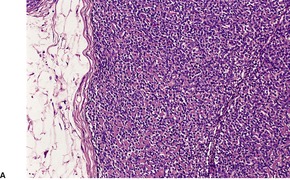
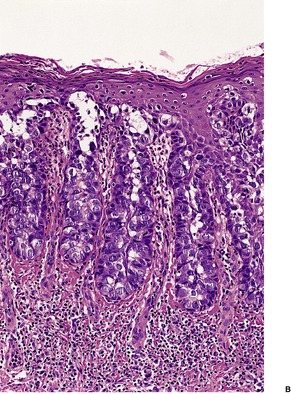
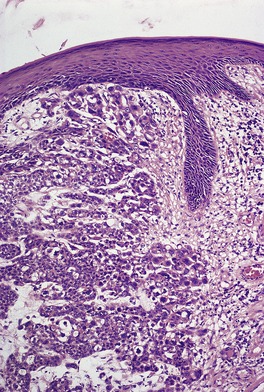
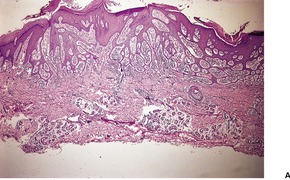
Histopathology
Trichoepitheliomas are dermal tumors with focal continuity with the epidermis in up to one-third of cases. They are composed of islands of uniform basaloid cells, sometimes showing peripheral palisading. This may lead to a mistaken diagnosis of basal cell carcinoma if features of pilar differentiation are not noted. In addition, there are usually branching nests of basaloid cells. Epithelial structures resembling hair papillae or abortive hair follicles may be seen (
Fig. 33.3). A tumor with
giant and multinucleated epithelial cells has been reported. 91 Small keratinous cysts lined by stratified squamous epithelium are quite common. Rupture of these cysts with liberation of the keratinous debris results in a small foreign-body granuloma in the stroma. Foci of calcification are often present; amyloid is generally considered to be uncommon although it was present in 33% of cases in one study.
92. and 93. Melanocytes are common in most pilar neoplasms.
94 The stroma is prominent and loosely arranged. Aggregations of fibroblasts, representing abortive attempts to form papillary mesenchyme (papillary-mesenchymal bodies), are characteristic of trichoepithelioma.
95 A trichoepithelioma with ‘monster’ stromal cells resembling an atypical fibroxanthoma has been reported.
96 A trichoepithelioma-like basal cell carcinoma has also been documented.
97 LeBoit has editorialized on the morphological overlap of these two tumors.
98Immunohistochemical staining of trichoepitheliomas resembles that seen in the outer root sheath, with strong reactions for keratins (CK) 5/6 and CK8. Some CK17 is present in cells surrounding horn cysts.
99 Conflicting views have been expressed on the specificity of cytokeratin types in the distinction between trichoepithelioma and trichoblastoma. Although one study concluded that there were no differences,
100 another study found CK7 in trichoblastomas but not trichoepithelioma.
101 Trichoepitheliomas express CK15, whereas only a subset of basal cell carcinomas do this.
102 Basal cell carcinomas differ from trichoepitheliomas by having stronger and more diffuse expression of PCNA and Ki-67.
103 They also show greater expression of p53 than trichoepitheliomas.
104CD10 expression varies but a small number of cases show overlap features. As a rule, trichoepithelioma shows CD10 stromal immunoreactivity while in basal cell carcinoma the staining involves basaloid cells, but some stromal staining is sometimes seen.
105 Both basal cell carcinomas and trichoepitheliomas express p27
kip1 but staining tends to be patchy in trichoepithelioma and more diffuse in basal cell carcinoma.
106 Peritumoral stromal cells expressing CD34 are almost invariably present in pilar tumors.
94
Electron microscopy
In trichoepitheliomas there is a proliferation of basaloid cells similar to basal cell carcinoma.
107 Abortive hair papillae and keratinous cysts may be seen. The cells sometimes contain paranuclear glycogen, a feature not seen in basal cell carcinomas.
107
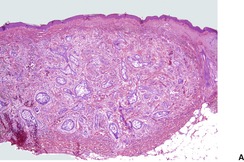
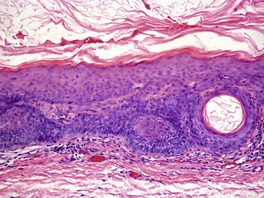
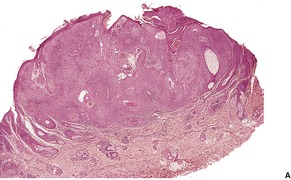
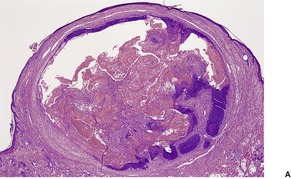
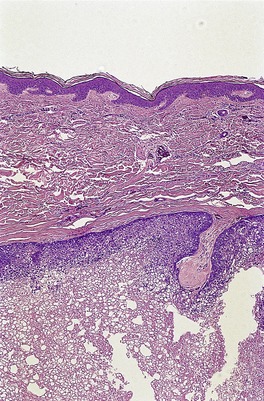
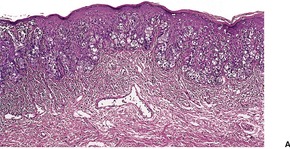
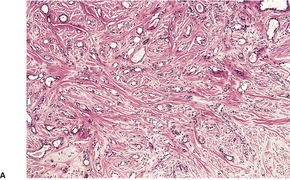
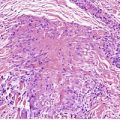
DESMOPLASTIC TRICHOEPITHELIOMA
Desmoplastic trichoepithelioma is a histological variant of trichoepithelioma that occurs almost exclusively on the face.
108.109.110.111.112.113.114. and 115. There is a predilection for females and relatively young adults. A congenital example has been reported.
116 Solitary familial desmoplastic trichoepithelioma,
108. and 117. and multiple familial
118. and 119. and non-familial tumors
109 have been reported. Desmoplastic trichoepitheliomas usually present as asymptomatic solitary hard annular lesions with a raised border and a depressed center. They vary from 3 to 8 mm in diameter. Atypical clinical presentations have been reported.
120The finding of Merkel cells as an integral constituent of this tumor raises the possibility of a bulge-derived origin because of the high number of Merkel cells known to reside in this area of the follicle.
121
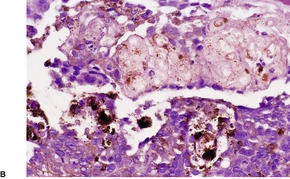
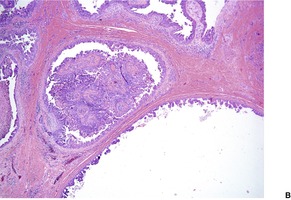
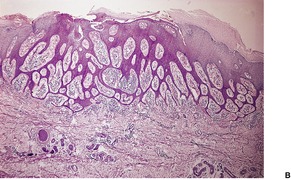
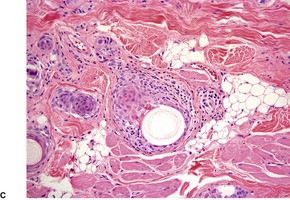
Histopathology122
Desmoplastic trichoepitheliomas are reasonably well-circumscribed tumors in the upper and mid dermis, with an overlying central depression. They are composed of cords and small nests of basaloid cells with scanty cytoplasm (
Fig. 33.4). Tumor strands may be attached to the epidermis. There are usually many keratinous (horn) cysts, with a peripheral basaloid layer and several layers of stratified squamous epithelium with central loosely laminated horn.
123 Sometimes this epithelial lining is attenuated and one cell thick. Tadpole- or comma-shaped epithelial projections extend from the peripheral layer of some of the keratinous cysts. Sometimes, structures resembling eccrine ducts are present.
The desmoplastic stroma is dense and hypocellular, with fewer elastic fibers and more acid mucopolysaccharides than in the normal dermis. Other features which are frequently present include foreign-body granulomas, usually related to ruptured cysts, and calcification. Stromal ossification is rare. Occasionally foci of sebaceous cells or shadow cells are present. Rarely, a nevocellular nevus is also present.
110. and 124.The tumor cells do not contain carcinoembryonic antigen, unlike the cells in a syringoma, which are usually positive.
111 In contrast, involucrin is expressed in desmoplastic trichoepitheliomas but not in syringomas.
112 The spindle-shaped cells surrounding the cellular islands in desmoplastic trichoepithelioma are focally strongly positive for CD34, whereas the stromal cells around basal cell carcinomas and microcystic adnexal carcinomas are usually negative.
125.126. and 127. Epithelial membrane antigen (EMA) is negative in desmoplastic trichoepitheliomas.
97 Whereas bcl-2 is expressed in most basal cell carcinomas, it is found only in the basal layer of trichoepitheliomas. Ber-EP4 is positive at least focally in most trichoepitheliomas, contrasting with the usual diffuse staining in basal cell carcinoma.
97 Up to 80% of basal cell carcinomas express androgen receptors, whereas trichoepitheliomas are typically negative.
128.129. and 130. Merkel cells (CK20
+) are present in
desmoplastic trichoepitheliomas but uncommon in basal cell carcinoma.128. and 131. Unfortunately these markers do not always differentiate reliably between basal cell carcinoma and desmoplastic trichoepithelioma as Merkel cells are sometimes sparse in desmoplastic trichoepitheliomas.129.130. and 132. The cord-like basaloid cells in desmoplastic trichoepitheliomas express CK1/5/10/14, CK5/8, CK14, and CK15, but not CK6. 133The differential diagnosis includes syringoma and morphea-like basal cell carcinoma.
114.122. and 134. Syringomas rarely have horn cysts, foreign-body granulomas, or calcification.
122 Narrow strands of tumor cells are also unusual in syringomas. Furthermore, syringomas are usually periorbital and multiple. Basal cell carcinomas of morpheic type may form clefts between the nests and the stroma.
114 There is often coexisting nodular basal cell carcinoma. Mitoses and apoptotic bodies are also quite common in basal cell carcinomas, whereas foreign-body granulomas and ruptured keratinous cysts are uncommon.
114 The staining pattern for CD34 and bcl-2 (see above) is also of use in the differentiation of these two tumors.
Electron microscopy
Ultrastructural studies
108. and 113. have shown basaloid cells surrounded by a basal lamina. Individual cells contain tonofilaments and are connected to adjacent cells by desmosomes.
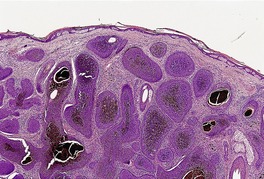
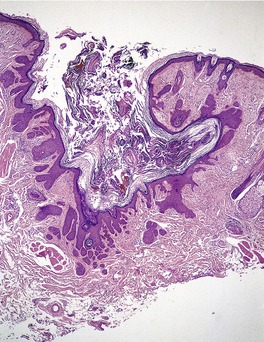
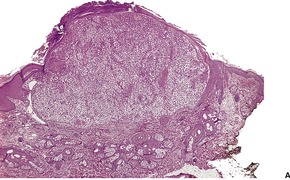
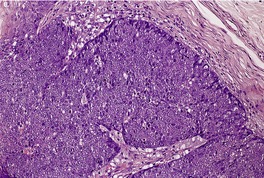
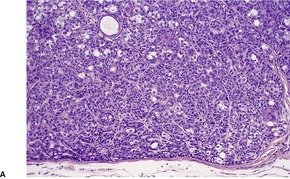
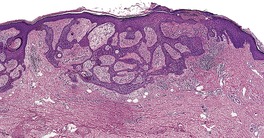
TRICHOBLASTOMA
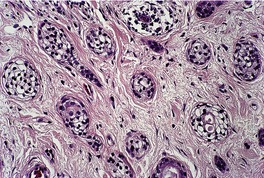
Trichoblastomas are extremely rare benign tumors of the hair germ in which follicle development may be partly or completely recapitulated.
46.135.136.137. and 138. They are constituted largely of follicular germinative cells.
139 They have been likened to the odontogenic tumors, which may also be epithelial and/or mesenchymal.
10 As initially reported by Headington, the trichogenic tumors were further classified on the basis of the relative proportions of epithelial and mesenchymal components: the predominantly epithelial trichogenic tumor was called a trichoblastoma and the predominantly mesenchymal variant was labeled trichogenic fibroma.
140 Other categories were included.
10 Other terms have also been used for tumors in this group, including solitary, giant and immature trichoepithelioma,
46.141. and 142. and trichogerminoma.
143 The cutaneous lymphadenoma is a distinctive variant of trichoblastoma which will be considered separately. The tumor reported as a ‘rippled-pattern trichomatricoma’ is also a trichoblastoma.
144Trichoblastomas are usually greater than 1 cm in diameter and involve the deep dermis and subcutis. Lesions confined to the subcutis are seen occasionally.
145 They usually present as a slowly growing nodule. A case with a keratin-filled dilated pore in the center of the lesion has been reported.
146 The head, particularly the scalp, is a common site.
147 The eyelid is a rare site.
148 Multiple lesions have been reported.
149 Trichoblastoma and syringocystadenoma papilliferum are two common tumors that arise in organoid nevi.
150 Trichoblastomas are not aggressive, unless they have been misdiagnosed or contain an element of basal cell carcinoma.
13. and 151.Whereas basal cell carcinomas and trichoepitheliomas are characterized by mutations in the
PTCH gene, such mutations are not common in sporadic trichoblastomas.
152 The genetic basis of trichoblastomas remains elusive.
152
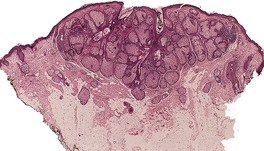
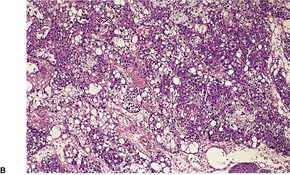
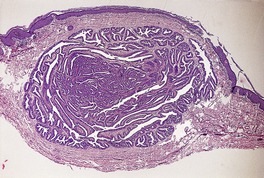
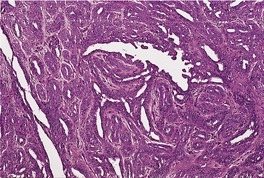
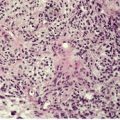
Histopathology10. and 137.
The low-power view is usually quite striking: a large, circumscribed basaloid tumor with no epidermal connection. It is usually situated in the mid and lower dermis, with extension into the subcutis. The tumor shows irregular nests of basaloid cells resembling a basal cell carcinoma, but with variable stromal condensation and pilar differentiation (
Fig. 33.5). The variant reported as
trichogerminoma was composed of closely packed lobules of basaloid cells resembling hair bulbs, with little intervening stroma.
143. and 153. It has also been regarded as a variant of (large nodular) trichoblastoma with less overt follicular differentiation.
97 Foci of necrosis may be present in this variant.
153 At the other end of the spectrum is the
trichoblastic fibroma, with an intimate relationship between basaloid nests and strands, and a fibrocellular stroma.
154.155. and 156. A desmoplastic stroma has been reported.
157 So-called ‘stromal induction’, with the formation of primitive hair bulbs, is present. Keratinous cysts may be present in this group of trichoblastomas but they are not seen in the cellular, basaloid variants that may resemble basal cell carcinoma.
Ackerman and colleagues have used ‘trichoblastoma’ as a generic term for all neoplasms of the skin and subcutaneous fat that are composed mostly of follicular germinative cells. Trichoepitheliomas are included in this definition.
13 They have reported nodular, retiform, cribriform, racemiform, and columnar patterns of trichoblastoma.
13Stromal amyloid and Merkel cells are quite common in trichoblastomas.
46.97. and 158. Rare variants include clear cell,
159 pigmented (pigmented trichoblastoma),
160. and 161. and adamantinoid lesions (
Fig. 33.6).
139 A variant of pigmented trichoblastoma colonized by abundant dendritic melanocytes has been called a
melanotrichoblastoma.
162 Focal sebaceous and apocrine differentiation are rare.
163.164. and 165. It is generally agreed that the cutaneous lymphadenoma is an adamantinoid variant of trichoblastoma with lymphocytic infiltration of the basaloid nests (see below). Trichoblastomas with basal cell carcinoma-like foci have been reported. Such
tumors do not express CK15 as do other trichoblastomas. Furthermore, they lose their Merkel cells. 166 A trichoblastoma arising within an apocrine poroma, which also showed sebaceous differentiation, has been reported.
167 The cells in a trichoblastoma express CK8 and CK19.
168 CK7 is often expressed, in contrast to trichoepithelioma.
101 Both trichoblastomas and trichoepitheliomas are characterized by papillary mesenchymal bodies which express CD10.
97 Trichoblastomas and trichoepitheliomas do not express androgen receptors; they are expressed in basal cell carcinomas.
169 Neither trichoblastomas nor basal cell carcinomas express hair keratins.
170The
rippled-pattern trichoblastoma has a palisaded arrangement of epithelial ribbons with areas of nuclear palisading resembling Verocay bodies. Focal sebaceous differentiation may occur. The expression of cytokeratins is similar to the more usual trichoblastoma.
171
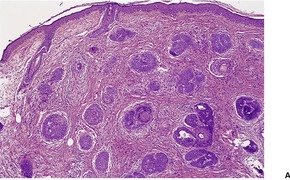
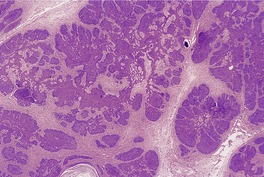
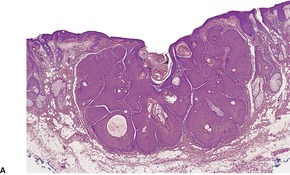
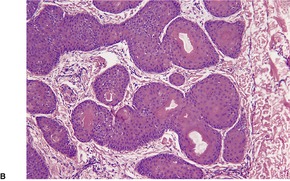
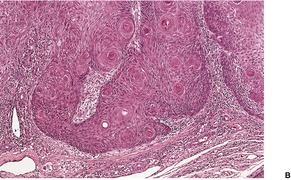
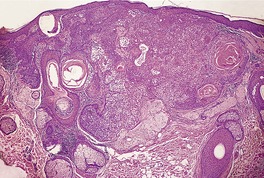
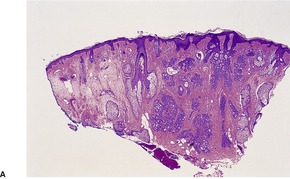
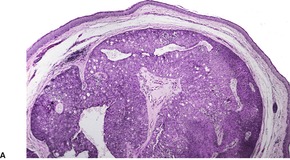
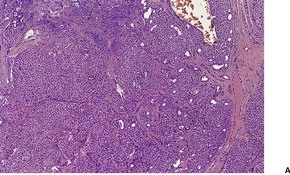
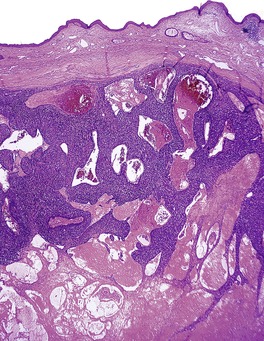
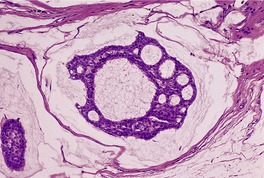
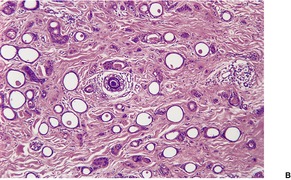
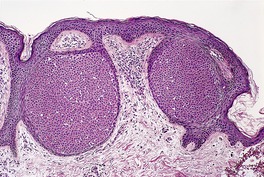
CUTANEOUS LYMPHADENOMA (TRICHOBLASTOMA VARIANT)
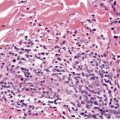
Cutaneous lymphadenoma is a rare adnexal tumor with a prominent lymphocytic infiltrate in the tumor nests.
172.173.174.175.176.177.178.179. and 180. It is a distinctive variant of trichoblastoma.
173 It has also been called ‘lymphotropic adamantinoid trichoblastoma’.
181 The tumor presents as a small nodule on the face or legs. The lesion has usually been present for many months or years. Most cases develop in adults; onset in adolescence is rare.
181. and 182.Local excision is curative.
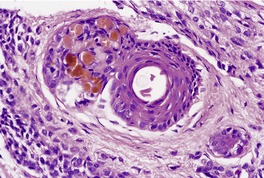
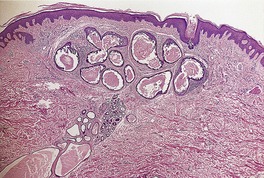
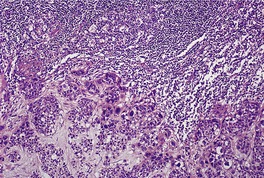
Histopathology
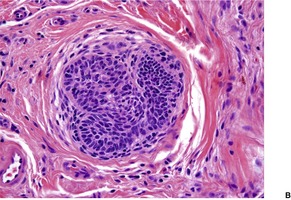
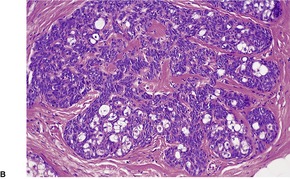
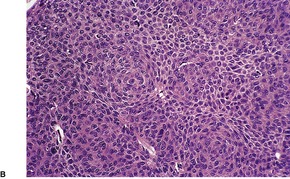
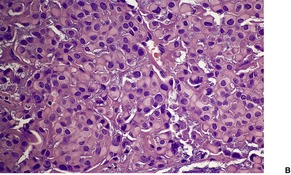
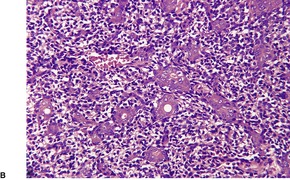
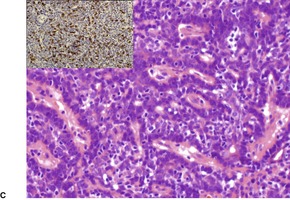
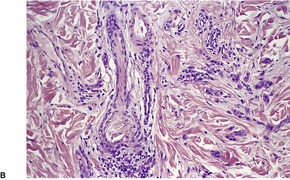
Cutaneous lymphadenoma is composed of multiple rounded lobules of basaloid cells with some degree of peripheral palisading, embedded in a fibrous stroma of variable density (
Fig. 33.7). The stroma may rarely be desmoplastic.
183 There is an intense infiltrate of small mature lymphocytes within the lobules, with some spillage into the stroma.
176 A hint of follicular differentiation and focal sebaceous differentiation is sometimes present.
177 Some nests may show adamantinoid features, but this does not appear to be a universal change, a strong point against cutaneous lymphadenoma being synonymous with adamantinoid trichoblastoma.
184 Focal stromal mucinosis is an uncommon finding.
179Both T and B lymphocytes are found within the lobules, as well as some S100-positive dendritic cells.
174. and 182. The pattern of staining with CD34 and bcl-2 is similar to that seen in trichoblastomas and trichoepitheliomas.
185 There are scattered CD30-positive cells within the tumor nests.
185.186. and 187. Focal staining with CD15 has also been reported.
187 The epithelial cells are usually positive for cytokeratin and EMA.
188
TRICHOBLASTIC CARCINOMA/SARCOMA/CARCINOSARCOMA
These three variants of malignant trichoblastoma are considered together, because of their extreme rarity. The
trichoblastic carcinoma (malignant trichoblastoma) is a high-grade carcinoma arising in a trichoblastoma. Two of the four cases reported up to 2005 had died with metastatic disease.
143. and 189. One of the trichoblastic carcinomas arose in the base of a trichoepithelioma in an elderly female with the Brooke–Spiegler syndrome.
190 Low-grade trichoblastic carcinomas are synonymous with basal cell carcinomas in some classifications.
13 All cases of trichoblastic carcinoma have been characterized by an undifferentiated carcinoma with numerous mitoses and some necrosis. Basaloid and spindle-cell areas are sometimes present.
190The term
trichoblastic sarcoma has been applied to a high-grade stromal tumor arising in a trichoblastoma.
191 The lesion, which was located on the posterior neck, had been present for many years, but there had been rapid growth in the months preceding its removal. The tumor consisted of a multifocal proliferation of basaloid follicular cells with a retiform growth pattern surrounded by a stroma resembling the perifollicular sheath.
191 In places the stroma showed abrupt transition into a pleomorphic proliferation of large sarcomatous cells with frequent and often atypical mitoses.
191 The stromal cells expressed CD10 while the basaloid cells were positive for cytokeratins and 34βE12.
191Both a high-grade and a low-grade
trichoblastic carcinosarcoma of the skin have been reported.
192. and 193. The authors believed that the two cases were authentic carcinosarcomas and not examples of metaplastic carcinoma.
193 Both tumors were composed of two discrete components: the first was epithelial with some basaloid cells with frequent mitotic figures, nuclear atypia and focal nuclear crowding; the second was a stromal component which was composed of pleomorphic spindle-shaped cells with some bizarre multinucleated cells in this high-grade lesion.
