Introduction810
INTRODUCTION
Dermal dendrocytes
Myofibroblasts
ACRAL ANGIOFIBROMAS
ADENOMA SEBACEUM
Histopathology32. and 70.
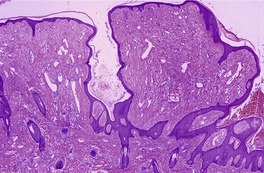
Fig. 34.1
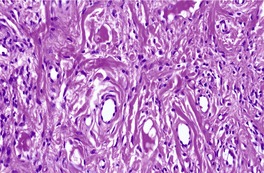
Fig. 34.2
Electron microscopy
ANGIOFIBROMAS IN OTHER SYNDROMES
FIBROUS PAPULE OF THE FACE
Histopathology32.70.86. and 87.
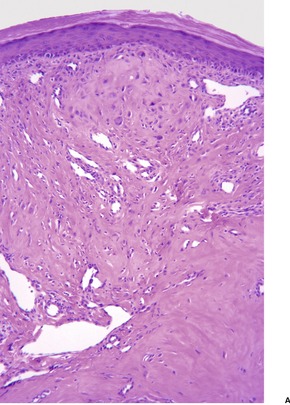
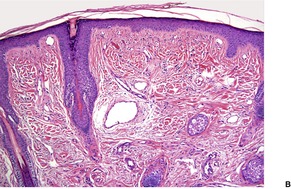
Fig. 34.3
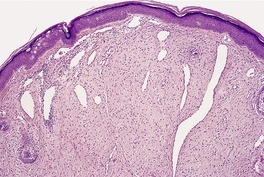
Fig. 34.4
PEARLY PENILE PAPULES
Histopathology31
ACRAL FIBROKERATOMA
Histopathology
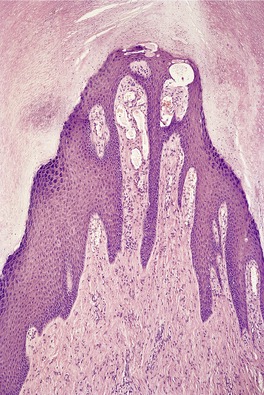
Fig. 34.5
FAMILIAL MYXOVASCULAR FIBROMA
FIBROUS OVERGROWTHS, FIBROMATOSES, MYOFIBROBLASTIC PROLIFERATIONS, AND FIBROSARCOMA
SKIN TAGS
Histopathology
PREPUBERTAL VULVAL FIBROMA (ASYMMETRIC LABIUM MAJUS ENLARGEMENT)
Histopathology
GARDNER FIBROMA
Histopathology
PLEOMORPHIC FIBROMA
Histopathology
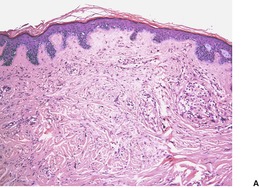
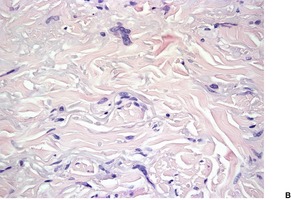
Fig. 34.6
SCLEROTIC FIBROMA (STORIFORM COLLAGENOMA)
Histopathology
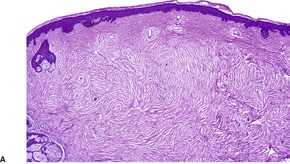
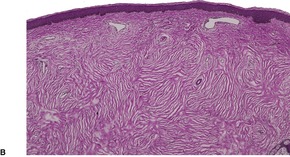
Fig. 34.7
COLLAGENOUS FIBROMA (DESMOPLASTIC FIBROBLASTOMA)
Histopathology
KNUCKLE PADS
Histopathology256
PACHYDERMODACTYLY
Histopathology
NODULAR FASCIITIS
Histopathology272.294. and 295.
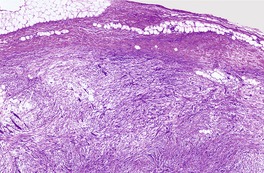
Fig. 34.8
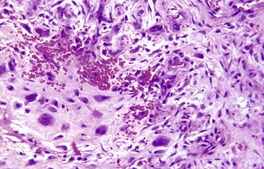
Fig. 34.9
Electron microscopy
ATYPICAL DECUBITAL FIBROPLASIA (ISCHEMIC FASCIITIS)
POSTOPERATIVE SPINDLE-CELL NODULE
SOLITARY FIBROUS TUMOR
Histopathology
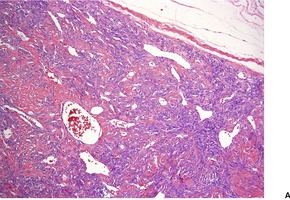
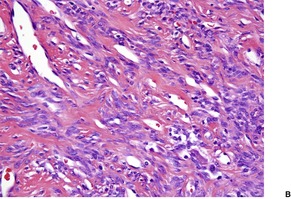
Fig. 34.10
FIBROUS HAMARTOMA OF INFANCY
Histopathology334. and 346.