192. and 193. The epithelial cell component stained for cytokeratin (AE1/AE3) and the stromal component for vimentin but not cytokeratin.
193 There was no recurrence of either tumor at follow-up. Further cases have since been reported. An underlying B-cell chronic lymphocytic leukemia was present in one case of trichoblastic carcinosarcoma, and also in a trichogenic carcinoma in the same series.
194
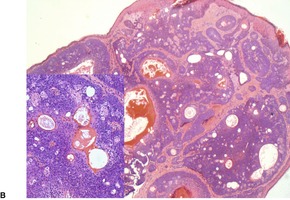
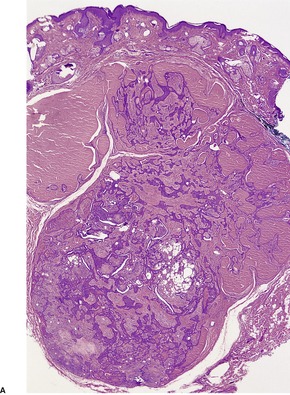
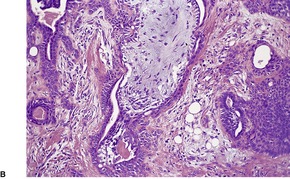
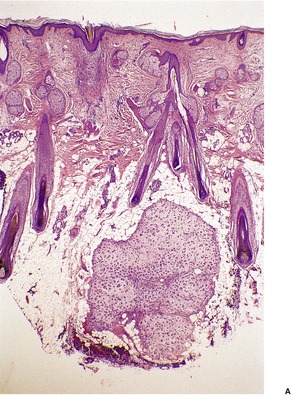
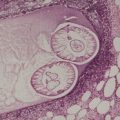
PANFOLLICULOMA
Panfolliculoma is an exceedingly rare, but distinctive tumor, with advanced follicular differentiation. It was described by Ackerman and colleagues.
13 It has overlap features between a trichoblastoma (see
above) and a matricoma (see p. 770), but it differs from the former by the presence of differentiation towards all elements of the follicle, including cystic structures containing corneocytes. Matrical differentiation is less conspicuous than it is in matricoma. A cystic variant has been described (
Fig. 33.8).
195
FOLLICULAR HAMARTOMA SYNDROMES
Several extremely rare syndromes are characterized by the development of localized, zosteriform or linear tumors with overlapping histological features. Cases have been both familial and sporadic, and the associated clinical features have been varied. Ackerman has challenged the validity of these syndromes, believing them to be different expressions of infundibulocystic basal cell carcinomas occurring in variants of the nevoid basal cell carcinoma syndrome, although this has been disputed.
196 The lesions exhibit fewer mitoses and apoptotic cells than basal cell carcinomas.
197 Their Ki-67 labeling is lower than basal cell carcinomas.
197 Some of the cases to be mentioned here have only a vague resemblance to infundibulocystic basal cell carcinomas. It is acknowledged that they may not all be discrete entities.
Generalized hair follicle hamartoma
Generalized hair follicle hamartoma has been applied to patients with papules and plaques on the face, progressive alopecia, and myasthenia gravis.
198. and 199. Cystic fibrosis is a further association of this follicular hamartoma.
200 Involved skin shows a spectrum of changes, with some lesions resembling trichoepithelioma and others ‘basaloid follicular hamartoma’, whereas uninvolved skin may show small islands of basaloid cells.
199 A case with diffuse sclerosis of the face has been reported.
201
Basaloid follicular hamartoma
The term ‘basaloid follicular hamartoma’ was coined by Mehregan and Baker for three patients with localized or systematized lesions in which individual hair follicles were replaced or were associated with solid strands and branching cords of undifferentiated basaloid cells with some intervening fibrous stroma.
202 Although they regarded the condition as a localized variant of generalized hair follicle hamartoma, in their cases there was less resemblance to trichoepithelioma and more to a premalignant fibroepithelial tumor of Pinkus.
202 A linear variant has also been reported.
203. and 204.Similar cases, both solitary and multiple familial cases, have been reported by Brownstein and others.
205. and 206. The lesions remain stable for many years.
197 They are 1–2 mm smooth facial papules composed of anastomosing strands of basaloid and squamoid cells in a loose stroma. Horn cysts and pigmentation are common. It seems that there are four distinctive clinical forms: a solitary papule, a localized plaque of alopecia, a localized linear and unilateral papule or plaque, and generalized papules often with associated alopecia and myasthenia gravis.
207.208.209. and 210. Increased sonic hedgehog signaling pathways with increased Gli-1 transcription is present in some cases. This may explain the response of one patient to retinoid therapy, as retinoids decrease Gli-1 transcriptional activity.
208The family reported by Wheeler and colleagues as dominantly inherited
generalized basaloid follicular hamartoma syndrome had clinical and histological differences to Brownstein’s cases.
211 The lesions were composed of nests of bland basaloid cells surrounded by scant fibrous stroma. They were usually associated with hair follicles. There were infundibular cysts and comedone-like lesions but they had no resemblance to the infundibulocystic basal cell carcinoma.
211 It has since been suggested
212 that this family may have a form of Bazex–Dupré–Christol syndrome (Bazex syndrome – OMIM 301845). Notwithstanding this view, Smoller and colleagues have accepted generalized basaloid follicular hamartoma syndrome as an autosomal-dominantly inherited disorder that presents with disseminated milia, palmoplantar pitting, hypotrichosis, and basaloid follicular hamartomas.
210 Their studies of this syndrome using bcl-2, CD34, and CD10 found similarities in differentiation with trichoepithelioma.
210
Linear unilateral basal cell nevus with comedones
Linear unilateral basal cell nevus with comedones refers to linear or zosteriform lesions, some with comedone plugs, that are present at birth or soon after.
213 Although there is some clinical resemblance to nevus comedonicus (see
p. 669), the histology resembles a basal cell carcinoma. Some cases have had a lattice-like growth of basaloid cells attached to the undersurface of the epidermis and vague follicular differentiation. These features were also present in the case reported as
localized follicular hamartoma.
214
INFUNDIBULAR AND ISTHMIC TUMORS
Two tumors arise from the infundibulum, the uppermost portion of the hair follicle above the opening of the sebaceous duct, and a further two arise from the segment below, the isthmus, which extends from the origin of the sebaceous duct to the level of the bulge. The infundibular tumors are the dilated pore of Winer and the inverted follicular keratosis, while the isthmic tumors are the tumor of the follicular infundibulum (better renamed the tumor of the follicular isthmus) and the pilar sheath acanthoma.
13All are characterized by a superficial location, connection with the epidermis and pilar structures, and infundibular (epidermoid) keratinization. The epidermal cyst could be included here, but for convenience it has been considered with other cysts (see
Ch. 16,
p. 442).
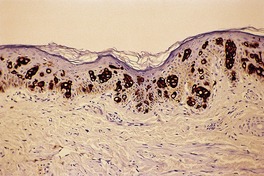
TUMOR OF THE FOLLICULAR INFUNDIBULUM
Despite its name, the tumor of the follicular infundibulum is really of isthmic origin.
215 It usually occurs as a solitary, asymptomatic, smooth or slightly keratotic papule on the head and neck or upper chest.
216 Multiple lesions, including a mantle distribution on the upper trunk (infundibulomatosis, eruptive infundibulomas),
217. and 218. have been reported.
219 The tumor of the follicular infundibulum may also occur in Cowden’s disease, the Schöpf–Schultz–Passarge syndrome and in
an organoid nevus. 220 It has been reported coexisting with an unusual tricholemmal tumor, which had some resemblance to a desmoplastic tricholemmoma.
221The treatment of this tumor is usually a ‘watch and wait’ approach with long-term supervision. Symptomatic tumors have been treated with salicylic acid, etretinate, and cryotherapy, with only slight improvement.
222
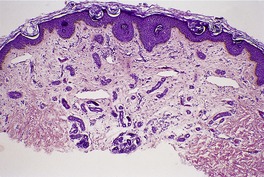
Histopathology216
The growth pattern resembles that of a superficial basal cell carcinoma. There is a plate-like fenestrated subepidermal tumor composed of pale or pink-staining glycogen-containing cells, with a peripheral palisade of basal cells. Some of the cords are basaloid without pale-staining cytoplasm. The tumor connects at intervals with the undersurface of the epidermis by slender pedicles. Hair follicles entering the tumor from below lose their identity and merge with it. Small follicular bulbs and papillary mesenchymal bodies may be found. Focal sebaceous differentiation is rare.
223 Ductal structures, resembling eccrine ducts, have been reported in the tumor strands.
224 The surrounding loose connective tissue stroma contains a network of elastic fibers.
216. and 217. The histological features sometimes overlap those of tricholemmoma, but the low-power architecture is quite different.
The tumor of the follicular infundibulum appears to be the same as that reported as a basal cell hamartoma with follicular differentiation.
225 However, it is histologically dissimilar to the cases reported as
multiple infundibular tumors of the head and neck.
226 In these latter cases clusters of enlarged follicular infundibula comprised each lesion, somewhat similar to the appearances of prurigo nodularis. The illustrations in the report of a tumor called an
infundibular keratosis showed some features of the multiple infundibular tumor referred to above, and others of an inverted follicular keratosis (see below) but without squamous eddies.
227
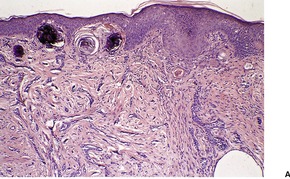
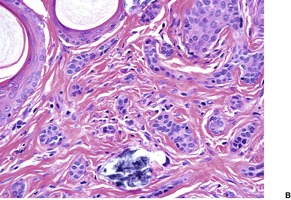
DILATED PORE OF WINER
The dilated pore of Winer
228 is a relatively common adnexal lesion which occurs predominantly on the head and neck, but also on the upper trunk of elderly individuals.
229 Clinically and histologically it is a comedo-like structure. Dilated pores may be acquired as a sequel of inflammatory cystic acne, or of actinic damage.
10
Histopathology216
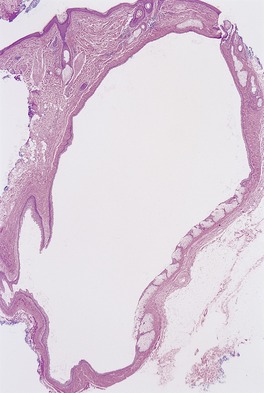
There is a markedly dilated follicular pore, which may extend to the mid or lower dermis. The follicle is lined by outer root sheath epithelium in which there is infundibular keratinization with the formation of keratohyaline granules. The epithelium shows acanthosis and finger-like projections that radiate into the surrounding dermis (
Fig. 33.9). There is sometimes heavy melanin pigmentation of the follicular wall; pigmentation may also involve the central horny plug. The pilary unit of the involved follicle and the sebaceous gland are absent or rudimentary.
PILAR SHEATH ACANTHOMA
Pilar sheath acanthoma is a rare, benign follicular tumor found almost exclusively on the upper lip of older individuals.
230.231. and 232. The tumors, which measure 5–10 mm in diameter, have a central pore-like opening plugged with keratin.
There is a central, cystically dilated follicle containing keratinous material which opens onto the surface (
Fig. 33.10). Tumor lobules composed of outer root sheath epithelium extend from the wall of the cystic cavity into the adjacent dermis. Lobules sometimes reach the subcutis. Occasional abortive hair follicles may be present. The tumor epithelium shows infundibular keratinization, although it is of isthmic origin. There may be abundant glycogen in some of the tumor cells.
The condition may be distinguished from the dilated pore of Winer, in which there is a patulous follicle with only small projections of epithelium extending into the surrounding connective tissue. In trichofolliculoma, well-formed follicles radiate from the central keratin-filled sinus and there is a well-formed stroma, which is absent in pilar sheath acanthoma.
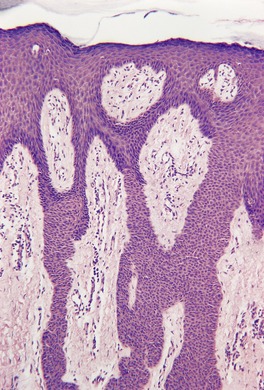
INVERTED FOLLICULAR KERATOSIS
Inverted follicular keratosis is a benign tumor of the follicular infundibulum that was first described by Helwig in 1954. It is a somewhat controversial entity, having been regarded by some as a variant of seborrheic keratosis or verruca vulgaris.
233. and 234. It occurs as a solitary flesh-colored nodular or filiform lesion which measures from 0.3 to 1 cm in diameter.
216 In about 90% of cases the tumor occurs on the head and neck, the cheeks and upper lip being the sites of predilection.
234 The eyelids may be involved.
235 It is more common in males and older individuals. Human papillomavirus has not been detected in most cases, suggesting that the inverted follicular keratosis is not a variant of verruca vulgaris, as has been claimed.
236. and 237. However, some cases of verruca vulgaris do develop areas with features of inverted follicular keratosis and/or tricholemmoma. In one case, pigment was increased considerably, leading to a mistaken dermoscopic and clinical diagnosis of malignant melanoma.
238
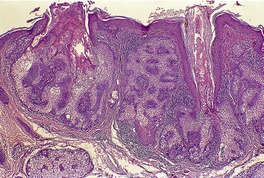
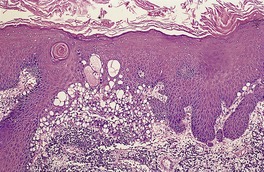
Histopathology216
Inverted follicular keratoses are predominantly endophytic tumors with large lobules or finger-like projections of tumor cells extending into the dermis. An exophytic growth pattern is present in some lesions, and it is occasionally the dominant feature. Mehregan has described four growth patterns:
1. a papillomatous wart-like variant, which is largely exophytic with overlying hyperkeratosis and parakeratosis
2. a keratoacanthoma-like pattern with marginal buttress formation and a central exo-endophytic mass of solid epithelium
3. a solid nodular form, which is largely endophytic with solid, lobulated masses of epithelium
4. an uncommon cystic type, with irregular clefts within the tumor and the formation of small cysts.
216 Each tumor lobule is composed of basaloid and squamous cells, with the basaloid cells at the periphery and larger keratinizing cells toward the center. Mitoses are not uncommon in the basaloid cells.
A characteristic feature is the presence of squamous eddies, which are formed by concentric layers of squamous cells in a whorled pattern and which may become keratinized with the formation of keratohyalin and sometimes keratin at the center of these islands (
Fig. 33.11). There is sometimes clefting at the periphery of the squamous eddies, and even focal acantholysis. Melanin pigment is usually inconspicuous. The surrounding dermis sometimes contains a mild inflammatory cell infiltrate which is predominantly lymphohistiocytic. Telangiectatic vessels may be found in the dermal papillae in the filiform lesions.
Overlying the tumor there is variable hyperkeratosis and parakeratosis. Funnel-shaped keratinous plugs may form. Occasionally a prominent cutaneous horn is present.
Bcl-2-positive dendritic cells are present in increased numbers in the suprabasal areas of inverted follicular keratoses compared to seborrheic keratoses. This density of cells in inverted follicular keratoses correlates with the density of CD1a-positive cells.
239
TRICHOLEMMAL (EXTERNAL SHEATH) TUMORS
This group of tumors is characterized by cells which differentiate toward those of the outer sheath of the hair follicle.
10 As such, they show a variable extent of clear cell change resulting from cytoplasmic accumulation of glycogen. There are also cells that are intermediate between those of the outer sheath and infundibular keratinocytes. These cells may have anisotropic tonofibrils, a characteristic feature of tricholemmal keratinization and one that is also seen in both tricholemmal cysts and proliferating tricholemmal cysts.
OUTER ROOT SHEATH ACANTHOMA
TRICHOLEMMOMA
Tricholemmomas (trichilemmomas) are small solitary asymptomatic papular lesions found almost exclusively on the face.
10.135.216. and 241. Multiple tricholemmomas are a cutaneous marker of Cowden’s disease (see below).
Although they appear to arise from the follicular infundibulum, they differentiate toward the outer root sheath. Ackerman regards tricholemmomas as old viral warts,
13. and 242. a view not supported by most dermatopathologists
243. and 244. or by immunoperoxidase studies to detect viral antigens.
245
Histopathology246
Tricholemmomas are sharply circumscribed tumors composed of one or more lobules which extend into the upper dermis and are in continuity with the epidermis or follicular epithelium at several points. In small lesions follicular concentricity is apparent.
247 The tumor is composed of squamoid cells showing variable glycogen vacuolation, this change being most marked deeply (
Fig. 33.12). Centrally, there may be foci of epidermal keratinization and occasionally small squamous eddies. Keratinous microcysts may form in large lesions. There is a peripheral layer of columnar cells with nuclear palisading resembling the outer root sheath of hair follicles. A thickened glassy basement membrane surrounds the tumor in part. This is PAS positive, and diastase resistant. The stroma is occasionally hyalinized or desmoplastic.
241. and 248. The term ‘d
esmoplastic tricholemmoma’ is used for this histological variant. A case of desmoplastic tricholemmoma arising in an organoid nevus has been reported.
249 Another case, on the eyelid, was misdiagnosed as sebaceous carcinoma.
250The overlying epidermis shows hyperkeratosis, mild acanthosis, and sometimes a prominent granular layer. Uncommonly, a cutaneous horn will form (
tricholemmomal horn).
251 This differs from the
tricholemmal horn, in which a horn showing tricholemmal keratinization overlies a depression in the epidermis which usually has a prominent basement membrane.
252 Warts, basal and squamous cell carcinomas, and inverted follicular keratoses and seborrheic keratoses sometimes contain areas of tricholemmal differentiation.
242. and 253.CD34 has been reported in tricholemmomas and also in the external root sheath of normal hair follicles.
254 Its presence in desmoplastic tricholemmoma may be of diagnostic value; it is not present in basal cell carcinoma
255 or sebaceous carcinoma.
250 The cytokeratin expression of tricholemmomas has been studied.
256 CK1 and CK10 are present in keratinizing ductal epithelium. CK14 is present in the whole layer, while CK16 is present in the suprabasal layer but absent in keratinizing ductal epithelium.
256
COWDEN’S DISEASE
Cowden’s disease (Cowden’s syndrome,
PTEN hamartoma tumor syndrome, multiple hamartoma and neoplasia syndrome – OMIM 158350) is a rare multisystem condition with autosomal dominant inheritance.
257.258.259.260. and 261. The eponym is the surname of the propositus of the report by Lloyd and Dennis in 1963.
262 The mucocutaneous features are the most constant findings. These include multiple tricholemmomas,
263 which are usually on the face, acral keratoses, palmar pits, and mucocutaneous papillomatous papules.
258. and 264. They usually appear in late adolescence. The tumor of the follicular infundibulum may also arise in Cowden’s disease.
220 Multiple inverted follicular keratoses were present in one affected person.
265 There is also a high incidence of visceral hamartomas and tumors, including fibrocystic disease of the breast,
266 thyroid adenomas, ovarian cysts, subcutaneous lipomas and neuromas, gastrointestinal polyps,
267 and carcinomas of the breast and thyroid.
260 Also documented are eye changes,
268 skeletal abnormalities,
269 acromelanosis,
268 non-Hodgkin’s lymphoma,
263 and carcinomas of the skin, tongue,
270 testis,
271 and cervix. Impaired T-cell function has been reported in this syndrome.
269Cowden’s disease involves a mutation in the
PTEN gene on chromosome 10q23.31 (10q22–q23), a tumor suppressor gene with tyrosine phosphatase and tensin homology.
272.273. and 274. A similar genetic abnormality occurs in the Bannayan–Zonana syndrome, suggesting that they are different phenotypic expressions of the one disease.
275 This syndrome has also been reported as the Bannayan–Riley–Ruvalcaba syndrome (OMIM 153480), its preferred name,
276.277. and 278. and the Ruvalcaba–Myre syndrome.
275 Macrocephaly, genital lentiginosis, and lipomatosis are the hallmarks of this syndrome.
279 It may present with tricholemmomas and lipomas. Germline mutations in the
PTEN gene have been found in 65% of individuals with this syndrome, and in 85% of individuals with classic Cowden’s disease.
278 It is thought that the rest may harbor pathogenic variants that have escaped detection
by standard methodologies. 280 Lhermitte–Duclos disease (OMIM 158350) also has mutations in the
PTEN gene. It may present with cerebellar ataxia and other manifestations of Cowden’s disease.
281. and 282. A Proteus-like syndrome has also been reported.
283. and 284. The pathogenesis of malignant tumors in Cowden’s disease appears to require complete inactivation of the
PTEN gene.
285 Analysis of the tricholemmomas in this disease show loss of heterozygosity at the
PTEN locus.
286
Most of the facial lesions are tricholemmomas or tumors of the follicular infundibulum. The tricholemmomas are cylindrical or lobular in configuration.
287 Sometimes the lesions keratinize with squamous eddies, resembling an inverted follicular keratosis. Non-specific verrucous acanthomas and lesions resembling digitate warts may also be present.
287The extrafacial lesions are mostly hyperkeratotic papillomas that resemble either verruca vulgaris, acrokeratosis verruciformis, or a nondescript hyperkeratotic acanthoma.
288 This latter change may take the form of hyperplasia of the follicular infundibulum.
288 No evidence of a viral etiology has been found on electron microscopy,
269. and 291. or with immunoperoxidase studies.
Distinctive dermal fibromas (sclerotic fibromas) with interwoven fascicles of coarse collagen, sometimes showing marked hyalinization, are not uncommon in Cowden’s disease.
288. and 292. They may have a plywood-like appearance histologically (see
p. 815).
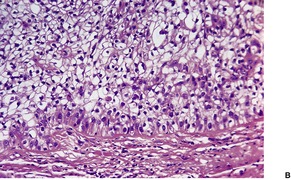
TRICHOLEMMAL CARCINOMA
Headington has defined tricholemmal carcinoma (tricholemmocarcinoma)
293 as a ‘histologically invasive, cytologically atypical clear cell neoplasm of adnexal keratinocytes which is in continuity with the epidermis and/or follicular epithelium’.
10. and 294. Only a small number of cases purporting to be this entity have been reported.
10.293.295.296.297.298.299.300. and 301. The illustrations in some of the reported cases resemble invasive intraepidermal carcinoma, a tumor which often shows adnexal differentiation in its dermal component.
293. and 302. Ackerman believes that true tricholemmal carcinoma is a rare expression of basal cell carcinoma (trichoblastic carcinoma) and that what ‘conventionally is called tricholemmal carcinoma is a clear cell variant of squamous cell carcinoma’.
303The tumors reported as tricholemmal carcinoma have usually arisen in the sun-exposed skin of the face and extremities of elderly patients.
304 They are usually diagnosed clinically as a basal cell carcinoma. Their development in Cowden’s disease is surprisingly rare.
305 A case has been reported in an organoid nevus, in continuity with a trichoblastoma that was also in the nevus.
306 The existence of tricholemmal carcinoma has been questioned (see
p. 695). It has been suggested that they may represent a clear cell variant of squamous cell carcinoma.
The hypercalcemia of malignancy was present in one extensive lesion that arose in a burn scar.
307Treatment is wide surgical excision.
Histopathology
The tumors are multilobulate, infiltrative growths connected to the epidermis and pilosebaceous structures and showing features reminiscent of the outer root sheath.
297 The lobules often show peripheral palisading, hyaline basement membranes, and tricholemmal keratinization (
Fig. 33.13).
297 A high mitotic rate is often present.
296. and 298. Neuroendocrine differentiation and melanocyte colonization were present in one case. This tumor expressed EMA, cytokeratin 15/20, chromogranin, synaptophysin, and CD56.
308 The usual tricholemmal carcinoma expresses CK7,
306 and podoplanin, detected by the D2-40 antibody.
309The tumor reported as ‘
clear cell pilar sheath tumor of scalp’ had some histological similarities to the tricholemmal carcinoma. The reported case was composed of small nests of glycogen-containing clear cells infiltrating the dermis and subcutis.
310 There was no underlying cyst or evidence of tricholemmal keratinization.
310
TUMORS WITH MATRICAL DIFFERENTIATION
In this group there is differentiation toward cells of the hair matrix and hair cortex and cells of the inner sheath. The prototype tumor is the pilomatrixoma. A rare malignant variant, the pilomatrix carcinoma, has also been described. A solitary case of ‘pilomatrical carcinosarcoma’ was reported in 1994.
311 Matrical differentiation can also be seen in other tumors:
• melanocytic matricoma (see below)
• complex adnexal tumors.
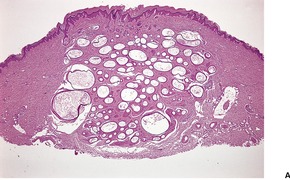
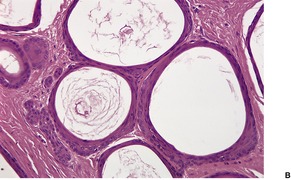
PILOMATRIXOMA
Pilomatrixoma (pilomatricoma, calcifying epithelioma of Malherbe – OMIM 132600), which accounts for almost 20% of pilar tumors, is a benign lesion with differentiation toward the matrix of the hair follicle.