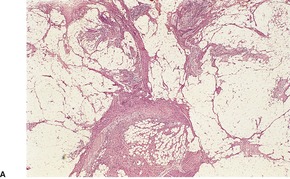
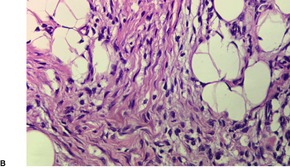
Fig. 34.11
DIGITAL FIBROMATOSIS OF CHILDHOOD
Histopathology357.359. and 375.
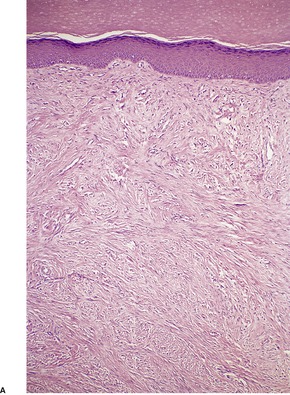
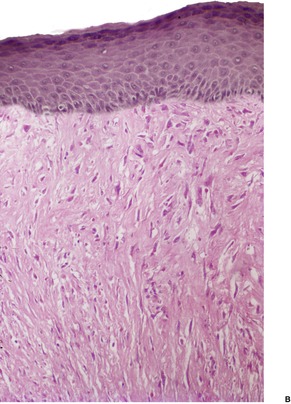
Fig. 34.12
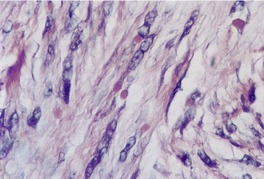
Fig. 34.13
Electron microscopy
ANGIOFIBROBLASTOMA
ANGIOMYOFIBROBLASTOMA OF THE VULVA
Histopathology392. and 394.
CELLULAR ANGIOFIBROMA
Histopathology
DERMATOMYOFIBROMA
Histopathology415
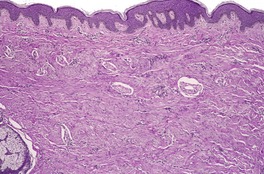
Fig. 34.14
INFANTILE MYOFIBROMATOSIS
Histopathology419. and 424.
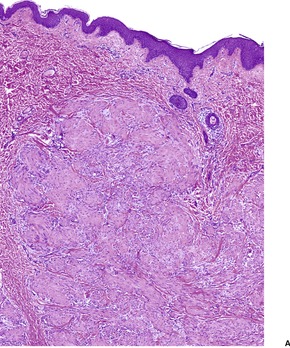
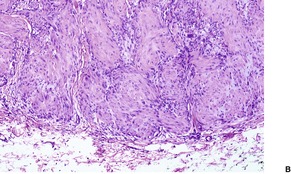
Fig. 34.15
Electron microscopy
PERIVASCULAR MYOMAS AND RELATED ENTITIES
Myopericytoma
Histopathology
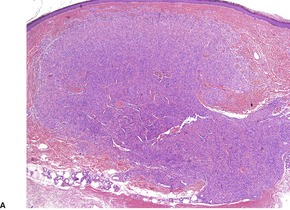
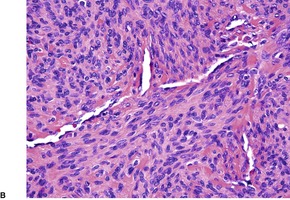
Fig. 34.16
Fig. 34.17
PEComa
Histopathology
Clear cell myomelanocytic tumor
Histopathology
Distinctive dermal clear cell mesenchymal neoplasm
INFLAMMATORY MYOFIBROBLASTIC TUMOR
Histopathology
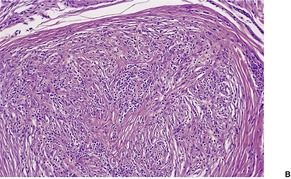
Fig. 34.18
CUTANEOUS MYXOID FIBROBLASTOMA
FIBROMYXOID SARCOMA (LOW GRADE)
Histopathology498. and 499.