313 It is found particularly on the head and neck and upper extremities.
11. and 314. A case has arisen in a BCG vaccination site.
315 About 60% develop in the first two decades of life.
316. and 317. They are mostly solitary, but multiple lesions – usually less than five in all – are sometimes found.
318.319.320.321. and 322. Some patients with multiple lesions have myotonic dystrophy.
11.323.324. and 325. They have also been reported in Turner’s syndrome,
326 trisomy 9,
327 Sotos syndrome (cerebral giantism – OMIM 117550),
328 and with other abnormalities.
329.330. and 331. A familial occurrence is rarely noted.
332. and 333. A pilomatrixoma-like change is not uncommon in the epidermal cysts found in Gardner’s syndrome.
334 This syndrome has also been described in a patient with multiple pilomatrixomas.
335Activating mutations in β-catenin, a constituent of the adherens junctions, have been found in sporadic pilomatrixomas.
352.353.354. and 355. A similar defect is also found in colonic carcinomas.
352 The β-catenin gene (
CTNNB1) maps to chromosome 3p22–p21.3.
Most tumors, even if inadequately excised, will not recur. However, local recurrence and aggressive forms have been documented.
356. and 357. The malignant variant, pilomatrix carcinoma, is discussed below.
Surgical excision is the usual method of treatment. The author has seen numerous incompletely excised lesions that have not recurred, and several that have. Recurrence does not seem to occur in ‘burnt-out’ lesions devoid of a basaloid component.
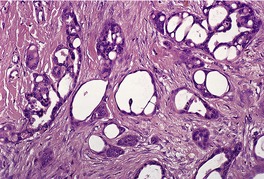
Histopathology
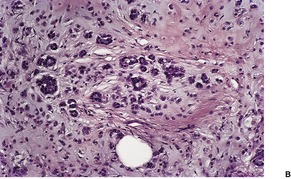
The appearances vary according to the age of the lesion. Established lesions are sharply demarcated tumors in the lower dermis, extending quite often into the subcutis. There are masses of epithelial cells of various shapes, with an intervening connective tissue stroma containing blood vessels, a mixed inflammatory cell infiltrate, foreign-body giant cells, and sometimes hemosiderin, melanin, bone, and rarely amyloid.
10. and 358.There are two basic cell types, basophilic cells and eosinophilic shadow cells (
Fig. 33.14). The basophilic cells tend to be at the periphery of the cell islands and have little cytoplasm, indistinct cell borders, hyperchromatic nuclei and plentiful mitoses. They resemble the cells of a basal cell carcinoma. They are sometimes the predominant cell in lesions removed from elderly patients. The term ‘
proliferating pilomatricoma’ has been proposed for this variant (
Fig. 33.15).
359. and 360. Follicular germinative cells have been reported in a palisaded arrangement at the edges of collections of matrical cells in cases of pilomatrixoma.
361 The eosinophilic shadow cells in the usual form are found toward the central areas of the cell masses. They have more cytoplasm and distinct cell borders, but no nuclear staining. These shadow (mummified) cells form from the basophilic cells, and the transition may be relatively abrupt or take place over several layers of cells (transitional cells). The intermediary cells develop progressively more eosinophilic cytoplasm and the nucleus becomes pyknotic. The mode of cell death has been reported as apoptotic
362. and 363. but it is more likely that the shadow cells represent terminal differentiation rather than apoptosis.
364. and 365. Hyalinization of the cells, squamous change, or disruption into amorphous debris may result.
Calcification occurs in more than two-thirds of the tumors and is usually in the shadow cells. Ossification of the stroma occurs in about 13%;
366 hemosiderin is found in about 25% of cases;
367 and melanin is present in nearly 20% of lesions and may be in the shadow cells as well as in the stroma.
367. and 368. Extramedullary hematopoiesis may occur adjacent to the spicules of bone.
369 Bone morphogenetic protein (BMP)-2, which plays an important role in ectopic bone formation, has been found in the shadow cells, suggesting that it may play a role in generating bone formation in pilomatrixomas.
370 It results in the deposition of type II collagen at the dermoepidermal junction.
371 Dendritic cells are sometimes seen among the basophilic cells in the cases with pigmentation.
The bullous (lymphangiectatic) lesions have marked dilatation of superficial lymphatics overlying the tumor.
345 There may be some loss of elastic fibers, as seen in the anetodermic variant.
343. and 347. The surrounding dermis may be edematous in these bullous variants.
347The immunohistochemical pattern indicates a tumor differentiating into both the hair cortex and outer root sheath.
382 The hard keratins hHa1, a2, and a5 are expressed in pilomatrixomas but not in other tumors of follicular origin.
383. and 384. Maturation to shadow cells is associated with a gradual loss of differentiation-specific hair keratins.
385 CK15, found in trichoepitheliomas, some basal cell carcinomas and proliferating tricholemmal cysts, inter alia, is not found in pilomatrixomas.
102 The basaloid cells of all pilomatrixomas express β-catenin, but the shadow cells do not stain.
386.387. and 388. The transitional cells show strong staining for involucrin
382 and bcl-2.
389 The S100 proteins S100A2, S100A3, and S100A6, which specifically label certain cells within the normal hair follicle, are all expressed in different parts of a pilomatrixoma.
390Pilomatrix dysplasia was the term used for a histologically distinctive pattern seen in a patient with facial dysmorphism and follicular papules who was receiving four immunosuppressive drugs.
391 Hair follicles were dilated and contained hyperkeratotic and parakeratotic debris in place of hair shafts. There were hyperplastic areas of differentiation into hair matrix with cellular disorganization and loss of nuclear polarity.
391 These changes are now known as trichodysplasia spinulosa (see
p. 405). This description is left in this chapter in case readers unfamiliar with the entity read this section looking for clues to the diagnosis of a problem case.
Matricoma is the designation used by Ackerman and colleagues for a tumor with the same constituent cells as a pilomatrixoma but with a different silhouette.
13 The lesions are well circumscribed, not fundamentally cystic (although cystic areas can be present in some lobules), and composed of discrete aggregations. Some of these cases have probably been included with the ‘proliferating pilomatricoma’ (see above).
Pilomatricomal horn is a verruca-like horn characterized by replacement of the epidermis by basaloid cells, with masses of cornified material containing shadow cells that form a cutaneous horn.
392 This lesion is the matrical equivalent of the tricholemmal horn. Anetodermic changes, as seen in some cases of pilomatrixoma, were present in the dermis in one case.
392
Electron microscopy
The cells differentiate and keratinize in a manner analogous to the cells that form the cortex of the hair.
393 The fully developed shadow cells contain interlacing swirls of keratin that form a mantle around the nuclear remnants.
393
PILOMATRIX CARCINOMA
More than 50 cases of pilomatrix carcinoma (matrical carcinoma) have now been reported.
394.395.396.397.398.399.400.401.402.403.404. and 405. It usually arises, de novo, as a solitary lesion, but some cases arise in a pilomatrixoma, or at the site of a previously excised pilomatrixoma.
406 In one case, the patient had multiple pilomatrixomas.
407 Although they are most common in the elderly, cases in young adults and even children have been reported.
408 There is a male predominance. Lesions are usually situated on the scalp and face, but they have been reported on most body sites.
409. and 410. They may measure from 0.6 to 10 cm in diameter.
Mutations in the
CTNNB1 gene, that encodes β-catenin have been reported in most cases of pilomatrix carcinoma.
411 Similar mutations are found in benign pilomatrixomas. Neither molecular nor immunohistochemical methods distinguish pilomatrix carcinomas from pilomatrixomas. Their distinction remains a histological one.
411Pilomatrix carcinomas have a high capacity for local infiltration; local recurrences are common. Metastases are infrequent. It can spread to regional lymph nodes,
412 and to visceral organs, particularly the lungs.
409 A
pilomatrical carcinosarcoma of the cheek with pulmonary metastases has been reported.
311 The author has seen a further case, in consultation.
Treatment is by wide local excision. Follow-up is necessary because local recurrence and metastases may be delayed.
413 Radiation therapy has been used for recurrences and nodal metastases, but its role in treatment is unclear.
409
Histopathology
Pilomatrix carcinoma is a poorly circumscribed, asymmetrical tumor that is often ulcerated.
410 It is centered on the dermis but extension into the subcutaneous tissue often occurs. The tumor is composed of pleomorphic basaloid cells with prominent nucleoli and frequent mitoses. Necrosis is sometimes present. Keratotic material and shadow cells may be present in the center of the basaloid islands. Vascular and/or lymphatic invasion are rare.
394. and 398.Melanin pigment and intralesional melanocytes have been present in several cases.
414. and 415. Such cases can be regarded as the malignant counterpart of the melanocytic matricoma (see below).
414 The case reported as a
pilomatrical carcinosarcoma was composed of a large pilomatrixoma lying within a spindled, sarcomatoid stroma.
311It should be remembered that shadow cell differentiation can occur in some visceral tumors, such as tumors of the colon and uterus.
416Immunohistochemical studies have demonstrated the hair matrix and precortex keratins hHa5 and hHa1.
417β-catenin is also expressed, as in pilomatrixomas.
417
MELANOCYTIC MATRICOMA
Melanocytic matricoma was first reported in 1999 by Carlson et al.
418 It recapitulates the bulb of the anagen hair follicle. The lesion presents as a small circumscribed papule, usually on the face.
419. and 420. Resnik has challenged the validity of this entity, claiming it to be a morphological expression of matricoma.
421. and 422. This has not been accepted by some.
423Examples of pilomatrix carcinomas with melanin pigment and intralesional melanocytes have been reported.
414. and 415. Such cases can be regarded as the malignant counterpart of melanocytic matricoma (see above), if indeed this is a discrete entity.
414
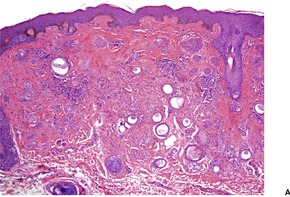
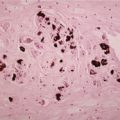
Histopathology
Melanocytic matricoma is a well-circumscribed dermal nodule composed of variably melanized, pleomorphic, and mitotically active matrical and supramatrical cells with islands of shadow cells (
Fig. 33.16).
418 The shadow cells are not always plentiful. Admixed with these cells are numerous dendritic melanocytes containing melanin pigment. There has been no discernible connection with the overlying epidermis or a hair follicle. Pigmented matrical cells are sometimes seen in normal hair follicles (
Fig. 33.17).
The melanocytes express S100 protein and HMB-45, while the basaloid cells express β-catenin.
424
TUMORS WITH PROMINENT PERIFOLLICULAR MESENCHYME
The
perifollicular fibroma is a controversial entity of doubtful existence. This term has been used for angiofibromas
425. and 426. and fibrofolliculomas.
427. and 428. It should no longer be used. A recently reported perifolliculoma-like lesion was thought to have resulted from trauma.
429
FIBROFOLLICULOMA/TRICHODISCOMA
Although fibrofolliculomas and trichodiscomas are quite different at first glance, Ackerman and colleagues have presented evidence that they are different stages in the development of a single entity.
13 For this reason the composite designation fibrofolliculoma/trichodiscoma will be used here.
Both variants are found as asymptomatic skin-colored papules 1–3 mm in diameter, usually on the face. Other sites include the arms, trunk, and thighs. A hair follicle may be present within the lesion. The lesions may be solitary or multiple with up to several hundred lesions present. Most cases develop in the third decade of life and persist thereafter.
13 A linear and a congenital annular variant have been described.
430. and 431. Multiple lesions may be found in pure form,
432. and 433. or they may be associated with the Birt–Hogg–Dubé syndrome (see below).
434.435.436.437.438.439.440. and 441. Fibrofolliculomas have also been associated with nevus lipomatosus. The lesions were present at birth.
442The origin of these tumors is uncertain. Ackerman and colleagues regard these tumors as hamartomas, but not one related to the hair disk (Haarscheibe), as was originally proposed for trichodiscoma.
13 The hair disk, a specialized component of the perifollicular mesenchyme, is a richly vascular dermal pad which serves as a slowly adapting mechanoreceptor.
432.443. and 444.
Birt–Hogg–Dubé syndrome
The Birt–Hogg–Dubé syndrome (OMIM 135150) is an autosomal dominant genodermatosis characterized by cutaneous fibrofolliculomas,
445 and an increased risk of multiple lung cysts,
446 spontaneous pneumothorax,
447 and renal tumors.
448. and 449. The renal tumors include hybrid oncocytic neoplasms and chromophobe renal cell carcinomas.
450.451. and 452. Colonic polyps and neural tumors have also been reported in this syndrome.
452.453. and 454. The syndrome is caused by mutations in the
FLCN (BHD) gene on chromosome 17p11.2, which encodes the protein folliculin. Several different mutations in this gene, which has a tumor suppressor role, have been reported.
455 Initially, it was thought that the syndrome was characterized by three distinct cutaneous tumors – fibrofolliculomas, trichodiscomas, and acrochordons, but these three lesions are now all regarded as being fibrofolliculomas.
456 As a consequence, the existence of this syndrome was, for a period, called into question.
457 Likewise, the perifollicular fibroma is also a fibrofolliculoma. Accordingly, it seems that some, or all of the patients with the Hornstein–Knickenberg syndrome, in which ‘perifollicular fibromas’ are a feature, actually have the Birt–Hogg–Dubé syndrome.
456.458. and 459.
Histopathology
It is now accepted that the fibrofolliculomas and trichodiscomas of the Birt–Hogg–Dubé syndrome are the same entity (fibrofolliculoma), but with a spectrum of morphological changes. It is inferred, but not proven, that sporadic cases may have either of these two morphologies. Furthermore, some cases reported as trichodiscomas have illustrations showing fibrofolliculomas.
460The
fibrofolliculoma end of the spectrum consists of cords and strands of epithelial cells, 2–4 cells thick, radiating from a follicular structure with infundibular features. This may be dilated and contain keratin. The strands may anastomose or rejoin the infundibulum at several points. One or more sebocytes may be seen within the epithelial cords. They may form tiny lobules. Sebaceous ducts may also be present. The term ‘
mantleoma’ has also been used for these tumors. Around the epithelial cords there is a well-circumscribed proliferation
of loose connective tissue composed of fine fibers with some intervening mucin. Elastic fibers are scant or absent.The
trichodiscoma component is usually a well-demarcated and non-encapsulated tumor, which often has a folliculosebaceous collarette of variable maturity. Areas resembling fibrofolliculoma may be seen, particularly at the periphery. Trichodiscomas are composed of fascicles of loose, finely fibrillar connective tissue with intervening mucinous ground substance (
Fig. 33.18). There is a moderate increase in fibroblasts, with occasional stellate forms. Elastic fibers are sparse or absent. There are prominent small vessels, some of which are telangiectatic. Sometimes, blood vessels with a concentric arrangement of PAS-positive collagen, forming a thickened wall, have been present toward the lower edge of the tumor. The term ‘perivascular fibroma’ has been used for these changes.
461 Nerve fibers have been described at the periphery of the lesions, and also extending into the base. Neurofollicular hamartoma (see below) is now regarded as a spindle-cell predominant trichodiscoma.
462Both syndromic-associated and sporadic types of fibrofolliculomas and trichodiscomas have identical immunophenotypic features with perifollicular vimentin and CD34, but not factor XIIIa.
463.464.465. and 466.Multiple giant tumors with the appearances of a trichodiscoma have been reported as ‘giant fibromyxoid tumors of the adventitial dermis’.
Electron microscopy
Trichodiscomas have shown deposits of fibrillar-amorphous material between the collagen bundles.
443 The significance of this material is uncertain. Banded structures and a Merkel cell-neurite complex in the basal layer of the epidermis have also been noted, although Merkel cells are not seen in the dermis.
443
NEUROFOLLICULAR HAMARTOMA
The neurofollicular hamartoma is a rare tumor found on the face, usually near the nose. It presents as a pale papule, usually solitary, that measures 3–7 mm in diameter.
467.468.469. and 470. Kutzner and colleagues have recently proposed that the neurofollicular hamartoma (or at least S100-negative variants of it) should be recategorized as a spindle-cell predominant trichodiscoma.
462
Histopathology
The lesion consists of hyperplastic pilosebaceous units with an intervening stroma of spindle cells arranged in broad, haphazard fascicles. The stroma has features of both an angiofibroma and a neurofibroma. It also resembles a trichodiscoma.
It has been suggested that the neurofollicular hamartoma, trichodiscoma, and fibrofolliculoma are part of the same spectrum of hamartomas,
467 and, as stated above, that the neurofollicular hamartoma should be recategorized as a spindle-cell predominant trichodiscoma. The folliculosebaceous cystic hamartoma is closely related (see
p. 773).
471The spindle-cell predominant trichodiscoma and the usual trichodiscoma express an identical CD13
+/CD34
+ fibrocytic immunophenotype without co-expression of neural/perineural, myogenic, or melanocytic markers.
462 It is composed of cellular fascicles of spindle cells set in a loosely textured stroma with a moderate mucinous background.
462Immunohistochemistry of the neurofollicular hamartoma shows scattered cells that are positive for S100 and factor XIIIa. Diffuse staining for S100 has also been reported.
470 Most of the connective tissue cells are positive for vimentin.
467
SEBACEOUS TUMORS
Sebaceous tumors are relatively uncommon tumors of the skin. They are derived from the sebaceous gland, which begins its development as a bulge or collar at the junction of the infundibulum and isthmus of the hair follicle. The sebaceous gland may be derived from CK15-positive stem cells located in the hair follicle bulge.
472 Early in life, small cords of basaloid cells extend downward on either side of the follicle, forming the so-called ‘mantle’. Maturation of the mantle occurs slowly in childhood with the accumulation of lipid in some of the cells, forming sebocytes at the base of the mantle. Sebocytes increase in number and size such that a fully developed sebaceous lobule is present by puberty. Mantles are best seen around vellus follicles on the face, but they also develop in association with terminal follicles. Later in life, the sebaceous glands undergo progressive involution so that mantles are again seen, this time as vestiges. Initially, Steffen and Ackerman suggested that sebaceous glands had several cycles of growth, involution, and rest, independent of the cycle of the hair follicle.
473 Ackerman has since modified this view by suggesting that the cycle occurs ‘but twice in a lifetime (involution early in infancy, evolution at puberty, and involution again at menopause)’.
474 The latest theory may not be absolute as one occasionally sees a mantle in mid-adult life.
The synthesis and accumulation of lipids is a key step in the differentiation of sebaceous gland cells.
475 Proteins involved in adipocyte differentiation such as galectin-12, resistin, SREBP-1, and stearoyl CoA desaturase (SCD) have also been detected in sebaceous glands of human scalp skin.
475 The periphery of normal sebaceous glands contains podoplanin, a sialoglycoprotein found also in lymphatics and identified by the D2-40 monoclonal antibody.
476The following categories of sebaceous tumors will be considered:
• ectopic sebaceous glands
• hamartomas and hyperplasias of sebaceous glands
• benign sebaceous tumors
• malignant sebaceous tumors
• tumors with focal sebaceous differentiation.
ECTOPIC SEBACEOUS GLANDS
FORDYCE’S SPOTS AND RELATED ECTOPIAS
Sebaceous glands are usually found in association with hair follicles, the so-called ‘pilosebaceous unit’. Ectopic sebaceous glands without attached follicles (
Fig. 33.19) may be found as tiny yellow papules near
mucocutaneous junctions, particularly the upper lip, and in the buccal mucosa (Fordyce’s spots). 313 They may also be found in the areolae of the breasts, where they are known as
Montgomery’s tubercles. They are said to be restricted to the female breast, but the author has seen a case from a male breast. Like Fordyce’s spots, the sebaceous gland in a Montgomery’s tubercle opens directly onto the surface.
Ectopic sebaceous glands can also be found on the penis (Tyson’s gland),
477. and 478. labia minora and, very rarely, in the esophagus and vagina.
479 An ectopic sebaceous gland has been reported within the hair matrix epithelium of an anagen hair follicle on the chin.
480
HAMARTOMAS AND HYPERPLASIAS
By far the most common abnormality of the sebaceous gland is sebaceous hyperplasia. The hamartomas are quite uncommon and include folliculosebaceous cystic hamartoma and steatocystoma. Organoid nevus (nevus sebaceus) involves other appendageal components and is considered with the complex adnexal tumors (see
p. 806).
FOLLICULOSEBACEOUS CYSTIC HAMARTOMA
The folliculosebaceous cystic hamartoma is a distinctive hamartoma composed of folliculosebaceous structures surrounded by a stroma consisting of various mesenchymal components.
471.481.482.483. and 484. It presents as a solitary papule or nodule 0.5–2 cm or more in diameter, usually on the face.
485 Other rare sites of involvement include the scalp,
486. and 487. upper arm,
488 nipple, and genital region.
489 A giant variant, replacing the scrotum, has been reported.
490 Another giant lesion was of congenital origin.
491 In one case, the lesion arose in a port-wine stain.
492 Lesions are usually removed in adulthood, although they have often been present for many years. Schulz and Hartschuh have presented convincing evidence that this lesion is a trichofolliculoma at its very late stage with the follicular structures in a state of involution, corresponding to the normal hair cycle.
32 Others agree with this concept but suggest the designation sebofolliculomas as a pole of the spectrum of tricho-sebo-folliculoma.
33 This entity is discussed here because the sebaceous elements are usually the most prominent feature.
Lesions on the face are usually excised or shaved. The giant variant described on the scrotum was successfully treated by CO
2 laser and acitretin therapy.
490
Histopathology
The lesion is composed of infundibular structures, sometimes cystic, with numerous radiating sebaceous lobules. Occasional rudimentary hair structures and even apocrine glands may be present. The pilosebaceous units are embedded in a mesenchymal stroma composed of variable proportions of fibrous, adipose, vascular and neural tissue. There are usually spindle-shaped cells in the stroma, and some of these stain for CD34 or factor XIIIa.
484 Smooth muscle was the only mesenchymal component in one case, leading to the diagnosis of
folliculosebaceous smooth muscle hamartoma.
493The case reported with a prominent neural component in the stroma has some features in common with the neurofollicular hamartoma (see
p. 772), although this latter entity lacks the haphazard cystic infundibular structures.
471 The morphologically related sebaceous trichofolliculoma (see
p. 759) lacks a mesenchymal stromal component.
487 Trichofolliculoma differs from folliculosebaceous cystic hamartoma by the presence of secondary follicles in trichofolliculoma.
494
SEBACEOUS HYPERPLASIA
Sebaceous hyperplasia occurs as asymptomatic, solitary or multiple yellowish papules, often umbilicated, on the forehead and cheeks of elderly, and sometimes younger individuals.
495.496.497.498. and 499. An infant with congenital papules and plaques on the face has been reported.
500 Clinically sebaceous hyperplasia may mimic basal cell carcinoma. Rare variants include a ‘giant’ form,
501. and 502. a linear or zosteriform arrangement,
503. and 504. a plaque composed of multiple papules,
505 a diffuse form,
506 a familial occurrence,
507. and 508. and involvement of the areola
509.510.511. and 512. penis,
504 scrotum,
513 or the vulva.
514. and 515. Sebaceous hyperplasia is seen in heart transplant recipients. It is thought to be related to the process of dysplastic epithelial proliferation in transplant recipients and not to the effects of cyclosporine (ciclosporin),
516 although this has been disputed.
517 Sebaceous hyperplasia was present in 30% of renal transplant recipients in one series.
518 These patients had a higher incidence of non-melanoma skin cancer than transplant recipients without sebaceous hyperplasia. It has also been seen in a bone marrow recipient; cyclosporine was implicated in this case.
519 Highly active anti-retroviral therapy (HAART) has been implicated in the causation of sebaceous gland hyperplasia in a patient with HIV infection.
520Juxtaclavicular beaded lines – tiny papules arranged in closely placed parallel rows, resembling ‘strands of beads’ – are a variant of sebaceous gland hyperplasia.
521.522. and 523. They are asymptomatic and skin-colored to slightly yellow. They are said to be arranged along skin tension lines in the supra- and subclavicular area; they may be seen at other sites, such as the face,
524 and penis.
525 The condition is said to be more common in dark-skinned people.
526
The etiopathogenesis of sebaceous hyperplasia is unknown. Interestingly, sebaceous hyperplasia can be produced in rats by the topical application of Citrol (3,7-dimethyl-2,6-octadienol), a chemical used in foods as a flavoring agent.
527The dermoscopic features of sebaceous hyperplasia consist of the ‘cumulus sign’ (because of the resemblance to cumulus clouds) and the ‘bonbon toffee sign’ in which the cumulus sign surrounds a central umbilication.
528
Histopathology
Large, mature sebaceous lobules are grouped around a central dilated duct, which is usually filled with debris, bacteria and occasionally a vellus hair. The lobules usually lack the indentations by fibrous septa that characterize the normal gland (
Fig. 33.20). The sebocytes are smaller than usual and there are more basal cells per unit basement membrane length than in normal glands. Autoradiographic studies have shown a lower labeling index.
529 Sebaceous hyperplasia may be a clue to an underlying dermatofibroma. In shallow shave biopsies, sebaceous glands may be the most conspicuous feature.
530In
juxtaclavicular beaded lines there may be isolated sebaceous lobules in the upper dermis, not obviously connected with hair follicles.
521
STEATOCYSTOMA
The steatocystoma is a cystic structure lined by epithelium resembling the sebaceous duct (
Fig. 33.21). Sebaceous lobules and individual sebaceous cells are present within the lining epithelium. Steatocystoma has been considered in detail with other cysts (see
p. 448).
BENIGN SEBACEOUS TUMORS
The four tumors in this category are sebaceous adenoma, sebaceoma, mantleoma, and reticulated acanthoma with sebaceous differentiation. As sebaceous tumors are an important component of the Muir–Torre syndrome, it is considered here also.
SEBACEOUS ADENOMA
The sebaceous adenoma is an uncommon benign tumor which usually presents as a slowly growing, pink or flesh-colored solitary nodule, predominantly on the head and neck of older individuals.
531 Rarely, it may involve the buccal mucosa
532 or penis.
533 Sebaceous adenomas are usually about 0.5 cm in diameter but larger variants, up to 9 cm in diameter, can develop. Occasionally they ulcerate and bleed or become tender. Sebaceous adenomas, either solitary or multiple, may be associated with visceral cancer, usually of the gastrointestinal tract – the Muir–Torre syndrome (see below).
Ackerman has been suggesting for some time that sebaceous adenoma is really a carcinoma.
534 However, the evidence for this is not convincing. Recent immunohistochemical studies also provide no support for this view.
535Treatment is usually by excision or shave biopsy.
Histopathology531
On immunohistochemistry, CD10 staining of the cytoplasmic membrane occurs in nearly half of the cases.
538 CK15 is expressed in the basal cells; it is also present in other sebaceous tumors and sebaceous hyperplasia.
539
MUIR–TORRE SYNDROME
Muir–Torre syndrome (OMIM 158320), the first examples of which were reported in 1967,
540 is characterized by the development of sebaceous tumors, often multiple, in association with visceral neoplasms, usually gastrointestinal carcinomas.
541.542.543.544.545.546.547.548. and 549. Keratoacanthomas, epidermal cysts, and colonic polyps may also be present.
550 The sebaceous tumors are sometimes difficult to classify,
551 but they most resemble either sebaceous adenoma or sebaceoma, and occasionally sebaceous carcinoma.
552.553. and 554. Multiple sebaceous tumors, and sebaceous tumors occurring before the age of 50 years are strong indicators of the syndrome.
555 Sebaceous hyperplasia may also be present,
556 but most reports have indicated that sebaceous hyperplasia is not indicative of Muir–Torre syndrome.
557 The cutaneous tumors may precede or follow the first direct manifestation of the visceral cancer, and they may occur sporadically in other family members.
558 The sebaceous neoplasms generally develop after the visceral cancer.
559 The visceral tumor is usually of the gastrointestinal tract, particularly adenocarcinoma or polyps of the large bowel (see below), but other sites, such as the larynx, the genitourinary system in men,
560 and the ovary and uterus may be involved.
561 Lymphoma has been reported. The visceral tumors may behave in a less aggressive fashion than would be expected from the histology.
541.562. and 563. This is particularly so for tumors displaying widespread microsatellite instability (MSI).
563 Detection of MSI in cutaneous tumors of various types may form the basis of a non-invasive screening technique for hereditary non-polyposis colon syndrome (Lynch syndrome), of which the Muir–Torre syndrome is regarded as an allelic variant.
564.565.566.567. and 568. It represents 1–2% of cases of Lynch syndrome.
The Muir–Torre syndrome is inherited as an autosomal dominant trait. Mutations in one of the DNA mismatch repair genes
MLH1,
MSH2, and
MSH6, have been found in these patients.
569. and 570. Mutations in
MSH2 account for the majority (90%) of cases,
571 while
MSH6 mutations are limited to a few cases.
572PMS2 mutations have not been reported so far in the Muir–Torre syndrome but they have been in the Lynch syndrome. A colleague of the author has recently seen two such cases in the Muir–Torre syndrome.
MSH6 and
PMS2 mutations are said to be associated with a milder phenotype than mutations in the other two genes.
572 While most abnormalities involve gene deletions, partial duplication of the
MSH2 gene has resulted in truncation of the gene product and a non-functional state.
557 The sebaceous tumors may show widespread microsatellite instability.
570 In one study, loss of one of the mismatch repair genes (either
MSH2 or
MLH1) was found in 80% of the benign sebaceous lesions associated with visceral malignancy, but in only 23% of sebaceous lesions not associated with visceral malignancy.
556 A more recent study found no difference in the incidence of mutations in the mismatch repair genes in these two groups.
573 It has been suggested that treatment with isotretinoin and interferon-α2A can prevent tumor development.
549
Histopathology541. and 551.
The sebaceous tumors resemble to varying degrees those already described.
574 Often they appear ‘unique’ and difficult to classify.
551. and 555. There may be solid sheets of basaloid cells in some lobules, or an intermingling of these cells and sebaceous cells without any orderly maturation. It has been suggested that these atypical features are predictive of malignant transformation, if not completely removed.
575 Sometimes the tumors resemble a basal cell carcinoma, but with focal sebaceous differentiation. Mucinous and cystic areas may be present (
Fig. 33.23). Cystic lesions are an important component of this syndrome (see above).
576 Other tumors may connect with the surface and have a central debris-filled crater resembling, in part, a keratoacanthoma. Typical keratoacanthomas also occur. The lesion reported as a keratoacanthoma-like squamous cell carcinoma on the basis of its venous invasion was probably a keratoacanthoma with venous invasion.
577 All sebaceous tumors should be screened for the presence of MSH-2, MSH-6, MLH-1, and PMS-2, the respective gene products of
MSH2,
MSH6,
MLH1, and
PMS2. The diagnosis of Muir–Torre syndrome is made when nuclear staining for the particular gene product is absent in the tumors.
578 As MSH-2 and MSH-6 proteins normally form a stable heterodimer, mutations in the
MSH2 gene can produce an instability of this heterodimer, with secondary lack of expression of MSH-6 protein.
559 False positive and negative results are uncommon.
579The cells express epithelial membrane antigen and nuclear androgen receptor.
580
SEBACEOMA
The term ‘sebaceoma’ was coined by Ackerman for a distinctive sebaceous tumor, 581 examples of which have been reported in the past as basal cell carcinoma with sebaceous differentiation
531 or sebaceous epithelioma.
313. and 582. These tumors are usually solitary, flesh-colored to yellowish papulonodules on the face or scalp, but they are sometimes multiple, particularly in the Muir–Torre syndrome (see above), or associated with organoid nevi.
531 Sebaceoma of the external ear canal has been reported.
583 They measure 0.5–3 cm in diameter, and tend to occur in older adults, although they may be seen in younger adults as well. They grow slowly and do not usually recur after treatment.
Histopathology531. and 581.
There are multiple nests of basaloid cells with a random admixture of sebaceous cells, either solitary or in clusters (
Fig. 33.24). The tumor is centered on the upper and mid dermis, but some nests may be continuous with the basal layer of the epidermis. The small basaloid cells of the tumor outnumber the mature sebaceous component. Cysts and duct-like structures containing the debris of holocrine degeneration may be present.
582 There are scattered mitoses, but the tumor lacks the atypia of sebaceous carcinoma.
The sebaceoma can exhibit an amazing diversity of patterns.
473 Sebocytes and sebaceous ducts may be plentiful or scarce, and the sebocytes may show variable vacuolation. Apocrine differentiation has also been reported.
584. and 585. Adenoid, reticulated, cribriform, cystic, and cornified areas may be present.
586. and 587. A rippled or carcinoid-like architecture in the basaloid component has also been described.
499. and 588. The cytokeratin expression in this variant (CK19) indicates an undifferentiated and pluripotent component.
589. and 590. Trichoblastoma can also have a rippled pattern,
591 although Ackerman believes this pattern is always indicative of a sebaceoma.
592 Sometimes, areas resembling a seborrheic keratosis with sebaceous differentiation may be present.
593 Furthermore, a sebaceoma has been reported arising in association with a seborrheic keratosis.
594 Such cases merge with the variant of sebaceoma with surface changes of verruca/seborrheic keratosis.
595There exist tumors with close morphological resemblance to basal cell carcinoma which may show focal sebaceous differentiation. Such tumors are best classified as basal cell carcinomas with sebaceous differentiation. It is acknowledged that there is some overlap of this tumor with what has been designated sebaceoma,
581 but this basal cell carcinoma variant shows peripheral basal-cell palisading, and sometimes tumor–stroma separation, and a loose fibromucinous stroma.
499 Rare variants have been reported with sweat gland differentiation in part of the lesion.
596Cytokeratin 7 (CK7) is present in most sebaceous tumors, but it is usually absent in basal cell carcinomas.
499 Sebaceous cells express EMA, but not carcinoembryonic antigen (CEA). Only mature sebaceous cells express EMA, not the germinative cells. In contrast, expression of EMA is uncommon in basal cell carcinomas; they invariably express Ber-EP4 whereas sebaceomas do not.
597 Sebaceomas express CK15, and D2-40 in the basaloid, germinative cells.
472. and 598.
RETICULATED ACANTHOMA WITH SEBACEOUS DIFFERENTIATION
The term reticulated acanthoma with sebaceous differentiation is a rare tumor with a predilection for the face of elderly individuals. It is usually solitary,
599 but multiple papules have been recorded.
600 It behaves in a benign fashion.
601 It was previously called superficial epithelioma with sebaceous differentiation, but Steffen and Ackerman believed that the use of the word ‘epithelioma’ may give an impression of malignancy and suggested the alternative title used above.
473 The lesions sometimes discharge yellowish material. Sánchez Yus and colleagues have suggested that the term ‘sebomatricoma’ should be applied to all benign neoplasms with sebaceous differentiation.
602. and 603. This latter term has not been used. In a recent article LeBoeuf and Mahalingam described six cases under the title ‘acanthomatous superficial sebaceous hamartoma’, even though the term superficial epithelioma with sebaceous differentiation was used throughout the text.
604
Histopathology
The tumor is characterized by a superficial plate-like proliferation of basaloid to squamoid cells with broad attachments to the overlying epidermis.
599 Clusters of mature sebaceous cells are present within the tumor. The low-power pattern is somewhat analogous to that seen in basaloid follicular hamartoma, an entity that lacks sebaceous differentiation.
605 There is also some resemblance to seborrheic keratosis but it lacks pigment and horn cysts, and infundibular tunnels stuffed with cornified cells are rare.
606
MANTLEOMA
Histopathology
The mantleoma consists of cords and columns of undifferentiated cells that radiate from the infundibulum of a hair follicle. The cords may interweave into a retiform pattern. Varying degrees of vacuolization of the cells, representing sebocyte formation, can be seen. Some sebaceous ductal structures may be present.
607The
folliculocentric basaloid proliferation of Leshin and White
608 is a hyperplasia of mantle epithelium that may become so pronounced that the term ‘mantleoma’ would be appropriate (
Fig. 33.25). It would probably be best to regard the two as variants of the same lesion. It may be confused with basal cell carcinomas, particularly on frozen section. It is a common and largely neglected entity.
MALIGNANT SEBACEOUS TUMORS
Although some tumors, particularly basal cell carcinomas, may show focal sebaceous differentiation, the sebaceous carcinoma is the only ‘pure’ tumor in this category.
SEBACEOUS CARCINOMA
Sebaceous carcinomas have traditionally been considered in two groups: those arising in the ocular adnexa, particularly the meibomian glands and glands of Zeiss, and tumors arising in extraocular sites.
609. and 610. The latter are less common and are usually found as yellow-tan firm nodules, often ulcerated, measuring 1–4 cm or more in diameter. They account for about 20% of all sebaceous carcinomas but this figure varies in different countries. They are found particularly on the head and neck of elderly patients. Rare sites include the leg,
611 foot,
612 nipple,
613 labia,
531. and 614. and penis.
615 Extraocular tumors are exceedingly rare in children.
616Those arising in the ocular adnexa are more common and comprise 1% of all eyelid neoplasms. They have a slight female preponderance and tend to involve the upper eyelid more than the lower.
617. and 618. They are rare in children.
619 They often masquerade clinically as a chalazion, delaying effective treatment. A cutaneous horn is rarely present.
620 Rarely, there is a history of radiation to the area.
618 Up to one-third develop lymph node metastases, usually to the preauricular and cervical nodes, and there is a 20% 5-year mortality. Extraocular cases with nodal and even visceral metastases have been reported, leading the authors to question the notion that extraocular tumors are less aggressive than sebaceous carcinomas of the eyelid.
615. and 621. Further examples of aggressive extraocular sebaceous carcinoma have been documented.
622Rarely, sebaceous carcinomas are associated with the Muir–Torre syndrome (see above),
609 organoid nevi (nevus sebaceus),
623. and 624. organ transplant recipients,
625 or a rhinophyma.
626
Treatment of sebaceous carcinoma
Wide surgical excision is the treatment of choice. Clearances of 5–6 mm have been recommended for eyelid tumors,
627 and an even wider clearance is preferable for extraocular lesions. Mohs micrographic surgery has been used with excellent results,
628 but no large series has been published. Adjuvant radiotherapy has been used, particularly for those with metastatic disease, but also for highly infiltrative or large tumors.
627 Neck dissection is used for operable cases of the eyelid, with regional lymph node metastases. Sentinel node biopsy for sebaceous carcinomas is subject to further study.
629 There is no published information on the best choice of chemotherapeutic agents for metastatic sebaceous carcinomas.
629 Because there are defects in the expression of retinoid acid receptors in sebaceous carcinomas, it is possible that this may affect the potential response of this tumor to retinoid therapy.
630
Histopathology531. and 618.
The tumor is composed of lobules or sheets of cells separated by a fibrovascular stroma. The cells extend deeply and often involve the subcutaneous tissue and even the underlying muscle. There is infiltration at the edges. The cells show variable sebaceous differentiation, manifest as finely vacuolated or foamy rather than clear cytoplasm (
Fig. 33.26). In one study, 48% of the cases had cytoplasmic vacuoles.
631 There is usually more differentiation at the center of the nests. The nuclei are large, with large nucleoli. There are scattered mitoses. Smaller basaloid cells and cells resembling those in a squamous cell carcinoma may be present; even focal keratinization does not negate the diagnosis. Less than 10% of cases resemble in some way a basal cell carcinoma.
631 Focal necrosis is not uncommon and this may have a ‘comedo-like’ pattern.
631 Sometimes pseudoglandular formation occurs. The vacuolated cells show abundant lipid if a frozen section is stained with oil red O or Sudan black. There may be a very small amount of PAS-positive diastase-resistant material in some cells. Focal apocrine differentiation is sometimes present.
584. and 632. This is not surprising in view of the common embryological origin of the
folliculosebaceous–apocrine unit. A carcinoid-like pattern is another rare histological variant. 588 It has also been reported in sebaceomas.
588The periocular lesions often show a pagetoid or, less commonly, a carcinoma-in-situ change in the overlying conjunctiva or epidermis of the eyelid (
Fig. 33.27).
617. and 618. This feature was seen in one-third of all cases in one series.
631 Such changes are not usually seen in extraocular cases. The presence of such a change above extraocular tumors should prompt reassessment of the lesion in case it is invasive Bowen’s disease, which may rarely mimic sebaceous carcinoma.
633 On the other hand, it is easy to misdiagnose intraepithelial sebaceous carcinoma of the eyelid as Bowen’s disease. Sebaceous carcinoma should be thought of in any lesion of the upper eyelid or if multivacuolated cytoplasmic clear cell changes are seen.
634Adverse prognostic features for tumors of the ocular adnexa include vascular and lymphatic invasion, orbital extension, poor differentiation, an infiltrative growth pattern, and large tumor size.
618Immunohistochemically, the tumor cells show positive reactions for epithelial membrane antigen (EMA) and androgen receptor (AR), but not for carcinoembryonic antigen (CEA), S100 protein, or gross cystic disease fluid protein-15 (GCDFP).
635 CAM5.2 and BRST-1 have also been positive.
631 In one report, two cases reacted with CD36.
636 It should be noted that EMA staining is often absent in poorly differentiated tumors but nuclear staining for AR is present, making it the more reliable marker.
580 Approximately 60% of basal cell carcinomas show focal positivity for AR.
580 A recent immunohistochemical review concluded that an EMA-positive, Ber-EP4-positive immunophenotype supports sebaceous carcinoma, an EMA-positive, Ber-EP4-negative profile supports squamous cell carcinoma, and an EMA-negative, Ber-EP4-positive result supports basal cell carcinoma.
637 Ocular sebaceous carcinomas contain cytokeratin 7 (CK7) but ocular basal cell carcinomas and squamous cell carcinomas do not.
638. and 639. Basaloid and undifferentiated cells express the keratin 15 (CK15) stem cell marker.
539 Cytokeratin 19 requires further studies before its use can be assessed.
589. and 640. Sebaceous carcinomas express statistically significant, increased levels of p53 and Ki-67 compared to benign lesions, and reduced levels of bcl-2 and p21 compared to adenomas.
535 Survivin, an inhibitor of apoptosis, is expressed more often in sebaceous carcinomas than adenomas and hyperplasia, but as the number of cases expressing this protein is low, it is not of any diagnostic use.
641 Bcl-X appears to be a marker of sebocytes.
642 Telomerase expression occurs in all sebaceous neoplasms and is of no value in distinguishing adenomas from carcinomas.
643
Electron microscopy
The tumor cells contain cytoplasmic lipid droplets and tonofilaments that insert into well-formed desmosomes.
615. and 617.
TUMORS WITH FOCAL SEBACEOUS DIFFERENTIATION
This category is included for completeness so that tumors considered in other sections of this book can be grouped together. Sebaceous differentiation or sebaceous glands can be seen in:
• squamous cell carcinoma
• inverted follicular keratosis
• apocrine poroma with sebaceous differentiation.
The occurrence of basal cell carcinomas with sebaceous differentiation is mentioned above. Sebaceous differentiation is very rare in squamous cell carcinomas, and such cases must be distinguished from sebaceous carcinomas and from invasive Bowen’s disease.
The
trichoblastoma with sebaceous differentiation is characterized by sebocytes and sebaceous duct-like structures within the basaloid aggregations of a large nodular type of trichoblastoma.
595The
verruca vulgaris with sebaceous differentiation is a rare entity. Sometimes, focal apocrine differentiation is also present. Several of the cases pictured in the excellent book by Steffen and Ackerman
473 would be regarded by many as examples of
inverted follicular keratosis. The induction of folliculosebaceous units by
dermatofibromas is mentioned elsewhere (see
p. 830). The
reticulated acanthoma with sebaceous differentiation is the term used by Steffen and Ackerman for what others have called superficial epithelioma with sebaceous differentiation (see
p. 776). The
apocrine poroma with sebaceous differentiation (sebocrine adenoma) at first glance resembles an eccrine poroma, but the epithelial proliferations show apocrine and sebaceous differentiation.
645.646. and 647. Sebaceous duct-like structures are observed within the basaloid aggregations.
595 A similar case with prominent cystic degeneration has been reported.
648
APOCRINE TUMORS
Normal apocrine glands
Apocrine glands, which are derived from the folliculosebaceous–apocrine germ, are restricted to the axillae, anogenital and inguinal regions, the periumbilical and periareolar regions, and, rarely, the face and scalp. Specialized apocrine glands are found on the eyelids (the glands of Moll) and in the auditory canal (ceruminous glands). The breast is sometimes regarded as a modified apocrine gland.
Apocrine glands are composed of a secretory coil in the lower dermis and subcutis, a straight ductal component which is indistinguishable from the eccrine duct, and a terminal intra-infundibular duct which opens into the follicular infundibulum. A conspicuous feature of apocrine secretory cells is their ‘decapitation’ mode of secretion whereby an apical cap, formed at the luminal border of the apocrine cells, separates off from the cell and is discharged into the lumen. Apocrine secretions produce a characteristic odor.
649Sclerosing adenosis, similar to the lesion of the breast, has been reported in apocrine glands of the scalp.
650
Alternate classification
Despite the relative paucity of apocrine glands in comparison with the ubiquitous eccrine gland, found everywhere in the skin, it would seem that many tumors arising in non-apocrine areas, and formerly regarded as being of eccrine origin, are probably apocrine in type. This reclassification of some tumors has taken place as a consequence of the identification of decapitation secretion, either as a regular or occasional feature, and the presence of, or association with, cells or other tumors showing follicular and/or sebaceous differentiation. The rationale is their common embryological origin from the folliculosebaceous–apocrine germ.
Using these and other criteria, Requena, Kiryu, and Ackerman, in a superbly illustrated book, have reclassified as apocrine the following tumors which were previously regarded as being of eccrine type:
651
• papillary eccrine adenoma
• hidradenoma (nodular, cystic)
• malignant variants of the above
• microcystic adnexal carcinoma
• signet-ring cell carcinoma
• adenoid cystic carcinoma.
This classification is likely to be regarded in some centers as controversial. Cynics will argue (a) does it matter whether they are called eccrine or apocrine, particularly when the ducts of either gland are indistinguishable, and (b) can the folliculosebaceous–apocrine germ be so unstable that it produces apocrine tumors at sites not normally frequented by apocrine glands? Immunohistochemical markers such as lysozyme, Leu M1, and gross cystic disease fluid protein-15 (GCDFP-15, AP-15, Brst-2)
652 are no longer regarded as being of much assistance in this debate.
653 Newer markers are offering some support for Ackerman’s views.
Of interest are the results obtained using a novel monoclonal antibody, IKH-4, which stains the eccrine secretory coil, but not the apocrine secretory segment.
654 Unfortunately, specific markers have a habit in dermatopathology of becoming less specific with time, and subsequent studies do not invariably confirm the initial ones. Notwithstanding these reservations, the study mentioned found staining that would support an eccrine origin for hidradenoma, poroma, spiradenoma, cylindroma, syringoma, and eccrine carcinoma and the occurrence of an eccrine variant of hidrocystoma, papillary adenoma, and syringocystadenoma papilliferum.
654 These tumors have also stained with the eccrine gland-associated monoclonal antibodies EKH-5 and EKH-6.
654 However, the association of cylindromas and spiradenomas with trichoepitheliomas in the Brooke–Spiegler syndrome is compelling evidence for their apocrine origin.
In the light of these studies, various changes have been made in the traditional subdivision of eccrine and apocrine tumors.
CYSTS AND HAMARTOMAS
Three entities will be considered in this category:
• apocrine hidrocystoma (apocrine gland cyst)
• syringocystadenoma papilliferum.
APOCRINE NEVUS
The apocrine nevus is a rare tumor composed of increased numbers of mature apocrine glands extending from the reticular dermis to the subcutis.
655.656.657. and 658. It has been reported on the upper chest, the temple,
659 and in the axilla; more often, it is an element of an organoid nevus (nevus sebaceus). It presents as a papule, or as a plaque studded with papules.
659 The lesion reported as congenital apocrine hamartoma was thought to represent a form of organoid nevus with pure apocrine differentiation on the basis of a deformed follicular structure present in the lesion.
660 Apocrine hamartoma has also been used interchangeably with apocrine nevus.
659
APOCRINE HIDROCYSTOMA (APOCRINE GLAND CYST)
Apocrine hidrocystoma has been regarded by some as an adenomatous cystic proliferation of apocrine glands, and by others as a simple retention cyst. For this reason, the term ‘apocrine cystadenoma’ is sometimes used for these cases. The tumor has a predilection for the head and neck, and is uncommon in the sites in which apocrine glands are usually found.
662 Clinically, it may have a bluish color. Apocrine hidrocystoma is considered further with other cutaneous cysts (see
p. 449).
In a recent study of 21 cases of apocrine hidrocystoma and apocrine cystadenoma, it was concluded that cases with true papillary projections containing a fibrous core are proliferative tumors (apocrine cystadenomas) that should be distinguished from apocrine hidrocystomas that are non-proliferative, cystic lesions.
663
Histopathology
The cysts are lined by two layers of cells: an inner lining of large columnar cells with eosinophilic cytoplasm often showing luminal decapitation secretion, and an outer flattened layer of myoepithelial cells. Papillary projections of epithelium into the lumen are found in about one-half of cases. The term ‘
papillary apocrine gland cyst’ has been used for these cases.
651 The cytokeratin expression suggests that it is a complex tumor, differentiating into each portion of the apocrine gland.
664 There is a suggestion from immunohistochemistry that lesions with a proliferative component (apocrine cystadenoma) are different from the pure cystic form.