MYXOFIBROSARCOMA
Histopathology
MYXOINFLAMMATORY FIBROBLASTIC SARCOMA
Histopathology
FIBROSARCOMA
Histopathology532. and 544.
MYOFIBROSARCOMA
Histopathology
FIBROHISTIOCYTIC TUMORS
DERMATOFIBROMA (FIBROUS HISTIOCYTOMA)
Histopathology552.554. and 555.
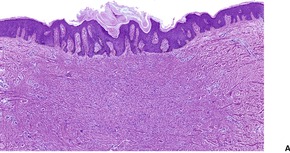
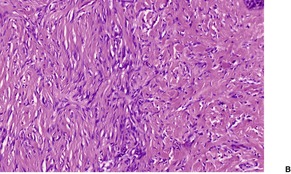
Fig. 34.19
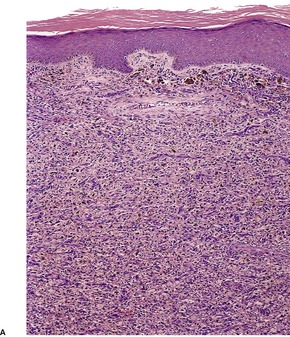
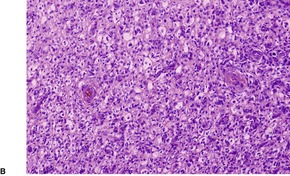
Fig. 34.20
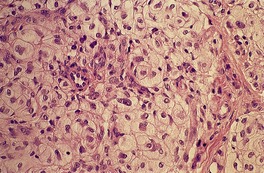
Fig. 34.21
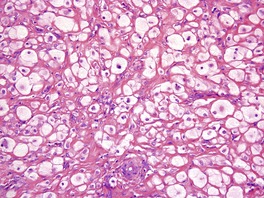
Fig. 34.22
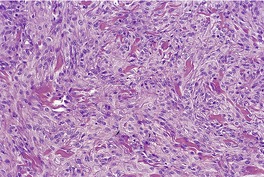
Fig. 34.23
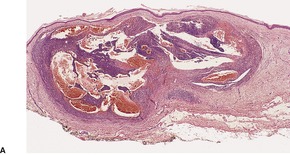
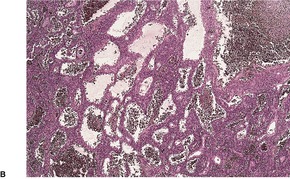
Fig. 34.24
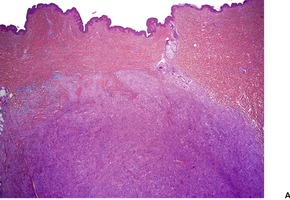
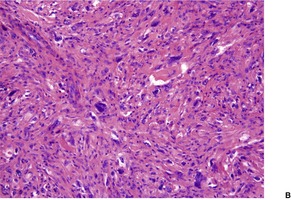
Fig. 34.25
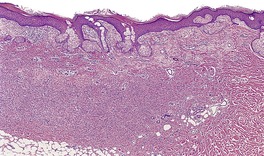
Fig. 34.26
Electron microscopy
EPITHELIOID CELL HISTIOCYTOMA
Histopathology
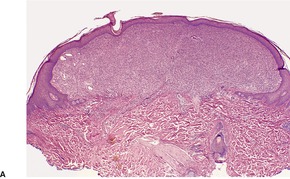
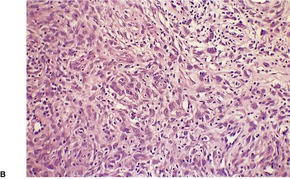
Fig. 34.27
PLEXIFORM FIBROHISTIOCYTIC TUMOR
Histopathology745
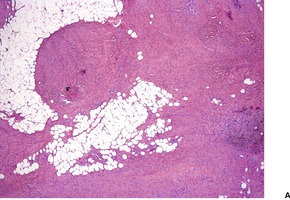
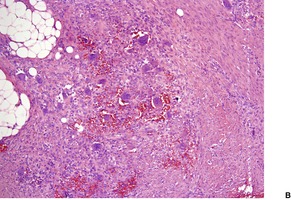
Fig. 34.28
Electron microscopy
GIANT CELL FIBROBLASTOMA
Histopathology753.755.767.768.769. and 770.