665 This was confirmed by the recent study (see above) which showed that apocrine cystadenomas, in which true papillae were found, had increased Ki-67 staining.
663Two cases of an apocrine gland cyst with a hemosiderotic dermatofibroma-like stroma have been reported.
666
SYRINGOCYSTADENOMA PAPILLIFERUM
Syringocystadenoma papilliferum is an uncommon benign tumor of disputed histogenesis,
667 with a predilection for the scalp and forehead.
668. and 669. Less common sites of involvement are the chest,
670 upper arms,
671 male breast,
672 eyelids,
673 scrotum,
674 and thighs. There is an associated organoid nevus in approximately one-third of cases,
668 and for this reason it is not always possible to be certain at what age the syringocystadenoma (syringoadenoma) papilliferum component developed.
675 Probably half are present at birth or develop in childhood.
676 A coexisting basal cell carcinoma is noted in 10% of cases,
668 and there are two reports of an associated condyloma acuminatum
674. and 677. and another of a verrucous carcinoma.
678 A contiguous verrucous cyst, and a tubulopapillary hidradenoma (apocrine adenoma) have also been present.
679. and 680. Other congenital lesions have been associated with the presence of the tumor. Localization to a postoperative scar has been reported, but no histology was performed on the congenital lesion that was removed from this site 7 years earlier.
681The tumor has a varied clinical appearance, most often presenting as a raised warty plaque or as an irregular, flat, gray or reddened area.
682 Linear papules and nodules are occasionally present.
671.683. and 684. The lesions measure from 1 to 3 cm in diameter and are usually solitary. Alopecia accompanies those on the scalp.
There is increasing evidence for an apocrine histogenesis,
685. and 686. but the possibility of an eccrine origin for a few cases seems likely. Another theory suggests the apoeccrine glands as the origin of this tumor.
687 It may be derived from pluripotent cells.
688 Allelic deletions have been reported at 9p21 (p16) and 9q22 (the patched gene).
689Several examples of a malignant variant,
syringadenocarcinoma papilliferum, have been reported.
690.691.692. and 693. They have arisen on the scalp, back, chest, and in the perianal region.
694. and 695. Some have been present for many years, suggesting the possibility of malignant transformation of a benign lesion.
Histopathology668
The tumor is composed of duct-like structures that extend as invaginations from the surface epithelium into the underlying dermis (
Fig. 33.28A). These may be lined by squamous epithelium near the epidermal surface, with a transition to double-layered cuboidal and columnar epithelium below. Sometimes this latter epithelium partly replaces the overlying epidermis. At other times the surface is composed of irregular papillary projections covered by stratified squamous epithelium. Rarely, there is no connection with the epidermis. Such cases are thought to be connected to the follicular infundibulum.
696 An unusual keratotic lesion, thought to be derived from the apocrine acrosyringium, has been reported in association with syringocystadenoma papilliferum.
697 It showed hyperkeratotic columns surrounded by acanthotic epidermis with features of tricholemmal keratinization. The term ‘
apocrine acrosyringeal keratosis’ was used for this associated proliferation.
697The stroma of the papillary processes contains connective tissue, dilated vessels, and, characteristically, numerous plasma cells admixed with a few lymphocytes. The underlying dermis also contains a few inflammatory cells.
There may be underlying dilated sweat glands and, occasionally, dilated apocrine glands. In those cases associated with an organoid nevus, apocrine glands are said to be always present.
699 This apocrine component may resemble a tubular apocrine adenoma. Sebaceous differentiation has been reported in an apocrine adenoma associated with syringocystadenoma papilliferum.
700Carcinoembryonic antigen is usually present in the epithelial cells.
701. and 702. Gross cystic disease fluid protein-15 (GCDFP-15) is variably positive in the tumor cells.
703 The luminal columnar cells express CK7 and more than 70% are positive for CK19. The basal cuboidal cells usually express CK7 also, but the expression of CK19 by these cells is variable.
688 IgA and secretory component have also been demonstrated in these cells, and it has been suggested that the cells attract plasma cells by a similar mechanism to that utilized by glands of the secretory immune system.
686 Further evidence for the existence of an eccrine variant comes from a study of the eccrine-specific marker IKH-4 which has labeled one case.
654Although Hashimoto has found evidence ultrastructurally for an eccrine origin,
704 Niizuma has suggested that the tumor differentiates toward the intrafollicular and intradermal duct of the embryonic apocrine gland.
685 Apocrine differentiation was also found in another ultrastructural study.
688Syringadenocarcinoma papilliferum lacks precise morphological definition, but some have a close but cytologically malignant similarity to their benign counterpart (
Fig. 33.28B).
692 Solid areas of tumor may be present, in addition to the cystic and papillary areas.
691. and 693. In one instance a ductal sweat gland carcinoma was reported arising in syringocystadenoma papilliferum;
705 another case has subsequently been challenged.
706. and 707. They express CEA and BRST-1.
695 GCDFP-15 may also be positive.
BENIGN APOCRINE TUMORS
This group is expanding as tumors previously regarded as being of eccrine origin are reclassified as apocrine tumors. The following tumors will be considered here:
• hidradenoma papilliferum
• tubular adenoma (apocrine adenoma)
• apocrine mixed tumor (apocrine chondroid syringoma)
• myoepithelioma (often apocrine associated)
Their association with follicular tumors and the occasional presence of sebocytes and other structures indicate that the cylindroma and its frequent associate the spiradenoma are also of apocrine origin, as stated by Ackerman and colleagues. There is less convincing evidence that syringomas are of apocrine origin. At this stage, syringomas will be considered with the eccrine tumors.
HIDRADENOMA PAPILLIFERUM
Hidradenoma papilliferum is a variant of apocrine adenoma with specific morphology. It is almost always found in the vulval and perianal regions.
708. and 709. There are reports of ectopic lesions developing on the face, scalp, chest,
710 back,
711 eyelid,
712 auditory canal,
713 and arm. Approximately 20 ectopic cases have been reported. It presents usually as a solitary nodule, usually less than 1 cm in diameter and usually in middle-aged women. Cases in males have been recorded.
710Hidradenoma papilliferum with a ductal carcinoma in-situ component, has been reported.
714 This differs from hidradenocarcinoma papilliferum, the malignant equivalent of hidradenoma papilliferum, in which invasive carcinoma is present.
714
Histopathology715
The tumor is usually partly cystic and has both papillary and glandular areas (
Fig. 33.29). The papillae often have an arborizing trabecular pattern; the glandular structures vary in size. Two types of epithelium are noted in both the papillary and glandular areas. Usually, the cells are tall and columnar with pale eosinophilic cytoplasm and nipple-like cytoplasmic projections on the surface. An underlying thin myoepithelial layer is often present. In about one-third of lesions, cuboidal cells with eosinophilic cytoplasm and small round nuclei, resembling apocrine metaplasia as seen in the breast, are present in some areas of the tumor. Oxyphilic metaplasia is uncommon; it may lead to a misdiagnosis of malignancy.
716 The mitotic index is variable, but can be high. However, it does not predict a more aggressive outcome.
717A case of hidradenoma papilliferum with mixed histological features of syringocystadenoma papilliferum and anogenital mammary-like glands has been reported.
718 It was postulated that the tumor might have developed from these mammary-like glands.
PAS-positive diastase-resistant granules are present in the apices of the large cells, and material in the glandular spaces stains with the colloidal iron method for acid mucopolysaccharides. GCDFP-15, a sensitive marker for apocrine differentiation, is usually positive.
718
Electron microscopy
Electron microscopy has shown characteristic secretory granules and ‘decapitation’ secretion.
719
TUBULAR ADENOMA (APOCRINE ADENOMA)
Tubular adenoma is a very rare tumor which may be found in the axilla,
720 cheek,
721 and breast,
722 or in association with an organoid nevus.
723. and 724. Lesions of the vulval and perianal area reported as apocrine adenoma
725 and apocrine fibroadenoma probably represent variants of what are now called ‘
adenomas of anogenital mammary-like glands’ (see
p. 793).
Requena, Kiryu, and Ackerman use the term ‘t
ubular adenoma’ for this spectrum of cases, and include tumors known as papillary eccrine adenoma.
651 The term ‘tubulopapillary hidradenoma’ has also been used for this spectrum.
680Apocrine cystadenoma, which differs from apocrine hidrocystoma by having true papillae with a fibrous core, is now included as part of this spectrum.
663Tubular adenomas are slowly growing, circumscribed nodules situated in the dermis or subcutaneous tissue. They are treated by surgical excision; Mohs micrographic surgery has been used for their removal.
729
The tumor is composed of circumscribed lobules of well-differentiated tubular structures situated in the dermis but sometimes extending into the subcutis (
Fig. 33.30). A transition to normal apocrine glands may be present. The tubules have apocrine features with an inner layer of cylindrical cells, often showing ‘decapitation’ secretion. Papillae devoid of stroma project into the lumina of some tubules. There is often an outer layer of flattened cuboidal cells. Follicular and sebaceous differentiation are rare, but their presence adds support to the view that this tumor is derived from the folliculosebaceous–apocrine germ.
733 Occasional comedo-like conduits extend into the epidermis and form a communication with some of the tubules of the tumor.
734 The epidermis may be hyperplastic. The stroma consists of fibrous tissue with only a paucity of inflammatory cells, in contrast to syringocystadenoma papilliferum, in which inflammatory cells, particularly plasma cells, are prominent. However, cases of tubular adenoma have been reported with features of syringocystadenoma papilliferum in the upper part of the lesion.
726.735.736.737. and 738.Apocrine cystadenomas are part of this spectrum. They are simpler lesions, always cystic, with one or several cystic apocrine structures with true papillae (papillae with a fibrous core) protruding into the lumen. They are more cystic and less complex than cases of tubular adenoma.
663These tumors differ from apocrine adenocarcinomas by lack of infiltration of surrounding tissues, and by less marked cytological atypia. Myoepithelial cells are usually present in adenomas, whereas they are absent in adenocarcinomas.
721Immunoperoxidase studies have shown that the tumor cells contain cytokeratin; human milk protein and CEA are localized to the apical region of the cells.
736
Electron microscopy
Ultrastructural features confirm the apocrine nature of apocrine adenoma; luminal cells show vacuolar change, lipid granules, and multiple microvilli projecting into the lumen.
731 Most of the cells are rich in organelles such as mitochondria and endoplasmic reticulum, and have a prominent Golgi apparatus. Cases with some eccrine features have been reported.
728
APOCRINE HIDRADENOMA
Most of the cases reported previously as eccrine hidradenoma and eccrine acrospiroma have been reclassified as apocrine hidradenoma on the basis of their presumed apocrine histogenesis. The terms ‘nodular hidradenoma’ and ‘clear cell hidradenoma’ are also used.
739. and 740. They usually present as solitary nodules 2–3 cm in diameter, but larger variants occur. They predominate in females. There is no site predilection. They can occur at all ages, including infancy.
741. and 742. Local recurrence can occur, particularly if the lesion is incompletely excised. The malignant variant, hidradenocarcinoma, is exceedingly rare (see
p. 791). It may arise ab initio or by transformation of a benign lesion.
A skin-type hidradenoma associated with a t(11;19) translocation has been reported in the breast parenchyma.
743 An identical translocation has also been reported in a cutaneous (clear cell) hidradenoma.
744 The fusion genes are the mucoepidermoid carcinoma-translocated 1 (
MECT1) gene on chromosome 19p13 and the mastermind-like family (
MAML2) gene on chromosome 11q21.
744
Histopathology745
Hidradenomas are usually circumscribed non-encapsulated multilobular tumors, centered on the dermis but sometimes extending into the subcutis. Epidermal connections are present in up to one-quarter of cases.
746 Mucinous syringometaplasia has been described in one case, overlying a clear cell variant of hidradenoma.
747 Hidradenomas may be solid or cystic in varying proportions. Uncommonly they are pedunculated. Sometimes, large cystic spaces are present and may contain sialomucin attached to the surface of the lining cells. The closely arranged tumor cells, which may be round, fusiform, or polygonal in shape, are biphasic in cytoplasmic architecture, with one type having clear and the other eosinophilic cytoplasm (
Fig. 33.31). There are variable proportions of each cell type in different tumors, but clear cells predominate in less than one-third.
741. and 746. Sometimes, only a few clear cells can be seen. They contain glycogen and some PAS-positive diastase-resistant material, but no lipid. The nuclei of the clear cells tend to be smaller than those in the eosinophilic cells. The nuclei of the non-clear cells are often elongated and about one-third of these cells often contain nuclear grooves.
739 Focal goblet-cell metaplasia is sometimes seen.
748 A diffuse proliferation of mucinous epithelium
(mucinous hidradenoma) is rare.
749 Mitoses are variable in number; their presence does not necessarily indicate malignancy.
750 However, in one study mitoses and atypical nuclear changes were associated with an increased local recurrence rate, and even subsequent malignant transformation.
751 Other cellular variations include an oncocytic variant,
752 an epidermoid variant
753.754. and 755. with large polyhedral cells having a squamous appearance, and a pigmented variant with some melanocytes and melanin pigment in cells and macrophages.
756. and 757. A racemiform and reticulated pattern of growth has been recorded.
758Immunohistochemistry demonstrates low molecular weight cytokeratin (CAM5.2) in most tumors. Some also express CEA and EMA.
759 There is some variability in the expression of the various keratin subtypes in different parts of the tumor.
760. and 761. Smooth muscle actin and S100 protein were co-expressed in one case.
762The malignant variant has an infiltrative growth pattern, frequent mitoses (although some overlap exists in the mitotic rate between benign and malignant variants),
750. and 763. and sometimes angiolymphatic invasion.
The cells composing the tumor are connected by desmosomes. The clear cells have abundant glycogen and few tonofilaments, whereas the other cell type has abundant tonofilaments and small amounts of glycogen.
APOCRINE MIXED TUMOR (APOCRINE CHONDROID SYRINGOMA)
More than 30 years ago it was suggested that chondroid syringomas (cutaneous mixed tumors), as they were called, had eccrine and apocrine variants.
765 In 1989, Ackerman’s group supported this contention.
766 This view is now universally accepted.
Apocrine mixed tumor (chondroid syringoma, apocrine type) is an uncommon tumor, usually occurring as a solitary, slowly growing nodule on the head or neck of the middle-aged and elderly.
767 It has also been reported in a child.
768 Uncommon sites include the vulva,
769 shoulder,
749 ear,
770 and extremities.
742 There is a male predilection. Most tumors are well circumscribed and measure 0.5–3 cm in diameter.
A series of 18 cases of apocrine mixed tumors of the skin with architectural and/or cytological atypia has been described (see below).
771 They presented as solitary nodules, usually 0.4–2 cm in diameter (although one was 12 cm in diameter), on the face, and less commonly the lower extremities and inguinal region. No recurrences or metastases were documented on clinical follow-up.
771 True malignant variants of apocrine mixed tumor are extremely rare.
Histopathology
Apocrine mixed tumors are circumscribed dermal tumors with an epithelial component distributed through a myxoid, chondroid, and fibrous stroma (
Fig. 33.32). The epithelial component includes clusters and solid cords of cells as well as ductal structures, sometimes branching, lined by two layers of cuboidal cells.
772 Some of the ducts are variably dilated, and keratinous cysts may form.
773 The finding of an apocrine duct in continuity with this tumor is further support for an apocrine origin.
774 Eosinophilic globules composed of collagen (collagenous spherulosis) may be found in the lumina of the glandular elements. This change was present in the stroma in one case.
775 Solid islands of squamous epithelium may be present. Focal calcification, ossification,
776 adipose metaplasia, and sebaceous and matrical differentiation are sometimes present.
777.778.779. and 780. When adipose tissue is a conspicuous component, the term
lipomatous (apocrine) mixed tumor is used.
781.782.783.784. and 785. Hyaline epithelial cells are uncommon.
786. and 787. Rare cases are composed almost exclusively of these hyaline cells, which have a ‘plasmacytoid’ appearance because of the peripheral displacement of the nucleus.
788. and 789. Such cases have been called hyaline-cell rich chondroid syringomas, but hyaline-cell rich apocrine mixed tumor would now be more appropriate (
Fig. 33.33).
In a large series of 244 cases of apocrine mixed tumor of the skin (mixed tumor of the folliculosebaceous–apocrine complex), all types of differentiation along the lines of the folliculosebaceous–apocrine unit were found.
780 The spectrum of metaplastic changes in the epithelial component included squamous, mucinous, oxyphilic, columnar, and hobnail metaplasia, as well as clear cell change, and cytoplasmic vacuolization.
780 Myoepithelial variability was also demonstrated. These changes have already been discussed.
790The term ‘
atypical mixed tumor’ has been suggested for tumors with borderline features of malignancy such as an infiltrative edge, but which do not develop metastasis after follow-up.
791 The series of 18 cases with architectural and/or cytological atypia, referred to above, was characterized by good circumscription, lack of capsular breach or hypercellularity.
771 Pushing borders were sometimes seen. All but one case were of the hyaline-cell rich type.
771 There were multinucleated, bizarre, hyperchromatic cells in hyaline cell areas. Cells were negative for p53. Ultrastructurally, hyaline cells exhibited features consistent with myoepithelial differentiation.
771The cells express CEA and cytokeratin, whereas cells in the outer layer of the tubular structures contain vimentin and S100 protein.
766.792.793. and 794. This co-expression of CEA and cytokeratins suggests that the keratin cysts may contain cells differentiating toward the intrafollicular portion of the apocrine duct.
795 Extracellular matrix components, such as type IV collagen, laminin, fibronectin, and tenascin, are prominently expressed in the chondromyxoid matrix.
796 Type II collagen, which is expressed almost exclusively in cartilage, has been
found not only in the stroma but also in epithelial portions of the tumor. 797 The chondroid matrix expresses BM-1.
798 Scattered Merkel cells are uncommon, as demonstrated by CK20 staining.
799 The monoclonal antibody G-81 which recognizes a component of dermcidin, a constituent of eccrine sweat glands, has stained some cases of apocrine mixed tumor. This suggests either dual eccrine/apocrine differentiation in some of these tumors or lack of specificity of the antibody.
800
Electron microscopy
Ultrastructural studies have shown tubuloalveolar spaces lined by ductal epithelium.
773. and 801. There are myoepithelial cells, which appear to be responsible for producing the chondroid material.
802 The epithelial cells in the solid nests of cells show intracytoplasmic luminal formation.
802
MYOEPITHELIOMA
Myoepitheliomas are exceedingly rare cutaneous tumors that present as dome-shaped, exophytic nodules on the face, neck, extremities, or trunk.
803.804.805. and 806. They may occur at any age, but in the study of 14 cutaneous cases by Hornick and Fletcher, the median age was 22.5 years (range, 10 to 63 years).
807 Myoepitheliomas also occur in salivary glands,
808 deep soft tissues,
809 and other organs. Parotid gland myoepitheliomas may present as a subcutaneous mass.
808 They are derived from myoepithelial cells which are found in the skin as a discontinuous peripheral layer around eccrine and apocrine glands.
804 Their contraction aids in the delivery of their secretory products. Focal myoepithelial proliferation is not uncommon in apocrine and eccrine mixed tumors. Sometimes the myoepithelial proliferation constitutes a significant component of the mixed tumor, justifying a designation of myoepithelioma.
806 Some of the mixed tumors and myoepitheliomas of soft tissue reported by Fletcher and colleagues were probably apocrine mixed tumors with areas of myoepitheliomatous change.
809 Myoepithelial tumors of soft tissue will not be considered further; Hornick and Fletcher have published 101 cases.
810The lesions are almost invariably benign; local recurrences have been recorded.
807 Malignant cases have been designated
myoepithelial carcinomas.
806 They have a very low metastatic potential.
806.807. and 811. Approximately five cases have been reported.
812
Histopathology
Myoepitheliomas are circumscribed, non-encapsulated tumors situated in the dermis or subcutis. Dermal tumors may extend into the subcutis. They are composed of spindle-shaped, epithelioid, histiocytoid, and plasmacytoid (hyaline) cells.
813 The cells usually have pale eosinophilic cytoplasm and relatively monomorphous ovoid nuclei. Mitoses are usually uncommon or absent. The mitotic rate is relatively high in cases that recur, or are frankly malignant.
807 Some tumors have very little stroma, while others have a myxoid or collagenous hyalinized stroma.
Immunohistochemistry shows that the cells express vimentin, S100 protein, EMA, and often, smooth muscle actin, muscle specific actin (HHF35), glial fibrillary acid protein (GFAP), and calponin.
804.806. and 807. Keratin staining is quite variable.
814 In the cutaneous cases reported by Hornick and Fletcher, all five cases without keratin staining were diffusely positive for EMA.
807The rare
adenomyoepithelioma combines glandular sweat gland elements with myoepithelial cells.
815. and 816. Adipose tissue is rarely a predominant element in another variant.
807The exceedingly rare
myoepithelial carcinoma is usually a larger tumor with cellular atypia and a high mitotic rate. Central necrosis is often present.
812
APOCRINE POROMA
Undoubted examples of a poroid neoplasm with apocrine features occur.
817 Often there is follicular and/or sebaceous differentiation as well, reflecting the common origin of the folliculosebaceous–apocrine unit.
818. and 819. Such tumors could also be regarded as complex adnexal tumors, examples of which have been reported in the past as poroma-like adnexal adenoma and sebocrine adenoma (see
p. 807). Apocrine poromas appear to originate from follicular infundibula.
Histopathology
Anastomosing lobules of small uniform basaloid cells form small ductular structures lined by eosinophilic cuticles.
817 Focal hair follicle and/or sebaceous differentiation are common.
820 In one case, follicular differentiation was a major feature of the lesion.
821 One case resembled hidroacanthoma simplex with underlying sebaceous glands.
644 Similar cases were regarded by Steffen and Ackerman as seborrheic keratoses with sebaceous differentiation.
473Cytokeratin expression has not assisted in the differentiation of apocrine and eccrine poromas.
822
CYLINDROMA
Cylindromas are usually found as small solitary lesions on the head and neck. There is a strong predilection for middle-aged and elderly females. Large variants, usually with multiple coalescing tumors (turban tumors), may arise on the scalp and forehead. A rare variant with multiple lesions in linear array has been reported.
823 Multiple cylindromas occur in familial cylindromatosis (OMIM 132700) and the Brooke–Spiegler syndrome (OMIM 605041) in which multiple trichoepitheliomas are associated with multiple cylindromas, and sometimes spiradenoma or spiradenoma/cylindroma overlap lesions.
54.55.56.824.825.826.827.828.829. and 830. It is autosomal dominant in inheritance.
831 This association is compelling evidence for the folliculosebaceous–apocrine origin of these various tumors. They are different phenotypic expressions of mutations in the same gene, the
CYLD gene on chromosome 16q12–q13 (see
p. 760).
832. and 833. Multiple cylindromas without the other components of the Brooke–Spiegler syndrome appear to be exceedingly rare, if they occur at all.
834 Cylindromas have been reported in association with monomorphic adenomas of the parotid gland.
835Local aggressive behavior and malignant transformation are uncommon and usually associated with long-standing turban tumors of the scalp.
836.837.838. and 839. However, metastasis of malignant tumors is very rare.
840The origin of this tumor has been controversial: some immunohistochemical studies suggest that cylindroma is linked to the secretory coil of the apocrine gland, rather than the coiled duct region of the eccrine gland as originally thought.
841.842. and 843. A more recent study using the monoclonal antibody IKH-4, thought to be eccrine specific, showed positive staining, again putting the histogenesis of cylindroma into dispute.
654 However, its recent association with mutations in the
CYLD gene, which may also occur in familial trichoepitheliomas would seem to implicate the folliculosebaceous–apocrine unit.
Histopathology
Cylindromas are poorly circumscribed dermal tumors composed of irregularly shaped islands and cords of basaloid cells surrounded by conspicuous eosinophilic hyaline bands which are PAS positive and diastase resistant (
Fig. 33.34). Droplets of similar hyaline material may be present in the cell nests. The hyaline basement membrane contains type IV and type VII collagen.
844. and 845. There is usually a thin band of uninvolved connective tissue beneath the epidermis; however, the large turban tumors will ulcerate. Subcutaneous extension may also occur.
Most of the tumor islands have two cell types: a peripheral cell with a dark-staining nucleus and a tendency for palisading, and a larger cell with a vesicular nucleus more centrally located. Small duct-like structures are sometimes present.
The stroma is composed of loosely arranged collagen containing an increased number of fibroblasts. A more compact stroma is occasionally present. Stromal ossification is a rare phenomenon.
846Areas resembling spiradenoma (see below) will sometimes be present, and there may also be tumor lobules with overlap features between these two entities.
829.847. and 848. A patient with the Brooke–Spiegler syndrome had a tumor with features of cylindroma, spiradenoma, and trichoepithelioma in the same lesion.