DERMATOFIBROSARCOMA PROTUBERANS
Treatment of dermatofibrosarcoma protuberans
Histopathology777
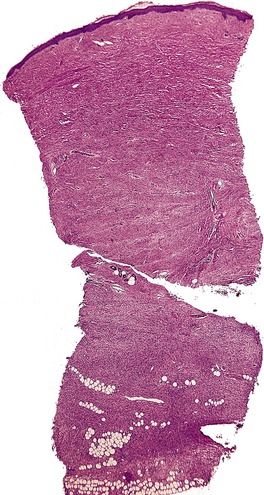
Fig. 34.29
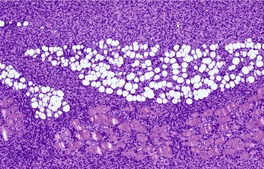
Fig. 34.30
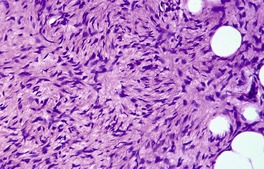
Fig. 34.31
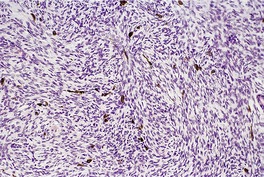
Fig. 34.32
Electron microscopy
ATYPICAL FIBROXANTHOMA
Histopathology934
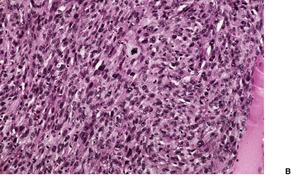
Fig. 34.33
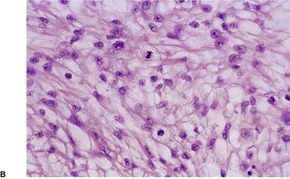
Fig. 34.34
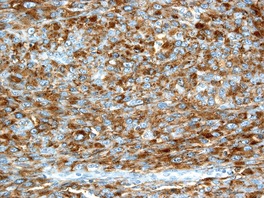
Fig. 34.35
Electron microscopy
ANGIOMATOID FIBROUS HISTIOCYTOMA
Histopathology
MALIGNANT FIBROUS HISTIOCYTOMA (PLEOMORPHIC UNDIFFERENTIATED SARCOMA)
Histopathology
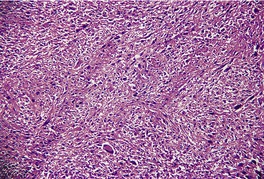
Fig. 34.36
GIANT CELL TUMOR OF SOFT TISSUE
Histopathology
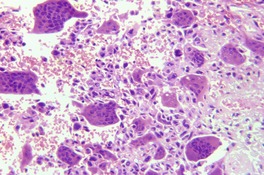
Fig. 34.37
PRESUMPTIVE SYNOVIAL AND TENDON SHEATH TUMORS
FIBROMA OF TENDON SHEATH
Histopathology1081. and 1085.
GIANT CELL TUMOR OF TENDON SHEATH
Histopathology1091. and 1100.
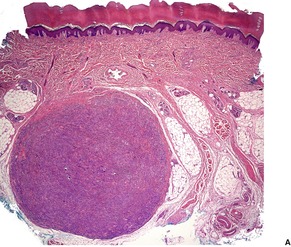
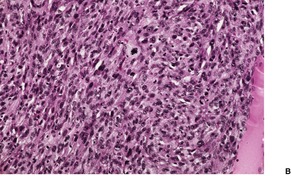
Fig. 34.38
Electron microscopy
EPITHELIOID SARCOMA
Histopathology1104. and 1106.
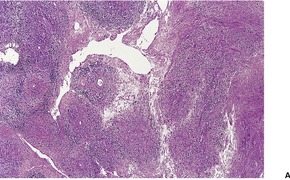
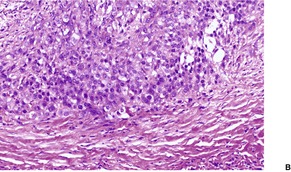
Fig. 34.39
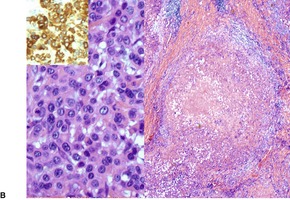
Fig. 34.40
Electron microscopy
SYNOVIAL SARCOMA
Histopathology
MISCELLANEOUS ENTITIES
FIBRO-OSSEOUS PSEUDOTUMOR OF THE DIGITS
OSSIFYING FIBROMYXOID TUMOR
Histopathology
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



Tumors and tumor-like proliferations of fibrous and related tissues
Fibrous overgrowths, fibromatoses, myofibroblastic proliferations, and fibrosarcoma813
Skin tags814
Prepubertal vulval fibroma (asymmetric labium majus enlargement)815
Gardner fibroma815
Pleomorphic fibroma815
Sclerotic fibroma (storiform collagenoma)815
Collagenous fibroma (desmoplastic fibroblastoma)816
Knuckle pads816
Pachydermodactyly817
Nodular fasciitis817
Atypical decubital fibroplasia (ischemic fasciitis)818
Postoperative spindle-cell nodule818
Solitary fibrous tumor818
Fibrous hamartoma of infancy818
Digital fibromatosis of childhood819
Angiofibroblastoma819
Angiomyofibroblastoma of the vulva820
Cellular angiofibroma821
Dermatomyofibroma821
Infantile myofibromatosis821
Inflammatory myofibroblastic tumor824
Cutaneous myxoid fibroblastoma825
Fibromyxoid sarcoma (low grade)825
Myxofibrosarcoma825
Myxoinflammatory fibroblastic sarcoma825
Fibrosarcoma826
Myofibrosarcoma826
Fibrohistiocytic tumors827
Dermatofibroma (fibrous histiocytoma)827
Epithelioid cell histiocytoma831
Plexiform fibrohistiocytic tumor831
Giant cell fibroblastoma832
Dermatofibrosarcoma protuberans833
Atypical fibroxanthoma835
Angiomatoid fibrous histiocytoma837
Malignant fibrous histiocytoma (pleomorphic undifferentiated sarcoma)838
Giant cell tumor of soft tissue839
Miscellaneous entities842
Fibro-osseous pseudotumor of the digits842
Ossifying fibromyxoid tumor842
Fibroadenoma843
Nodular fibrosis in elephantiasis843
Juvenile hyaline fibromatosis843
Multifocal fibrosclerosis843
Mesenchymal hamartoma843
Pleomorphic hyalinizing angiectatic tumor843
Phosphaturic mesenchymal tumor843
Cutaneous myxoma844
Superficial acral fibromyxoma844
Superficial angiomyxoma844
Recent immunohistochemical findings have assisted in the histogenetic classification of many of the soft tissue tumors. 1 However, as vimentin is positive in many tumors, it is of no value in the differential diagnosis of tumors discussed in this chapter. 2 However the ‘fibrohistiocytic’ tumors are still largely enigmatic with respect to their histogenesis, and the diagnosis is largely dependent on H & E inspection. 3 In addition to the ‘fibrohistiocytic’ tumors, this chapter includes those entities that have traditionally been grouped together on the basis of collagen production and/or the presence of fibroblasts or fibroblast-like cells forming an integral component of the tumor. It also includes tumors of presumptive origin from dermal dendrocytes and myofibroblasts.
Dermal dendrocytes are bone marrow-derived cells found in different parts of the dermis.4.5.6. and 7. They are closely related to mast cells in the perivascular space. 7 They express factor XIIIa and von Willebrand factor receptor, suggesting a possible role in tissue repair and hemostasis. An antigen-processing function has also been proposed.8.9. and 10. The subset of dendrocytes expressing factor XIIIa is found in some of the acral angiofibromas and in dermatofibromas. They are also increased in many other situations. Another subset of dendrocytes, comprising 10–30% of all interstitial cells in the reticular dermis, expresses CD34. 11 This antigen is expressed in vascular endothelial cells, some perivascular and interstitial dendritic cells in the dermis, as mentioned above, and spindle cells in the basement membrane zone of eccrine ducts and the bulge area of the hair follicle. It is also expressed in a wide range of tumors. 12 The two subsets of dendrocytes appear to interact in many situations. 13 A hamartoma composed of CD34-positive cells has been reported as a ‘dermal dendrocyte hamartoma’. 14 The cells in the three congenital lesions reported as ‘medallion-like dermal dendrocyte hamartoma’ were positive for CD34, factor XIIIa, and fascin. 15 Subsequent cases have been reported.16. and 17. A granular cell variant of CD34-positive cells, presenting as multiple papules and plaques in a baby, has been called ‘granular cell dendrocytosis’, 18 while a post-traumatic myxoid tumor of the thumb composed of both CD34+ and factor XIIIa+ cells has been called a ‘myxoid dermatofibrohistiocytoma’. 19 Yet another term, ‘CD34-positive eruptive fibroma’, has been used for the multiple papules composed of CD34+ spindle cells reported on the neck and upper chest of a female teenager. 20 The term ‘CD34-reactive myxoid dermal dendrocytoma’ was used for another unique case. 21 Some CD34+ dermal proliferations defy classification. 22 One such case had perivascular spindle cells that formed a dermal ‘fibroma’. 23 There is a lot to be said in favor of a simple diagnosis of ‘CD34+ dendrocytoma’ for all these variants.