57 In another patient with this syndrome, hybrid lesions containing two or more of these already mentioned components and/or trichoblastomatous, lymphadenomatous, and sebaceous features were present.
849Aggressive or malignant behavior is characterized by loss of the hyaline sheath and expanded cellular islands composed predominantly of larger cells, devoid of peripheral palisading.
839Although the expression of cytokeratins could be interpreted as indicating an eccrine origin, the presence of lysozyme, human milk factor globulin 1, α-smooth muscle actin, and α
1-antichymotrypsin favors an apocrine origin. CEA and EMA are also expressed.
842 Both cylindroma and spiradenoma express cytokeratins 7, 8, and 18.
848 In a comparative study of cylindroma of the skin and of the breast, both tumors did not express CK20, gross cystic disease fluid protein-15, or estrogen or progesterone receptor.
850 Both tumors expressed CK7 in the central basaloid cells, while ducts were highlighted by EMA and CEA.
850
Electron microscopy shows small dense basal cells, large light indeterminate cells, ductal cells, secretory cells containing secretory granules, and some Langerhans cells. Ductal structures are also present. The thick hyaline band is composed of thickened amorphous basal lamina
and a fibrous component consisting of anchoring fibrils. 851 Collagen fibrils of varying width are also present.
SPIRADENOMA
Spiradenoma is usually a solitary gray-pink nodule, less than 1 cm in diameter, arising on the head and neck, trunk or, less commonly, the extremities of adults.
853 Satellite tumors, multiple lesions,
854.855.856. and 857. giant variants,
858 a linear
859.860.861. and 862. or zosteriform
863. and 864. distribution and occurrence at birth,
865 and in infancy
866 have also been described. It may arise in an organoid nevus.
867 Pain and/or tenderness are frequently present, but one recent study has suggested that these features may have been overstressed in the past.
853Spiradenoma has been associated with cylindromas in the same patient (see above),
828.856. and 868. and tumors with overlap features between these two types have also been described.
829 Many of these cases have the Brooke–Spiegler syndrome (see
p. 760). Ackerman and colleagues regard both tumors as apocrine in type. Malignant transformation is a very rare event.
829.839.853.869.870.871.872. and 873. It has been reported in spiradenomas associated with the Brooke–Spiegler syndrome.
874 It appears to be accompanied by increased expression of p53 protein.
875
Histopathology853. and 876.
The tumor is composed of one or more large, sharply delineated basophilic nodules in the dermis, unattached to the epidermis and sometimes extending into the subcutis. Small satellite lobules may be present. The tumor nodules are composed of aggregates of cells in sheets, cords, or islands, or with a trabecular arrangement (
Fig. 33.35). Two cell types are present: small dark basaloid cells with hyperchromatic nuclei, and a more frequent larger cell with a pale nucleus which tends to be near the center of the clusters. The cells are PAS negative, but droplets of PAS-positive hyalin may be present in some areas of the tumor. A few duct-like structures are often present (
Fig. 33.36). Other findings include squamous eddies, small cysts, and lymphocytes infiltrating the tumor nests. Irregular, thin bands of fibrous tissue containing blood vessels are present within the tumor lobules.
Perivascular spaces, containing some lymphocytes, sometimes form between the blood vessels and the tumor cells. 877 The stroma between lobules may be edematous and sometimes there are prominent vessels with telangiectasia and even hemorrhage.
858. and 878. Dilated vessels rimmed by sclerosis have been called ‘ancient’ changes.
879 Cylindromatous areas are sometimes present.
829.847. and 848. Adenomatous elements have also been described.
880The strands of cells are cytokeratin positive and the lumina are CEA positive.
858 The pattern of cytokeratin expression mirrors that seen in the transitional portion between the secretory segments and the coiled ducts of sweat glands.
881 In another report there was co-expression of CK17 and SMA suggesting myoepithelial differentiation in this case.
882 Abundant T lymphocytes and Langerhans cells are found within the tumor lobules.
883
Electron microscopy
There is some variability in the ultrastructural findings.
858.876. and 884. The tumor is composed of sheets of cells separated into lobules by strands of amorphous and fibrillar material.
876 The most common cell is a clear polygonal or round cell with mitochondria, small vesicles, and sometimes glycogen in the cytoplasm.
858 A rare dark cell is present. Although lumina have not always been seen,
876 they have been reported, with microvilli on the lining cells. Intracytoplasmic lumina have also been seen within the epithelial cells.
884
MALIGNANT APOCRINE TUMORS
This group of tumors is expanding with the reclassification of some eccrine tumors. The following tumors will be considered:
• apocrine adenocarcinoma
• extramammary Paget’s disease
• adenoid cystic carcinoma
• endocrine mucin-producing sweat gland carcinoma
There is also some evidence that the microcystic adnexal carcinoma belongs to this group. They have been considered in their traditional eccrine category here.
APOCRINE ADENOCARCINOMA
Apocrine adenocarcinoma is a very rare tumor with no distinctive clinical features.
667.720. and 885. It usually presents as a single or multinodular mass, or plaque, in the axilla
720.886.887. and 888. or anogenital region,
889 and rarely in other sites such as the chest, nipple, finger,
890 and the scalp in association with organoid nevi.
723 In one case, the tumor presented as carcinoma erysipeloides.
891 Cases arising in apocrine hyperplasia (nevus) of the axilla have been reported.
892. and 893. Those in the anogenital region may be associated with extramammary Paget’s disease.
894. and 895. The age range in one series was 18 to 81 years (median, 61 years).
896 The tumors vary from 2 to 8 cm in diameter.
Metastasis is to regional lymph nodes in the first instance, but death, usually from visceral metastases, has been reported in 40% of the reported cases.
667 In the large series of 24 cases reported recently, four died with metastatic tumor (24%).
896 There is some correlation between survival and the differentiation of the tumor.
885Wide surgical excision is the treatment of choice. Adjuvant radiotherapy may be beneficial in advanced cases. Chemotherapy with 5-fluorouracil, tamoxifen, and capecitabine has been used for inoperable cases.
891
Histopathology667. and 720.
Tumor cells have PAS-positive diastase-resistant granules in the cytoplasm; in contrast to eccrine tumors, glycogen is virtually absent. Cytoplasmic granules of hemosiderin are present in about one-third of cases.
720 The tumor cells express cytokeratin and gross cystic disease fluid protein-15, but, not usually, CEA.
885. and 899. S100 protein and EMA have been reported in some cases,
885. and 891. but not others.
887 Tumor cells also express CK7.
888. and 901. Estrogen receptors (ER) are not usually found immunohistochemically, but ER mRNA may be demonstrated by the reverse transcription PCR method in many of these negative cases.
903 However, 62% of cases in a recent study expressed ER, and about the same percentage expressed androgen receptor.
896A tumor resembling a basal cell carcinoma but with histochemical and ultrastructural features of apocrine cells has been reported.
904 It is best regarded as a variant of basal cell carcinoma.
EXTRAMAMMARY PAGET’S DISEASE
The precise incidence of extramammary Paget’s disease is unknown; it is rarer than mammary Paget’s disease, accounting for 6.5% of all cases of Paget’s disease.
905 Extramammary Paget’s disease usually affects sites with a high density of apocrine glands, such as the anogenital region,
906. and 907. and less commonly the axilla.
908.909.910. and 911. It has also been reported in areas with modified apocrine glands, such as the external auditory canal in association with ceruminous carcinoma,
912 and possibly in the eyelid accompanying carcinoma of Moll’s glands. Rare sites of involvement include the glans penis,
913 buttock,
908 thigh, knee,
914 umbilicus,
915 abdomen,
916 scalp,
917 and lower anterior chest.
918. and 919. Rare presentations have included the concurrence of mammary and vulval Paget’s disease,
920 the concurrence of genital Paget’s disease with clear cells in the epidermis of the axilla,
921 its association with metastatic breast carcinoma in the arm,
922 the presence of superimposed herpes simplex virus infection,
923 and the concurrent involvement of both axillae and the genital region in an elderly male.
924. and 925. This has been called triple extramammary Paget’s disease.
926. and 927. Extramammary Paget’s disease presented as alopecia neoplastica in one case.
917 Paget’s disease has been reported in a supernumerary nipple,
928 and overlying a hidradenoma papilliferum.
929 Paget’s disease of the breast is sometimes quite extensive, involving a large part of the chest wall.
930Extramammary Paget’s disease presents as an erythematous, eczematoid, slowly spreading plaque. A rare pattern of erythema, localized to the underpants area, is associated with lymphatic permeation and a bad prognosis.
931 Rarely, the lesions may be focally depigmented.
932.933. and 934. Pigmented Paget’s disease also occurs.
935.936.937. and 938. The size of the lesion usually correlates with its duration. There is a predilection for older individuals. It has been reported in elderly siblings.
939. and 940. There is an overall female preponderance because the vulva is a common site of involvement.
941. and 942. Intractable pruritus is a common presenting symptom. In one study, nearly 50% of the patients had an elevated serum CEA.
943 Genetic studies of extramammary Paget’s disease are scarce. They have not shown any consistent abnormality.
944
Pathogenesis
Although it is generally accepted that mammary Paget’s disease results from the direct extension into the epidermis of an underlying intraductal adenocarcinoma in the breast,
945 the histogenesis of the extramammary type remains controversial.
946. and 947. The evidence would seem to indicate that extramammary Paget’s disease does not have a uniform histogenesis.
948 About 25% of all cases have an underlying cutaneous adnexal carcinoma, mostly of apocrine type,
910. and 949. but sometimes derived from eccrine,
950.951. and 952. periurethral, perianal, or Bartholin’s glands.
953 The underlying appendageal component may include mucinous carcinoma or porocarcinoma. A further 10–15% of patients have an internal carcinoma involving the rectum,
908 prostate,
906.954. and 955. bladder,
913.941.956. and 957. cervix,
908.941. and 958. or urethra, which appears to be of etiological significance.
953 A case that possibly arose in a non-invasive rectal adenoma has been reported.
959 Two cases have been associated with an underlying gastric adenocarcinoma.
960 Such tumors are not always the cause of an associated extramammary Paget’s disease.
961 In the case of perianal Paget’s disease, an underlying adnexal or visceral carcinoma is present in nearly 80% of cases.
962 Several explanations have been proposed for the pathogenesis of those cases in which no underlying carcinoma is found. They have included the presence of an underlying in-situ adnexal carcinoma which for technical (sampling) reasons has not been discovered, or an origin from the dermal or poral portion of sweat glands.
941 Alternatively, such cases could be derived from apocrine or eccrine cells or other pluripotential cells in the epidermis.
908.963. and 964. In the latter category are the pagetoid clear cells of the nipple, first described by Toker and thought to give rise to clear cell papulosis (see
p. 675).
965 They have been identified in genital skin.
966.967.968. and 969. These cells may give rise to a primary form of Paget’s disease, while epidermal spread of an underlying malignancy may constitute a secondary form.
970 Because the large cells in extramammary Paget’s uniformly express CK19, it has recently been suggested that the cell of origin might be follicular stem cells located in the hair follicle bulge region.
971Local recurrence of extramammary Paget’s disease is quite common because of histological extension beyond the clinically abnormal area.
972.973. and 974. The prognosis is generally good, except in those cases with an underlying adnexal or visceral carcinoma, in which the mortality is 50% or higher. Reduced survival is also associated with the presence of nodules in the primary lesion, elevated CEA levels, tumor invasion level, and lymph node metastases.
975 Spontaneous regression has been reported after partial surgical excision.
976 Metastatic extramammary Paget’s disease is relatively rare, although in one series it was 17%.
975 It may spread to regional lymph nodes and visceral organs.
977 In one case it metastasized to a renal cell carcinoma.
978
Treatment of extramammary Paget’s disease
Wide surgical excision is the treatment of choice. Sometimes this takes the form of vulvectomy, abdominoperineal resection, or other deforming procedures. Local recurrence rates for these procedures is still high, ranging from 33 to 60%.
979 In one study using Mohs surgery, the recurrence rate was 16% for primary extramammary Paget’s disease and 50% for cases with recurrent disease.
979 Margins of 5 cm were used in this study. In another report, no difference was found in the recurrence rate between cases with a clearance of more than or less than 2 cm.
975 The use of CK7 immunoperoxidase staining may assist in defining the margins of the disease.
911. and 979. Patients with clinical evidence of nodal involvement may benefit from therapeutic regional node dissection, but there is no evidence that elective nodal dissection in the absence of palpable nodes improves survival.
979 Postsurgical infections are not uncommon after extensive resections.
980Combination chemotherapy has been used for metastatic disease. In one case weekly docetaxel was used in a patient with multiple bone and lung metastases.
977 It kept the disease under control for a period.
Histopathology908. and 985.
The tumor cells in Paget’s disease have abundant pale cytoplasm and large pleomorphic nuclei, sometimes with a prominent nucleolus (
Fig. 33.37). Occasional cells have an eccentric nucleus and the appearance of a signet ring.
985 Mitoses are usually present. In early lesions the cells are arranged singly or in small groups, sometimes with glandular formation, in the basal and parabasal regions of the epidermis. Later, the entire thickness of the epidermis may be involved, although the greatest concentration of tumor cells is in the lower epidermis.
985 They usually spread into the contiguous epithelium of hair follicles and eccrine ducts. Uncommonly, Paget cells may invade the dermis.
908 Rarely, an invasive microacinar adenocarcinoma may develop.
986. and 987. The epidermis is usually hyperplastic and there is often overlying hyperkeratosis and parakeratosis. The epidermal hyperplasia has been categorized into three types – squamous hyperplasia, fibroepithelioma-like hyperplasia, and papillomatous hyperplasia.
988. and 989. The rare pigmented form has melanocytic colonization. The dendritic cells stain for both S100 protein and HMB-45.
933 A chronic inflammatory cell infiltrate is found in the upper dermis. An underlying in-situ or invasive adnexal carcinoma may be present. This may show apocrine differentiation, but in other cases it is not possible to determine the cell of origin. Attempts to divide this disease into cutaneous and endodermal types on the basis of their immunohistochemical profile have already produced non-conforming cases.
990 An unusual variant of anogenital Paget’s disease has the concurrence of a dermal mucinous carcinoma and fibroepithelioma (of Pinkus)-like changes. This latter phenomenon may represent an unusual form of eccrine duct spread of the Paget’s cells.
951 Another case mimicked pemphigus vulgaris with prominent acantholysis due to scant desmosomes in the tumor cells.
991Metastatic Paget’s disease is a rare occurrence in inguinal lymph nodes. In one case, the node contained a mixture of Paget’s cells and squamoid cells, probably derived by metaplasia of the Paget’s cells.
926Unlike the great majority of cases of mammary Paget’s disease, the tumor cells in extramammary Paget’s disease contain abundant mucin, which may be confirmed by positive staining with mucicarmine, Alcian blue at pH 2.5, Hale’s colloidal iron, and the PAS method.
985.992.993. and 994. Zirconyl hematoxylin is a useful alternative for Alcian blue when it is not available.
995 However, small ‘skip areas’, devoid of mucin, have been described.
996 Such areas may resemble Bowen’s disease.
963 The cells of clear cell papulosis (see
p. 675) do not contain mucin or show cytological atypia.
997 With immunoperoxidase techniques the Paget cells stain for CEA,
963.994.998.999. and 1000. CA15.3 and KA-93,
1001 CD5,
1002 CD23,
1003 low molecular weight cytokeratins,
994.996.1000.1004.1005.1006. and 1007. and epithelial membrane antigen.
963.972.1000. and 1008. In one study, all 15 cases of primary extramammary Paget’s disease had the cytokeratin immunophenotype CK7
+/CK20
−, while only some cases secondary to underlying carcinomas were CK7
+; furthermore, some were CK20
+.
1009 Interestingly, CK7 is also found in Toker cells, mammary Paget cells, and Merkel cells.
966.1010. and 1011. Others have confirmed these findings.
942.988.1012.1013. and 1014. The Paget cells also contain apocrine epithelial antigen,
998 MUC1, MUC2, and gross cystic disease fluid protein (GCDFP), which was thought to be specific for apocrine cells.
703.914.962. and 1014. However, GCDFP sometimes localizes in eccrine cells.
653. and 985. GCDFP is not always expressed in Paget cells. Variable staining of cells has been reported for human milk fat globulin.
1015 HER-2 protein is expressed in some cases.
1016 There is a consistent lack of estrogen and progesterone receptors but, in about half the cases, androgen receptors are expressed.
944. and 1017. However, they involve from only 1% of cells to more than 75% of them.
1018 Prostate-specific antigen is expressed in the pagetoid cells of many cases associated with an underlying adenocarcinoma of the prostate (
Fig. 33.38).
954 It may also be expressed in cases without any associated carcinoma of the prostate.
1017 The cells do not contain CD44 or S100 protein.
992.1000.1001. and 1006. Matrix metalloproteinases (MMP-7, MMP-19) are often expressed in cases with an underlying carcinoma.
1019 Recently, various other proteins have been demonstrated in this condition. They include insulin-like
growth factor-1 receptor, p-AKT, p-ERK1/2, Stat3, Stat5a, E-cadherin, cyclin D1, and Bcl-xL.1020.1021. and 1022. Overexpression of p53 is correlated with stromal invasion. 1023 There is also differential expression of two new members of the p53 family, p63 and p73. Overexpression of p73 occurs, while there is decreased expression of p63.
1024The various immunoperoxidase markers can be used to distinguish Paget’s disease from Bowen’s disease and superficial spreading melanoma in situ, should any difficulty be experienced on examining hematoxylin and eosin-stained sections.
1025 As some cases of Bowen’s disease express CK7, it has been suggested that Ber-EP4 should be added to the panel of markers as it labels all cases of extramammary Paget’s disease but none of the other pagetoid neoplasms.
1026 The possibility of combined Paget’s disease and Bowen’s disease should be kept in mind.
964. and 1027. Immunolabeling for syndecan-1 shows cell-membrane staining in Bowen’s disease, cytoplasmic expression in extramammary Paget’s disease, and no expression in melanoma in situ.
1028
Electron microscopy
There are secretory and non-secretory cells, the former having a prominent Golgi complex, numerous free ribosomes, and clusters of mucinous secretory granules.
1029 Some cells have a few surface microvilli, and a few may border a small lumen. Adjacent Paget cells are joined by small desmosomes, and these may also exist between Paget cells and keratinocytes.
985
ADENOID CYSTIC CARCINOMA
Over 50 cases have now been reported with the designation ‘adenoid cystic carcinoma’,
690.1030.1031.1032.1033. and 1034. excluding purported cases in the external auditory canal.
1035 Santa Cruz, in his major review,
692 appears to have accepted as adenoid cystic carcinomas all cases reported with this title. However, Cooper
1036 has pointed out that some of these cases
1035. and 1037. belong to a category of eccrine tumor which has been reported as eccrine carcinoma (eccrine epithelioma) and basal cell carcinoma with eccrine differentiation.
1038. and 1039. There are rare cutaneous tumors that do have a striking resemblance to adenoid cystic carcinoma of the salivary gland, and therefore it seems appropriate to accept the existence of adenoid cystic carcinoma of the skin.
1032The scalp and chest have been the sites of predilection.
1040.1041. and 1042. Most parts of the body have been involved, but usually only solitary cases have been reported at sites away from the scalp and chest.
1043 An exception is the vulva where adenoid cystic carcinoma of Bartholin’s gland is a recognized entity.
1044 Although local recurrence is common, only a few cases with distant metastases (usually to the lung), have been reported.
1032.1040.1041. and 1045. The histogenesis of these tumors has been disputed. They have been regarded in the past as eccrine tumors, although it has been acknowledged that many arise from ceruminous glands, which are modified apocrine glands. It seems best to regard these tumors as apocrine in origin.
1041Treatment is by wide surgical excision ensuring that margins are tumor free.
1045 Lymph node dissection has been carried out in a few cases. Tumors of Bartholin’s gland have been treated by simple local excision and, sometimes, radical vulvectomy.
1044
Histopathology
The tumor is composed of islands and cords of basaloid cells showing cribriform and some tubular areas. There is abundant basophilic mucin (hyaluronic acid) in the small cysts and between cells. Mitoses are uncommon. Perineural invasion is almost invariably present.
1046 The histological features closely resemble those of the corresponding tumor in salivary glands.
1047 The tumor cells express epithelial membrane antigen and, sometimes, CEA.
1030. and 1047. There is focal staining for cytokeratin, vimentin, and S100 protein.
1041.1042. and 1048.
MUCINOUS CARCINOMA
Over 150 cases of primary mucinous carcinoma of the skin have been reported.
692.1049.1050.1051.1052.1053. and 1054. The age-standardized incidence rate is less than 0.1 per million.
1055 This is a slowly growing tumor arising on the face (particularly the eyelids),
1056.1057. and 1058. scalp,
690.1059.1060. and 1061. axilla, and trunk of middle-aged and older individuals.
1055 The tumor nodules are often reddish and painless, measuring 0.5–7 cm in diameter. Larger variants have been recorded.
1062 Late recurrences are common. Metastases to regional nodes and widespread dissemination occur in about 15% of cases.
1049 One of the reported cases with multiple in-transit and pulmonary metastases has subsequently been challenged.
1063. and 1064. Only one case recurred, and one regional metastasis developed in the 15 cases reported from Denmark.
1055 Both an eccrine and apocrine derivation have been suggested.
1065. and 1066.
Treatment of mucinous carcinoma
The recommended treatment varies from standard excision to wide local excision, including dissection of regional lymph nodes.
1055 Mohs micrographic surgery has also been recommended.
1067 Adjuvant hormone treatment with antiestrogenic drugs for patients with estrogen receptor-positive tumors has been used. Recurrent tumors are not responsive to radiation treatment or chemotherapy.
1055
Histopathology
Mucinous carcinomas are dermal tumors that sometimes extend into the subcutis and deeper tissues. There are large pools of basophilic mucin separated by thin fibrovascular septa. Small islands of epithelial cells appear to float in these mucinous pools (
Fig. 33.39). The epithelial component is denser at the periphery of the lesion. The tumor cells are small and cuboidal, and some have vacuolated cytoplasm. Rarely, signet-ring cells comprise a major component of the tumor.
1054 The cell nests in some areas have a cribriform appearance, whereas other cells form small glandular or tubular spaces containing mucin. Some of these cases originate from an in-situ component, as seen in the corresponding mammary carcinoma.
1054 Focal neuroendocrine differentiation was present in a lesion removed from the vulva,
1068 suggesting that it may have been an endocrine mucin-producing sweat gland carcinoma (see below). Another uncommon component may mimic infiltrating mammary carcinoma.
1069 A sarcomatous component was present in one case.
1070 Epidermotropism, resembling Paget’s disease, has been reported.
951Microcalcification is sometimes present. Psammoma bodies are rare.1054. and 1071.The mucin is PAS positive and stains with mucicarmine and colloidal iron. It is hyaluronidase resistant and sialidase labile, indicating that it is a sialomucin. This feature assists in differentiating this tumor from a metastatic mucinous carcinoma, which it may closely mimic on histological examination.
1072 Metastatic mucinous carcinomas of intestinal origin show characteristic ‘dirty necrosis’.
1054 Some mucinous carcinomas express apocrine markers.
653. and 1073. MUC1 and MUC2 stain various components of the tumor.
1054 They also express low molecular weight cytokeratins (CK7, CAM5.2), CEA, epithelial membrane antigen and, sometimes, S100 protein.
1054.1061.1073.1074. and 1075. CD15 and 34βE12 are sometimes positive.
1076 There is strong nuclear expression of estrogen receptors, but a more variable pattern for progesterone receptors.
1077. and 1078. Focal expression of human milk fat globulin 1 (HMFG) in the luminal or outer surface of the nests is seen in one-third of all cases.
1079 CK20 is not expressed but it may be present in metastatic colorectal tumors.
1079 The myoepithelial cells in any in-situ component express p63, CK5/6, calponin, smooth muscle actin, and HHF-35.
1080 Lymphatic permeation is best visualized with the D2-40 antibody.
1081
Electron microscopy
The tumor is composed of peripheral dark cells, some of which contain mucin-like material, and inner pale cells which are less well differentiated.
1082
ENDOCRINE MUCIN-PRODUCING SWEAT GLAND CARCINOMA
Endocrine mucin-producing sweat gland carcinoma is an exceedingly rare but distinctive tumor reported on the eyelids; it is analogous to a similar tumor reported in the breast.
1083.1084.1085. and 1086. There is a predisposition for elderly females.
1086 There is a multinodular, solid, and cystic dermal tumor. Some nodules contain mucin pools with adjacent areas resembling mucinous carcinoma. The cells express estrogen and progesterone receptors as well as chromogranin and synaptophysin.
1083 CD57 and NSE are expressed in most cases. Also expressed are CK7, CAM5.2, and EMA.
1086Its origin may be the apocrine glands of Moll.
1084 However, the cases of mucinous carcinoma reported by Hanby et al appeared to include both mucinous carcinomas and the lesion reported here. These authors listed tumors on the eyelids and other sites but it is not possible from their paper to correlate site with histological pattern.
1077In the series of 12 cases reported by Zembowicz and colleagues, there were no recurrences or metastases, consistent with low-grade carcinoma.
1086
MALIGNANT MIXED TUMOR
Malignant mixed tumor (malignant chondroid syringoma, malignant apocrine mixed tumor) is a rare tumor of the skin, most often found on the trunk and extremities, which are not the usual sites of the benign variant.
692.1087.1088.1089.1090. and 1091. Fewer than 50 cases have been reported.
1092 Sometimes, juxtaposed areas of benign and malignant tumor are found, evidence for malignant transformation of an apocrine mixed tumor, at least in some cases.
692 Local or distant metastases are common, although there is usually a prolonged course.
1093 The most common site for distal metastases is the lung, followed by bone.
1092 Nodal metastases occur in more than a third of all cases.
Early and wide surgical excision with a broad margin (3 cm clearance has been suggested) is the treatment of choice.
1092 Radiotherapy and/or chemotherapy have not been effective in diminishing the tumor mass.
1092
Histopathology
The tumors have a lobulated appearance. They are composed of an epithelial and a mesenchyme-like component, the latter consisting of myxomatous and cartilaginous areas.
1087. and 1089. The epithelial component predominates at the periphery of the tumor, where there are cords and nests of cuboidal or polygonal cells with some glandular structures. There is variable pleomorphism (
Fig. 33.40). Scattered mitoses are present. Mesenchymal elements are progressively more abundant toward the center.
1088 They may also be present in lymph node metastases (
Fig. 33.41). Ossification is occasionally present.
1087 The histological appearance may be a poor indicator of the biological behavior of a particular tumor in this category.
692. and 1094.The epithelial cells express cytokeratins (AE1/AE3).
1092 The luminal epithelial cells show binding to the lectin
Ulex europaeus; intraluminal cells are CEA positive.
1095 Androgen receptors have not been detected.
1096 The chondroid areas express S100 protein.
1095 GFAP may also be expressed.
1092
HIDRADENOCARCINOMA (NODULAR)
Hidradenocarcinoma (malignant (nodular) hidradenoma,
1097 malignant acrospiroma
1098. and 1099.) is a rare sweat gland tumor of the skin, which may have both apocrine and eccrine variants.
1097. and 1100. Although most cases arise de novo, in rare cases, it may arise in a pre-existing hidradenoma.
1097. and 1101. It has a predilection for the face and extremities.
1102 The trunk is sometimes involved.
1097 It usually presents as an ulcerated reddish nodule in older individuals, but cases have been recorded in children,
1103. and 1104. and at birth.
1105 They measure 1–5 cm in diameter; some are ulcerated.
1097The tumors have an aggressive course, with eventual distant metastasis to lymph nodes, bones, and lungs.
1099.1102. and 1106. In a recent report of seven cases from Italy, six patients died within 15 to 45 months of diagnosis.
1097 Local vascular invasion with hemorrhage caused death in three patients, while distant metastases occurred in a further two cases.
1097 Survival time was inversely proportional to the size of the tumor.
Treatment of hidradenocarcinoma (malignant hidradenoma)
Wide local excision is the treatment of choice with clearances of 2 cm recommended.
1097 Despite this, local recurrence is common. Selective lymph node dissection is often used. The value of adjuvant
radiotherapy has not been confirmed. 1097 A case expressing
Her-2/neu, a proto-oncogene on the long arm of chromosome 17 that encodes a tyrosine kinase receptor protein structurally related to epidermal growth factor receptor, has been successfully treated with chemotherapy and trastuzumab (Hercepten), a humanized IgG
1 murine antibody to the extracellular portion of the Her-2/neu receptor.
1101 Sweat gland tumors that express this protein are usually associated with a poor prognosis.
1101 Chemotherapy has not been successful in several cases, although capecitabine, a fluorouracil analogue was successful in one case.
1107
The tumor is composed of sheets of cells with glycogen-containing pale cytoplasm and distinct cell membranes.
1108 The term ‘clear cell eccrine carcinoma’ has been used in the past for those with a prominent clear cell component.
1109 In some cases there is little clear cell change and the cells have a basaloid or even squamoid appearance. Sometimes, squamous differentiation is widespread.
1110 Cytoplasmic vacuoles, representing intracellular lumina formation, are an important feature.
692 Peripheral lobules of the tumors are often irregular and appear invasive.
1111 Focal necrosis is sometimes present.
1112 An occasional malignant tumor is deceptively bland in appearance
1105 whereas others have numerous mitoses.
692 Melanocytes are sometimes present in the tumor lobules.
692The cells express the high molecular weight cytokeratins CK5 and CK7, as well as p63, androgen receptor, estrogen receptor, and sometimes Her-2/neu.
1101 In a larger series, all cases stained positively for keratin AE1/3, and cytokeratin 5/6.
1100 Ki-67 and p53 staining was strongly positive in five of six tumors, but the neoplasms expressed CEA, S100 protein, GCDFP-15, and EMA in no consistent pattern.
1100
Electron microscopy
The tumor cells contain cytoplasmic glycogen and form intracellular lumina,
692 around which tonofilaments are arranged in a circumferential pattern. Desmosomes are usually well developed.
MALIGNANT CYLINDROMA
Malignant cylindroma is a very rare tumor of apocrine type that usually arises in a long-standing cylindroma of the scalp.
836.838.839. and 1113. It develops more often in patients with multiple cylindromas.
1114.1115. and 1116. Ab initio variants probably also occur.
692 This tumor has also arisen in an organoid nevus (nevus sebaceus).
1117 Multiple malignant cylindromas have been reported in a patient who also had a basal cell adenocarcinoma of minor salivary gland.
1118 The tumors are aggressive, although subsequent metastasis is rare.
840Wide surgical excision is the treatment of choice.
Histopathology
The tumor is composed of nests and cords of basaloid cells showing frequent mitoses, focal necrosis and loss of the PAS-positive hyaline membrane. It has been suggested that the rare variants that have partially preserved hyaline membranes have a good prognosis.
1119 Foci of squamous differentiation occur. A contiguous benign cylindroma is usually present.
1120
MALIGNANT SPIRADENOMA (SPIRADENOCARCINOMA)
The malignant transformation of spiradenoma is a rare event, heralded by the rapid enlargement of a cutaneous nodule of long-standing.
871.872.1121. and 1122. Malignant transformation appears to occur slightly more often in multiple spiradenomas than in solitary lesions.
1123 Many different sites have been involved, several of these tumors occurring around the elbow and on the digits.
692. and 1124. The trunk is the favored site.
1125 The tumors are quite aggressive, with fatal metastasis developing in at least 20% of cases.
692 A low-grade variant with systemic metastases has also been reported.
1126Treatment is wide surgical excision. In cases with a large underlying benign (usually linear) spiradenoma there is controversy as to whether the benign lesion needs excision in its entirety as this may lead to mutilating surgery.
1123
Histopathology
There are solid islands of tumor cells, which may show either a squamous or a basaloid pattern. Glandular and sarcomatous areas have also been reported.
692.870. and 1131. The diagnosis depends on finding a contiguous spiradenoma, as the malignant component usually lacks any distinguishing features. Sometimes, an abrupt transition between the benign and malignant components is present.
1125The tumor cells express cytokeratins, epithelial membrane antigen,
1124 and p53.
1132
TUMORS OF MODIFIED APOCRINE GLANDS
The glands of Moll on the eyelid, and the ceruminous glands of the external ear are examples of modified apocrine glands. The breast can also be regarded as a modified apocrine gland. Ectopic mammary glands do occur (see below).
ADENOCARCINOMA OF MOLL’S GLANDS
The glands of Moll are modified apocrine glands in the eyelids. Retention cysts (resembling apocrine cystadenoma), hidradenoma papilliferum,
712 and malignant tumors resembling apocrine adenocarcinoma have been reported.
667. and 1133. There are too few cases of adenocarcinoma in the literature for significant clinical comment.
Histopathology
The tumors resemble apocrine adenocarcinoma with architectural and cytological features of malignancy. There may be iron granules in the cytoplasm of the tumor cells. Cases reported in the past with pagetoid epidermal involvement have been regarded as possibly sebaceous rather than apocrine tumors.
1134
EROSIVE ADENOMATOSIS OF THE NIPPLE
Erosive adenomatosis (florid papillomatosis, papillary adenoma) is a rare benign tumor involving the nipple which may clinically mimic Paget’s disease.
1135. and 1136. It is thought to arise from the ducts of the nipple. It may rarely occur in males, and in children.
1137. and 1138.Total excision of the tumor, including the nipple, has been the usual treatment modality because of the high incidence of recurrence when removal is incomplete. Mohs micrographic surgery can be used, with preservation of the nipple, in cases that are not advanced.
1139
Histopathology1140
This dermal tumor is a well-circumscribed non-encapsulated lesion with an adenomatous configuration (
Fig. 33.42). Some of the ducts have papillary projections into the lumen, and a few show cystic dilatation. The lining epithelium is of apocrine secretory type and there is usually a backing of myoepithelial cells. Some ducts connect with the surface epithelium; squamous epithelium may extend into them.
The fibrous stroma sometimes contains a mild inflammatory infiltrate, which may be rich in plasma cells.
CERUMINOUS ADENOMA AND ADENOCARCINOMA
The ceruminous glands are modified apocrine glands in the external auditory canal. They give rise to rare tumors in which the distinction between adenoma and adenocarcinoma may be difficult on histological grounds.
1141.1142. and 1143. For this reason the term ceruminous gland tumor has been suggested.
1144 The term ‘ceruminoma’ has been used in the past, not only for the ceruminous adenoma and adenocarcinoma, but also for adenoid cystic carcinomas and mixed tumors of the auditory canal. The term is best abandoned.
A series of 41 cases of ceruminous adenoma was reported from the Armed Forces Institute of Pathology (AFIP) in 2004.
1145 There was a slight male predominance; the mean age was 54.2 years (range, 24 to 85 years).
1145 Patients usually presented clinically with a painless mass of the outer half of the external auditory canal. Hearing loss was present in approximately 25% of cases. Surgical excision was used for all lesions; recurrence occurred in four patients due to incomplete excision. No malignant cases were reported.
Histopathology
In the AFIP series (see above), 36 cases were classified as ceruminous adenomas, 4 as ceruminous pleomorphic adenomas, and 1 case as syringocystadenoma papilliferum.
1145 The tumors were composed of a tubuloglandular proliferation of inner ceruminous cells subtended by a spindled to cuboidal myoepithelial layer. The luminal cells expressed CK7 and CD117, while the basal cells were highlighted with CK5/6, S100 protein, and p63.
1145
TUMORS OF ANOGENITAL MAMMARY-LIKE GLANDS
Mammary-like glands are a newly recognized variant of cutaneous adnexal gland found in the anogenital region.
1146 They resemble mammary glands, although they have variously been regarded in the past as modified eccrine or apocrine glands. Adenosis,
1147 hyperplasia,
1148 adenomas
1146. and 1149. (including fibroadenomas),
1150.1151.1152.1153.1154. and 1155. and adenocarcinomas
1156 may arise in these glands. More than 20 cases of
adenocarcinoma of mammary-like glands of the vulva have now been recorded.1157. and 1158. An adenocarcinoma of tubulolobular type is a rare variant. 1159 Decapitation secretion is sometimes present in tumors of this region.
1160 These glands possess receptors for estrogen and progesterone proteins which help to distinguish them from classical eccrine and apocrine glands. Mammary-like sweat glands can occur in other parts of the body, outside the milk line. Cases of breast-like lesions arising in the skin of the thigh, scalp, eyelid, and umbilicus have been reported.
1161. and 1162. They presumably arose in ectopic mammary-like sweat glands. The origin of the facial apocrine fibroadenoma reported in a male is speculative.
1163 It did not express estrogen or progesterone receptors and may represent a second type of apocrine fibroadenoma.
1163Tumors of the anogenital mammary-like glands are comparatively common when compared to lesions of Bartholin’s gland, and minor vestibular glands.
1164. and 1165. Nodular hyperplasia and adenomas of Bartholin’s gland have been reported, although the distinction between the two is not always clear cut.
1164. and 1166.
ECCRINE TUMORS
In the past there has been some debate as to the eccrine or apocrine origin and differentiation of certain adnexal tumors. Histochemistry and electron microscopy sometimes gave conflicting results, with features suggestive of both apocrine and eccrine differentiation in some tumors. The recent development of various monoclonal antibodies, for use with immunoperoxidase techniques, has assisted marginally in the classification of the various adnexal tumors.
701.702.703. and 1167. The various markers have not lived up to initial expectations. The first of these markers to have diagnostic value was the finding of carcinoembryonic antigen (CEA) in sweat gland tumors.
1168 This does not represent a single oncofetal antigen, but comprises a family of homologous glycoproteins that include classic CEA-180, biliary glycoprotein (BGP), and non-specific cross-reacting antigens (NCA). If these monospecific antibodies are used, a rather consistent profile emerges: staining for both CEA-180 and NCA indicates ductal differentiation of both apocrine and eccrine type; co-expression of all three is consistent with differentiation toward the secretory portion of eccrine glands or the transitional portion of proximal ducts.
1169Other monoclonal antibodies have been developed with variable specificity and sensitivity for eccrine-related antigens. They include:
1. SKH1, which reacts with the secretory portion and coiled duct of the eccrine gland and the secretory portion of apocrine glands
1170
2. ferritin antibody, which demonstrates ferritin in the outermost layer of the eccrine and apocrine duct
1171
3. antibodies to IgA and secretory component, which detect antigen in the lumen and on the surface of the epithelium of sweat glands
1172
4. IKH-4, EKH-5, and EKH-6, which stain the eccrine secretory coil
654
5. Dako-CK1 and CAM5.2 (both commercially available), which react with two cytokeratins of different molecular weight.
1173 Whereas Dako-CK1 detects a cytokeratin in the intraepidermal eccrine duct and the inner layer of the intradermal portion of the duct but not other structures, CAM5.2 reacts with the apocrine gland and supposedly the duct, and the eccrine secretory coils but not the eccrine duct.
1173 Numerous other monoclonal antibodies have been prepared to various cytokeratins.
The monoclonal antibody MNF116, which detects the low and intermediate molecular weight keratins (5, 6, 8, 17, and 19), stains the basal cells of the epidermis and adnexae. It is found in all epithelial tumors, including adnexal ones.
1174 Antibodies to individual keratins have not been of much assistance in routine diagnosis, but they have given a valuable insight into the possible derivation and/or differentiation of various eccrine tumors.
99.1175. and 1176. For example, syringomas exhibit a pattern similar to normal dermal eccrine ducts (EMA in peripheral cells, CK10 in intermediate cells, and CK6, CK19 and CEA in luminal cells), and eccrine poromas exhibit a widespread reaction for CK5/6 and EMA, analogous to peripheral dermal duct cells.
99 A recent publication stated that syringomas were derived from luminal cells of the lowermost intraglandular sweat duct, and poromas tend to differentiate towards the cells of the upper acrosyringium.
1177 Cylindromas and eccrine spiradenomas have a more complex pattern.
99 They are now regarded as apocrine tumors.
Although CD44 is strongly expressed in the eccrine coil secretory cells, it has not proved a useful marker of sweat gland differentiation in tumors.
1178 It is expressed not only in syringomas and eccrine poromas but in tumors of undoubted apocrine origin such as hidradenoma papilliferum.
1178Some eccrine tumors contain estrogen receptor protein, a feature of some breast carcinomas.
1179 Myoepithelial cells (as determined by the expression of vimentin and α-smooth muscle actin) are seen in most sweat gland tumors considered to differentiate toward the secretory coil of sweat glands, and in most of the traditional apocrine tumors.
1180.1181. and 1182. The myoepithelioma has been considered with the apocrine tumors (see
p. 784). Another feature of some sweat gland carcinomas is the presence of p53 protein; it is rarely present in benign tumors.
1183 The mitotic rate is also an important indicator of malignancy.
1184It has been mentioned already that some of the tumors traditionally regarded as being of eccrine origin are apocrine in type, while the evidence for the histogenesis of a further group of these tumors is still being evaluated.
The eccrine hamartomas will be considered first, followed by a discussion of the benign eccrine tumors and the eccrine carcinomas. Hyperplastic and metaplastic lesions of the eccrine glands are discussed in
Chapter 15 (
pp. 438–439).
HAMARTOMAS AND BENIGN TUMORS
ECCRINE HAMARTOMAS
The term ‘eccrine hamartomas’ is used to cover the diverse group of nevoid conditions involving the eccrine sweat glands. The simplest lesion is an
eccrine nevus,
1185.1186.1187.1188. and 1189. a rare abnormality in which there is an increased number of normal-appearing eccrine coils or an increase in the size of the coils (
Fig. 33.43). A case presenting as a perianal skin tag has been reported.
1190 Sometimes an eccrine nevus produces localized hyperhidrosis.
1191. and 1192. A variant with a conspicuous mucinous stroma has been reported.
1193 In one case, extensive mucinous eccrine nevi were present following the lines of Blaschko.
1194 The eccrine nevus with an angiomyxoid stroma is a related entity.
1195Eccrine duct hyperplasia usually occurs as a reactive process (see eccrine syringofibroadenomatosis –
p. 799), but it has been reported in association with a nasal glioma, a hamartomatous lesion.
1196In
eccrine angiomatous hamartoma1197.1198.1199.1200.1201.1202.1203.1204.1205.1206. and 1207. there is an increase in the number of small blood vessels, and sometimes of nerve fibers, mucin, or fat,
1208 in addition to the increase in eccrine glands.
1209 Hair follicles are sometimes associated with this lesion. Overlying verrucous features have been recorded.
1210 In one case the verrucous features were associated with an underlying hemangiomatous component resembling an angiokeratoma or verrucous hemangioma.
1211. and 1212. In other cases components resembling spindle-cell hemangioma or arteriovenous hemangioma were present.
1208. and 1213. Eccrine angiomatous hamartoma can be present at birth or appear in early childhood. Even onset
in adulthood has been reported. 1214 Neurofibromatosis type 1 was present in one case.
1215 Lesions usually present as a bluish or brown nodule or plaque
1216 but multiple lesions can occur.
1217 The extremities are often involved.
1218 Hyperhidrosis and hypertrichosis may be present.
1219 The hyperhidrosis of an eccrine nevus has been successfully treated with botulinum toxin.
1220 Pain and/or tenderness are usually present.
1221 Spontaneous regression has been reported.
1219 Gross cystic disease fluid protein-15 was present in the eccrine glands in one case.
1222 The lesion reported as a
palmar cutaneous hamartoma had neurovascular glomic bodies in addition to fat, angiomatous vessels and eccrine glands.
1223The
acrosyringeal nevus consists of a proliferation of PAS-positive acrosyringeal keratinocytes, which extend down into the dermis as thin anastomosing cords from the undersurface of the epidermis (
Fig. 33.44).
1224 Some of these structures are recognizable as eccrine ducts. Stromal plasma cells may be prominent. Lesions may be linear,
1225 plaque-like or multiple.
1224 Diffuse lesions have been observed in ectodermal dysplasia.
1226 Although usually regarded as an identical lesion,
1227. and 1228. the solitary tumor reported as a syringofibroadenoma
1229 does have clinicopathological differences; it will be considered with the benign tumors (see
p. 799).
1230. and 1231. Incidentally, the term ‘acrosyringeal nevus’ was suggested to the author by the late Dr Hermann Pinkus, who agreed that the lesion had clinical and pathological features that distinguished it from the lesion reported earlier by Mascaro.
The
phakomatous choristoma is a benign congenital lesion of the eyelid consisting of lens tissue in an ectopic location.
1232 It is mentioned here because the irregularly branched ducts and cords, some cystically dilated, can mimic an adnexal tumor. Furthermore, there is no other suitable place for its inclusion. There is a densely fibrotic stroma, psammoma body-like calcifications and intraluminal degenerated ghost cells. There is strong staining for vimentin and weak focal staining for S100 protein.
1232There are three somewhat related lesions characterized by comedonal dilatation of eccrine ostia, with or without cornoid lamellae. These lesions are comedo nevus of the palm, linear eccrine nevus with comedones, and porokeratotic eccrine ostial nevus. In
comedo nevus of the palm1233 there are keratotic pits formed by parakeratotic plugs within dilated eccrine ostia. The lesion reported as
linear eccrine nevus with comedones1234 resembles nevus comedonicus, with the addition of basaloid nests in the dermis resembling eccrine spiradenoma in some areas and eccrine acrospiroma in others. In
porokeratotic eccrine ostial nevus there are cornoid lamellae associated with eccrine ducts.
1235.1236.1237.1238. and 1239.In
eccrine-centered nevus there are nevus cells intimately associated with eccrine sweat ducts.
1240. and 1241.
ECCRINE HIDROCYSTOMA
Eccrine hidrocystoma is usually solitary, but multiple lesions may occur.
1242.1243. and 1244. There is a predilection for the periorbital area.
1245 It is discussed further with the other cystic lesions in
Chapter 16 (
p. 449).
The existence of this entity has been challenged; many, if not all of these lesions are now regarded as being of apocrine type.
651. and 1246.
Histopathology
The cysts are unilocular and lined by two layers of cuboidal epithelium. Cytokeratins 7, 8, and 19 were present in some cases. On this basis they were regarded as being of eccrine origin.
1247
PAPILLARY ECCRINE ADENOMA
The benign papillary eccrine adenoma was first described by Rulon and Helwig in 1977.
1248 It presents as a slowly growing firm nodule with a predilection for the extremities of black people.
1248.1249.1250.1251.1252.1253.1254. and 1255. Ackerman and colleagues include this group with their tubular adenomas of apocrine origin.
651
Papillary eccrine adenoma is a circumscribed dermal tumor composed of multiple variably dilated duct-like structures lined by two or more
layers of cells. 1250 The inner layer often forms intraluminal papillations of variable complexity (
Fig. 33.45).
1249 This latter feature is not always prominent in all areas of the tumor. There is no decapitation secretion. The epithelial cells may show focal clear cell change and even focal squamous differentiation. Some of the lumina contain an amorphous eosinophilic material.
1255 Immunoperoxidase studies have demonstrated the presence of CEA, cytokeratins, particularly CK8 and CK14, and S100 protein.
1250.1252.1253.1256. and 1257. The unique pattern of some cases, which are devoid of apocrine features, together with the positive staining with the eccrine-specific antibody IKH-4 are reasons for the retention of an eccrine variant of tubular adenoma. It is acknowledged that apocrine variants are far more common. The stromal connective tissue may show hyalinized collagen and a focal increase in fibroblasts. Inflammatory cells are usually sparse.
Electron microscopy
The duct-like structures are composed of basal and luminal cells, the latter containing intracytoplasmic cavities but not secretory granules.
1251
SYRINGOMA
Syringomas are usually found as multiple small papules on the lower eyelids and cheeks of adolescent females.
854 Other variants include solitary and giant
1258 lesions, a plaque form,
1259 milia-like lesions,
1260.1261. and 1262. tumors limited to the vulva,
1258.1263.1264. and 1265. penis,
1266.1267.1268.1269. and 1270. axillae and groins,
1271 buttocks,
1272 moustache area
1273 or scalp,
1274 and acral,
1275 linear
1276. and 1277. or bathing-trunk distributions.
1278 Eruptive
1279.1280.1281.1282.1283.1284. and 1285. and disseminated forms,
1286 some of which may be familial,
1287 have also been described. The case reported as ‘linear syringomatous hamartoma’ was present at birth.
1288 It was characterized by syringoma-like glands in a desmoplastic stroma. The authors regarded it as a variant of eccrine nevus.
1288 An eccrine nevus has been reported in association with a clear cell syringoma.
1289 The clear cell variant of syringoma has been associated clinically with diabetes mellitus in many instances.
1290. and 1291. Syringomas appear to be more common in patients with Down syndrome.
1279.1292. and 1293. Their occurrence adjacent to a basal cell carcinoma is probably fortuitous.
1294 They are regarded as probable apocrine tumors by Ackerman and colleagues.
651In an award-winning Poster Display at The American Society of Dermatopathology Meeting in San Francisco (October, 2008), Chandler presented evidence that eruptive syringomas result from autoimmune destruction of dermal eccrine ducts. Eruptive syringomas have also developed in an area of skin subject to waxing.
1295 The lesions were thought to have resulted from reactive inflammation in the dermis.
1295As syringomas are benign and usually asymptomatic, treatment is usually offered for cosmetic reasons. Treatment options include surgical excision, dermabrasion, cryotherapy, electrodesiccation with curettage, and carbon dioxide laser, with or without trichloroacetic acid.
1296 Topical retinoids have also been used. Oral tranilast, an anthranilic acid derivative, has been used with some success to treat multiple syringomas.
1264
Histopathology854
Syringomas are dermal tumors composed of multiple small ducts lined usually by two layers of cuboidal epithelium (
Fig. 33.46). Sometimes, the ducts have a comma-like tail resembling those seen in desmoplastic trichoepithelioma. Solid nests and strands of cells, sometimes having a basaloid appearance, may be present. Some ducts are dilated and contain eosinophilic material. There is usually a dense fibrous stroma.
In the clear cell variant the ducts are lined by larger epithelial cells with pale or clear cytoplasm (
Fig. 33.47).
1290. and 1297. This clear cell change may involve only part of the tumor or be limited to the cells adjacent to the duct lumina. The clear cells contain abundant glycogen. It is regarded as a ‘metabolic’ variant of the conventional syringoma.
1298Staining with monoclonal anti-eccrine gland antibodies has shown positivity for EKH-6 (eccrine secretory and ductal structures), but the tumor cells are negative for EKH-5 and SKH1, which label eccrine secretory elements.
1170. and 1306. The cytokeratin pattern has been further characterized: the cells express CK1/5/10/11/19, and CK14 variably.
1298. and 1307. Syringomas usually contain CEA, whereas desmoplastic trichoepitheliomas do not.
111 Ferritin is present in the outermost layer of cells in the epithelial elements of the syringoma, in a similar pattern to that seen in normal eccrine ducts.
1171 Progesterone receptors are expressed in most syringomas, supporting the view that they are under hormonal control.
1308. and 1309. They were not expressed in all 15 cases on the vulva, in one series.
1296
Electron microscopy
There are numerous microvilli on the cells bordering the lumina and a band of periluminal tonofilaments.
851. and 1306. Intracytoplasmic lumen formation and keratohyaline granules in luminal cells have also been reported.
851
ECCRINE MIXED TUMOR
Eccrine mixed tumor (chondroid syringoma, eccrine type) is a rare tumor which was formerly included with the apocrine variant of mixed tumor as a chondroid syringoma. It is found as a solitary, slowly growing nodule on the head or extremities of the middle-aged and elderly. Local recurrence occurs only if the tumor is incompletely excised.
Histopathology
The eccrine mixed tumor has small, non-branching ducts resembling a syringoma, set in a myxoid and cartilaginous stroma (
Fig. 33.48). The tumor is well circumscribed and may extend into the subcutis. The cells express CEA and cytokeratin.
POROMA GROUP
Here the poroma group will be subdivided into eccrine poroma, dermal duct tumor (dermal eccrine poroma), hidroacanthoma simplex (intraepidermal poroma), syringoacanthoma, and syringofibroadenoma. Lesions have been described which contained two or three histologically distinct components: hidroacanthoma simplex, eccrine poroma, and dermal duct tumor.
1312. and 1313.Ackerman believes that some poromas are of apocrine origin, but they can only be inferred to be apocrine on the basis of a connection of one or more elongated tubules to infundibula.
651
ECCRINE POROMA
Eccrine poroma, a tumor derived from the acrosyringium, presents as a solitary, pink or red exophytic nodule, usually on plantar or palmar skin.
1313.1314. and 1315. It may also be found elsewhere on the lower extremities and hands, and at times on any other area of the body with sweat glands.
1316 It has also arisen on a burn site.
1317 Some of the lesions on the head and neck may be of apocrine origin.
1318 It usually arises in middle-aged or elderly people. Rare cases have been reported in childhood,
1319 including one congenital case.
1320 A rapidly growing eccrine poroma of the nose has been reported in a pregnant woman.
1321 Multiple lesions (eccrine poromatosis) are very rare and some purported cases appear to be examples of acrosyringeal nevus.
1224. and 1322. Origin in an area of chronic radiation dermatitis has been documented.
1323.1324. and 1325. In another patient, multiple poromas arose in the setting of total body irradiation and immunosuppression with allogeneic bone marrow transplantation for the treatment of acute lymphocytic leukemia.
1326
Histopathology1327
Eccrine poroma is a circumscribed tumor composed of cords and broad columns of uniform basaloid cells extending into the dermis from the undersurface of the epidermis (
Fig. 33.49). The cells are smaller than, and well delineated from, the epidermal cells with which they are in contact. They are PAS positive and much of the PAS-positive material is diastase sensitive. Melanin pigment is sometimes present and is sometimes visible clinically.
1328. and 1329. Several mechanisms have been proposed for the melanocyte colonization of various appendageal tumors.
1330.1331. and 1332. Increased endothelin-1 expression is one of these proposed mechanisms.
1333 Ducts and, less commonly, small cysts may also be seen within the tumor columns. The stroma is usually richly vascular with some telangiectatic vessels, contributing to the clinical appearance.
Uncommonly, divergent adnexal differentiation is seen in eccrine poromas, with focal sebaceous, pilar and possibly apocrine secretory differentiation.
1334 On the basis of the common embryological origin of follicular, sebaceous and apocrine structures, it has been suggested that some ‘eccrine’ poromas might be of apocrine origin (see above).
1318. and 1334. Keratins 1 and 10 are expressed in the tumor nests.
1335
Electron microscopy
The tumor cells have numerous connecting desmosomes, cytoplasmic tonofilaments, glycogen granules, and intracytoplasmic lumina. The latter appear to coalesce to form larger intercellular duct-like structures resembling embryonic intraepidermal sweat ducts.
1336
DERMAL DUCT TUMOR
The term ‘dermal duct tumor’ was coined by Winkelmann and McLeod in 1966 for a tumor composed of basaloid cells, like an eccrine poroma, but located in the dermis.
1337 Relatively few cases have been reported since, and this may simply reflect the hesitation of some dermatopathologists to make the diagnosis, particularly when multiple sections through a presumed dermal duct tumor often show a connection with the undersurface of the epidermis or areas resembling solid–cystic hidradenoma (poroid hidradenoma). Clinically, dermal duct tumor presents as a firm papule, plaque or nodule, particularly on the lower limbs or head and neck region.
1338 They measure from a few millimeters to several centimeters in diameter.
1339
Histopathology
The tumor is composed of islands of basaloid tumor cells within the dermis. Duct-like structures are prominent in many of the nests, and the tumor may itself connect with a normal-appearing eccrine duct. An epidermal connection is often found if multiple sections are examined.
1340 The cells are PAS positive, but this is usually not as striking as in eccrine poroma. Melanin pigment is sometimes present.
In the clear cell variant, there are multiple solid aggregations of clear cells involving the dermis. Ductal structures are also present.
1339 The neoplastic cells are immunoreactive for MNF116 and AE1/AE3
cytokeratins, but not for CAM5.2 or CK7. 1339 EMA, CEA, and GCDFP-15 decorate the ductal structures but not the neoplastic cells.
1339
Electron microscopy
The tumor is composed of clear, dark and luminal cells, the clear cell being predominant.
1338 There are microvilli on the luminal side of the cells lining the ducts within the cell nests.
HIDROACANTHOMA SIMPLEX
Hidroacanthoma simplex (intraepidermal eccrine poroma) is a solitary plaque or nodular lesion found particularly on the extremities and the trunk.
1341.1342. and 1343. It arises throughout adult life and there is an equal sex incidence. Clinically, it resembles a seborrheic keratosis or basal cell carcinoma. Many of the cases reported in the literature as hidroacanthoma simplex are probably examples of the clonal variant of seborrheic keratosis;
1344 two cases purporting to be hidroacanthoma simplex with porocarcinomatous transformation
1345. and 1346. are possibly other entities.
1347 A pigmented malignant hidroacanthoma simplex arising in a benign variant has been reported. Although mimicking seborrheic keratosis, the lesion was thought to be a distinctive entity.
1348 Such tumors are best called porocarcinoma in situ.
1349. and 1350.
Histopathology
Hidroacanthoma simplex is composed of well-circumscribed nests of cuboidal to oval basaloid cells within the epidermis, resembling those seen in eccrine poroma (
Fig. 33.50). The cells are smaller than neighboring epidermal keratinocytes and they contain some glycogen. Rarely, melanin pigment is present.
1351 Reports that mention squamous and spindle-cell variants
1342 probably included seborrheic keratoses.
1347A few ductal structures may sometimes be seen within the islands. The epidermis is usually acanthotic with some overlying hyperkeratosis. A dermal component resembling solid–cystic hidradenoma is sometimes present.
One immunohistochemical study has shown that the tumor cells stain intensely with antikeratin antibodies (using an AE1/AE3 cytokeratin ‘cocktail’), but not with CEA, which did stain adjacent normal acrosyringium.
1352 On the basis of these findings and the electron microscopy (see below) the authors concluded that the case studied ‘did not appear to arise from or differentiate toward the luminal cells of the acrosyringium’.
1352 Hidroacanthoma simplex may arise from the
outer cells of the intraepidermal eccrine duct. In another immunohistochemical study of hidroacanthoma simplex, the tumor cells expressed CK14 and CK17. There was an absence of CK1, CK5, and CK8.
1353Seborrheic keratoses of the clonal type may have islands of cells within the epidermis, the so-called ‘Jadassohn phenomenon’. However, the cells in seborrheic keratoses are not as small and basaloid and do not contain glycogen. Both clonal seborrheic keratoses and hidroacanthoma simplex have a similar expression of cytokeratin, although there are more Langerhans cells in clonal seborrheic keratoses than in hidroacanthoma simplex.
1354 Another tumor that shows the Jadassohn phenomenon of intraepidermal nesting is the so-called ‘intraepidermal epithelioma of Jadassohn’.
1355 This is perhaps the most controversial entity in dermatopathology.
1356 It is assumed to be derived from acrosyringeal keratinocytes (as are hidroacanthoma simplex, acrosyringeal nevus and syringofibroadenoma). Many believe that this entity is heterogeneous and includes seborrheic keratoses with atypia or bowenoid transformation, and variants of intraepidermal carcinoma.
1347. and 1357. It has not been mentioned in the recent literature.
The reported case of malignant hidroacanthoma simplex was composed of an intraepidermal proliferation of atypical polygonal poroid cells forming large, sharply demarcated nests with colonization of dendritic melanocytes.
1348 The intracytoplasmic lumina of the ductal structures expressed both EMA and CEA.
1348
Electron microscopy
The tumor cells contain few desmosomes, decreased numbers of tonofilaments, and abundant cytoplasmic glycogen.
1352
SYRINGOACANTHOMA
Syringoacanthoma was described by Rahbari in 1984 as a tumor derived from the acrosyringium.
1358 It is said to differ from hidroacanthoma simplex by the presence of prominent acanthosis and papillomatosis, and less orderly intraepidermal nests. Steffen and Ackerman believe that it is not a distinct entity.
1347
SYRINGOFIBROADENOMA
It has been suggested
1227 that eccrine syringofibroadenoma, as described by Mascaro in 1963,
1229 is identical to the acrosyringeal nevus of Weedon and Lewis
1224 (see
eccrine hamartomas, p. 795), but there do appear to be some clinicopathological differences. 1231 The eccrine syringofibroadenoma presents as a solitary, often large, hyperkeratotic nodular lesion with a predilection for the extremities.1227.1230.1359.1360. and 1361. Rare sites of involvement include the nail apparatus, 1362 and an organoid nevus. 1363 This term has also been applied to diffuse, zosteriform and multiple lesions resembling the case reported as an acrosyringeal nevus. 1364 One such case produced diffuse plantar hyperkeratosis. It may have been the result of a postsomatic mutation in the early embryonic stage. 1365 The terms ‘acrosyringeal adenomatosis’ and ‘eccrine syringofibroadenomatosis (Mascaro)’ have been suggested as appropriate designations for the more diffuse cases.2.1366.1367. and 1368. Eccrine syringofibroadenomatosis can occur in association with hidrotic ectodermal dysplasia.1369. and 1370. It has been said that the lesions are somewhat different in the two variants, Clouston’s syndrome and the Schöpf syndrome. HPV-10 has been detected in the lesions occurring in Clouston’s syndrome. 1371 Eccrine syringofibroadenomatosis has also been reported in a familial setting in association with ophthalmological abnormalities. 1372The occurrence of this phenomenon next to inflammatory dermatoses and tumors, often in an acral location, has been called
reactive eccrine syringofibroadenomatosis. As stromal changes are minimal in some instances, ‘eccrine duct hyperplasia’ would be a better term. This reactive change has been seen next to palmoplantar erosive lichen planus,
1373 bullous pemphigoid,
1374 a burn scar,
1375 pincer nail,
1376 an ileostomy stoma,
1377. and 1378. squamous cell carcinoma,
1379.1380. and 1381. and a chronic
diabetic foot ulcer. 1382 It has also occurred as multiple lesions on the foot in a patient with previous lepromatous leprosy.
1383It may present as a ‘mossy’ leg, resembling lymphedematous keratoderma.
1384 A polyp of the uterine cervix with eccrine syringofibroadenoma features has also been reported.
1385 The various subtypes of this condition are listed in
Table 33.1.
Table 33.1 Subtypes of eccrine syringofibroadenoma (ESFA)
|
• Multiple (diffuse, zosteriform, linear) ESFA (eccrine syringofibroadenomatosis) |
• Multiple ESFA with hidrotic ectodermal dysplasia |
• Familial ESFA with ophthalmological abnormalities |
• Reactive ESFA (reactive eccrine syringofibroadenomatosis) associated with inflammatory or neoplastic disorders including lichen planus, bullous pemphigoid, burn scar, pincer nail, ileostomy stoma, diabetic foot ulcer, leprosy, mossy foot, squamous cell carcinoma |
There are thin anastomosing epithelial cords and strands forming a lattice and connected to the undersurface of the epidermis (
Fig. 33.51). The cells are smaller and more basophilic than the epidermal keratinocytes. Nests of clear cells have been reported.
1387.1388. and 1389. Ducts are present within the tumor. Between the strands there is a rich fibrovascular stroma. The tumor does not have the strong PAS positivity or the abundant stromal plasma cells noted in the case reported as an acrosyringeal nevus.
1224 However, plasma cells have not been a feature in many of the other multiple/diffuse cases. In one of the reactive peristomal cases there was the formation of hybrid epidermal–colonic mucosa glandular structures, intraepidermal areas of sebaceous differentiation, and the induction of hair follicles.
1378The term ‘
syringofibrocarcinoma’ has been suggested for a single case in which a net-like arrangement of cytologically malignant cells showing ductal differentiation impinged on a benign component.
1390 Subsequently, other cases have been reported in which carcinomatous transformation of syringofibroadenoma was thought to have occurred rather than the development of this entity as a reactive phenomenon in a pre-existing carcinoma.
1391. and 1392. One of the patients had ectodermal dysplasia.
1392Immunohistochemical studies have not shown consistent results with respect to cytokeratin expression, but they seem to indicate differentiation toward the acrosyringium and dermal duct.
1359.1393.1394.1395.1396. and 1397. Expression of CK19 is regarded as indicating ductal differentiation, whereas the presence of CK1, involucrin, and filaggrin represents acrosyringeal differentiation.
1389 One clear cell variant expressed GCDFP-15, but not estrogen or progesterone receptors.
1389
Electron microscopy
Intracellular duct formation, characteristic of developing acrosyringia, has been observed. Abundant glycogen is also present.
1393. and 1398.
HIDRADENOMA
Considerable confusion exists in the literature concerning the most appropriate designation for this tumor. Wilson-Jones has called it a ‘nosological jungle’.
756 Terms used have included ‘solid–cystic hidradenoma’,
746‘eccrine acrospiroma’,
745‘clear cell hidradenoma’,
1111‘eccrine sweat gland adenoma’,
1399 and ‘clear cell myoepithelioma’.
1400 This topic is further confused by the separation of apocrine hidradenomas from a less common eccrine group, which has also been called poroid hidradenoma. The apocrine group (apocrine hidradenoma) are composed of clear, polygonal and mucinous cells while the eccrine lesion (hidradenoma, poroid hidradenoma) consists of poroid and cuticular cells.
758. and 1401.
HIDRADENOMA (POROID HIDRADENOMA)
Hidradenoma refers to a tumor that includes both an eccrine and apocrine variant. The two are clinically indistinguishable. Both usually present as a solitary, solid or partially cystic nodule with a slight preponderance in females of middle age,
1111 but with no site predilection. A case presenting on the vulva has been reported.
1402 Cases occurring in children are rare.
1403. and 1404. Hidradenoma averages 1–3 cm in diameter, but larger variants up to 6 cm or more in diameter have been recorded.
763.1313. and 1405. The behavior of the eccrine variant is probably no different from that of the apocrine variant (see
p. 782).
Histopathology
Despite their identical clinical appearances, the eccrine variant of hidradenoma differs histologically from the apocrine hidradenoma. Eccrine hidradenoma (poroid hidradenoma) is a circumscribed, non-encapsulated dermal tumor composed of poroid and cuticular cells.
1313 Ductal structures may form, particularly within the zones of cuticular cells. It has the architectural features of the apocrine hidradenoma but with the cytological characteristics of a poroma.
1310 It lacks the polygonal, clear, and mucinous cells of the apocrine variant,
651 although cases purported to be eccrine variants have contained clear cells.
1313 Sebaceous differentiation has been described,
819 again calling into question the eccrine origin of such cases.
MALIGNANT ECCRINE TUMORS
The classification of malignant eccrine tumors is one of the most confusing areas of dermatopathology, with identical tumors reported in the literature under three or more designations. Furthermore, tumors with heterogeneous histological features that defy classification are common. Some earlier reports give scant histological details, precluding reassessment.
1406 It is tempting to suggest that the term ‘eccrine carcinoma’ be applied to all malignant eccrine tumors, in much the same way that the term ‘basal cell carcinoma’ has proved adequate for a histologically diverse group of tumors.
In his major review of sweat gland carcinomas, Santa Cruz lists the synonyms and related terms used for each of the tumors he describes.
692 He also suggests a classification based on the malignant potential as currently understood. Some of the ductal adenocarcinomas are high-grade tumors. The malignant eccrine poroma is of intermediate
malignancy. The low-malignancy group includes microcystic adnexal carcinoma and eccrine epithelioma. 692 If there is any doubt about the potential malignancy of a sweat gland tumor, aneuploidy detected by DNA image cytometry is a clear and specific indicator of prospective malignancy.
1407The high frequency of positive sentinel lymph nodes in two small series of sweat gland carcinomas of different types, suggests that sentinel lymph node biopsy is a useful staging tool for patients with sweat gland carcinomas.
1408. and 1409. Further studies are needed to determine if this information leads to a survival benefit by selecting a group suitable for regional lymphadenectomy.
1409Rarely, a pleomorphic or mixed sarcoma may arise in a sweat gland adenoma, or de novo, that is assumed to be derived from the myoepithelial elements of eccrine tumors. Recognition of an underlying benign tumor is usually necessary to make the diagnosis.
1410 The carcinosarcoma of scrotal skin reported by Lin et al was composed of mucin-producing adenocarcinoma and solid spindle-cell sarcoma.
1070There has been much interest in assessing the various immunohistochemical markers of the eccrine carcinomas. In a study of 32 cases, epithelial membrane antigen and cytokeratin were present in all cases, CEA was detected in 25, and S100 in 19 cases.
1411 Diffuse staining for ferritin is another marker of eccrine carcinomas.
1171 Whereas cutaneous metastases from internal adenocarcinomas do not usually express p53 or CK 5/6, the majority of primary adnexal carcinomas (but not all apocrine carcinomas) strongly express p53 and CK5/6.
1412.1413. and 1414. D2-40, a monoclonal antibody to a component of podoplanin, which is highly expressed in lymphatic endothelium, is also a highly sensitive and specific marker that distinguishes primary skin adnexal carcinomas from adenocarcinomas metastatic to the skin.
309 The metastatic tumors do not express podoplanin.
309A study of genetic and other markers in a mixed group of 21 sweat gland carcinomas found loss of heterozygosity (LOH), confined mostly to chromosome arm 17p, in four cases.
1415 There was only a low frequency of p53 alterations.
MICROCYSTIC ADNEXAL CARCINOMA
Microcystic adnexal carcinoma was first reported in 1982 by Goldstein and colleagues.
1416 It has also been referred to as malignant syringoma,
1417 sweat gland carcinoma with syringomatous features,
1418 and sclerosing sweat duct carcinoma.
1419Syringomatous adenoma of the nipple is a closely related entity that is distinct from erosive adenomatosis (papillary adenoma) of the nipple.
1420. and 1421.Microcystic adnexal carcinoma is a slowly growing, locally aggressive tumor which presents as an indurated plaque or nodule, usually on the upper lip or elsewhere on the face.
2.1422.1423.1424. and 1425. It has a predilection for the left side of the face, thought to be related to sun exposure while driving.
1426 However, this hypothesis has not been tested, as the series of 44 cases reported from Australia (where driving is on the opposite side of the road to the United States) did not have the appropriate data to allow this comparison to be made.
1427 It may also be found in the axilla,
1428 the extremities,
1429 genital skin,
1430 and on the trunk and scalp.
1431.1432. and 1433. It affects adults of all ages. Childhood cases are rare.
1427. and 1434. It has been reported in all races, but it appears to be uncommon in African Americans.
1435 In several cases the patient has previously received radiotherapy for adolescent acne or cancers.
1419.1433.1436.1437.1438. and 1439. Other associations have included an underlying ‘systematized compound epithelial nevus’,
1440 and a primary immunodeficiency syndrome.
1441Local recurrence occurs in nearly 50% of cases,
1419 but this is much less likely if the excision margins are free of tumor in the initial excision.
1439 This can be achieved with Mohs micrographic surgery. The recurrence rate has been calculated as 1.98% per patient-year.
1426 Several cases have now been reported of a high (nuclear) grade adnexal carcinoma with features of a microcystic adnexal carcinoma that produced distant metastases.
1442 This is in addition to rare cases with lymph node involvement, almost certainly due to in-continuity extension.
1419 Mandibular bone marrow involvement occurred in a tumor of the mental region.
1443 Microcystic adnexal carcinoma is included within the spectrum of
locally aggressive adnexal carcinomas.
1444 It is regarded by many as an apocrine tumor.
651As already stated, the treatment of choice is Mohs micrographic surgical technique. If the diagnosis is made early, and if the anatomic location is accessible to excision by this technique, a favorable outcome can be expected.
1445 On the basis of several cases reported in the literature, it has been suggested that not only is radiotherapy an ineffective treatment, but this modality may induce conversion to a clinically and histologically less favorable tumor.
1445 In the series of 44 cases from Australia, treated by experienced Mohs surgeons, there was only one case of recurrence (5%) out of 20 patients who had completed a 5-year follow-up period after the Mohs surgery.
1427 Most of the cases (90.9%) in that series were on the head and neck and 31.8% of the tumors had already recurred prior to the Mohs surgery.
1427
Histopathology692. and 1419.
The tumor usually involves the subcutis as well as the dermis, and it may extend into the underlying muscle. There is some stratification of the various histological changes.
692 The superficial part is composed of numerous keratinous cysts and small islands and strands of basaloid and squamous epithelium showing variable ductal differentiation. Focal clear cell change may be present in some of the cells. Rarely, this is a prominent feature.
1446 Sebaceous differentiation has also been reported.
1444. and 1447.The deeper component has smaller nests and strands of cells in a dense, hyalinized stroma, giving a scirrhous appearance (
Fig. 33.52). Perineural invasion is frequently present.
1448. and 1449. The aggressive growth and perineural spread allow a distinction to be made from syringoma and desmoplastic trichoepithelioma.
Accurate examination of frozen tissue specimens of this tumor can be a challenging task. Staining with toluidine blue may provide additional help for detecting single cells, small nests, and perineural involvement on frozen sections.
1450 Normal nerve bundles have a light blue hue, whereas nerves with tumor involvement are colored magenta.
1450The microcystic adnexal carcinoma has become an expanding ‘entity’ with the inclusion of tumors that bear no relationship to those originally described. Cases of eccrine (syringoid) carcinoma are sometimes included. Notwithstanding these comments, there does appear to be a rare high-grade variant of microcystic adnexal carcinoma with marked nuclear pleomorphism and hyperchromasia, with squamous pearl formation and a widespread strong p53 immunoreaction.
1442The
solid carcinoma, previously included with either the eccrine (syringoid) carcinoma or the microcystic adnexal carcinoma, has now been regarded as a discrete entity by Requena, Kiryu, and Ackerman.
651 It consists of numerous small nests and strands of cells having a predominantly solid pattern. There is stromal desmoplasia and perineural spread.
In some of the reported cases of microcystic adnexal carcinoma the luminal cells have been CEA positive.
1423.1443. and 1451. The cells stain for epithelial membrane antigen and various keratins, particularly CK7.
1452.1453. and 1454. Ber-EP4 was not expressed in any of the 13 cases studied in one series, whereas all 28 cases of basal cell carcinoma expressed this marker.
1455 Some S100-positive cells are also present.
1456 S100A6 is expressed in more than half of the cases.
1457 The low level of Ki-67 supports a low proliferative rate.
1454 It has been suggested, on the basis of the electron microscopy and the immunophenotype, that microcystic adnexal carcinoma expresses both eccrine and pilar differentiation, but this is at variance with other views that it is of apocrine type.