Tumors of myofibroblasts are also considered in this chapter. They often coexist with ordinary fibroblasts in many of the lesions. There is still a lack of consensus regarding their exact role in the formation of the soft tissue tumors usually attributed to them. 3 It has even been suggested that myofibroblasts are merely a functional stage of fibroblasts, smooth muscle cells or pericytes, and that their identification requires the ultrastructural recognition of the so-called ‘fibronexus’ (microtendons).24. and 25. As well as an origin from fibroblasts, myofibroblasts have been derived experimentally from microvascular endothelial cells by the action of inflammatory cytokines. 26 Neoplastic myofibroblasts express vimentin, muscle actin, α-smooth muscle actin, and/or desmin, although the specificity of desmin has been questioned. Myofibroblasts are best defined ultrastructurally, as their immunohistochemical profile is not specific. 27
Acral angiofibromas are a clinically diverse group of entities that share distinctive histological features.31. and 32. They are thought by some to represent hyperplasias of the papillary and/or periadnexal dermis (the adventitial dermis). 33 Immunohistochemical studies have shown that the large stellate fibroblast-like cells that characterize these tumors express factor XIIIa.34. and 35. They are not mesenchymally derived fibroblasts. 36 Factor XIIIa appears to be important in the promulgation of fibroplasia. 37
Tumors derived from the perifollicular mesenchyme – the perifollicular fibroma, trichodiscoma and fibrofolliculoma – are usually considered separately from the acral angiofibromas. 38 They are discussed with the tumors of the hair follicle (see p. 771).
The following clinical conditions will be discussed:
• adenoma sebaceum (tuberous sclerosis)
• angiofibromas in other syndromes
• fibrous papule of the face
• pearly penile papules
• acral fibrokeratoma
• familial myxovascular fibroma.
The entity reported as linear papular ectodermal–mesodermal hamartoma has some features of this group. 39
‘Adenoma sebaceum’ is the misnomer (there is no adenomatous proliferation of sebaceous glands as the name implies) used for the angiofibromatous lesions found in most patients with tuberous sclerosis (OMIM 191100), an autosomal dominant neurocutaneous syndrome in which learning disability and epilepsy are often present.40. and 41. Major reviews of the tuberous sclerosis complex have been published in recent years.42. and 43. Other organ systems are often involved.42.44. and 45. Other cutaneous angiofibromatous lesions may accompany adenoma sebaceum, and these include plaque-like lesions of the forehead and scalp and ungual fibromas (seeacral fibrokeratomas on p. 812).46.47. and 48. ‘Shagreen patches’, with the histology of connective tissue nevi, are commonly found in tuberous sclerosis. 49 They are usually present by puberty. 50 Hypopigmented macules are a common finding. 51 Molluscum pendulum is less common. 52 Oral fibromas, mostly gingival in location, and dental pits are common findings in the mouth. 53 Genetic linkage studies initially indicated that about half of all families with tuberous sclerosis showed linkage to chromosome 9q34 (TSC1), and the remainder to chromosome 16p13 (TSC2).54. and 55. Subsequent studies have shown that TSC1 mutations account for only 10–30% of the families identified with tuberous sclerosis complex. In sporadic cases, there is an even greater excess of mutations in TSC2. This latter group is usually associated with more severe disease. 42 No identifiable mutations can be found in 15–20% of patients meeting the clinical criteria of tuberous sclerosis. 42 Hamartin is encoded by TSC1 and tuberin, a tumor suppressor, by TSC2. There is a wide spectrum of mutations. More than 200 TSC1 and nearly 700 TSC2 unique allelic variants have been reported. 42 Approximately two-thirds of all cases are sporadic and assumed to result from new mutations, many of which are in TSC2. 56 Other cases are inherited as an autosomal dominant trait. Mutation screening in tuberous sclerosis is labor intensive and expensive. 57 It is now available commercially. Prenatal or preimplantation genetic testing is becoming more widely available. 42
Adenoma sebaceum consists of several or multiple papules and nodules, sometimes grouped, with a predilection for the butterfly area of the face, particularly the nasolabial groove.44. and 46. They appear in early childhood as pink-red to yellow-brown lesions and their growth is usually progressive until adult life. A giant angiofibromatous plaque and a cluster growth of large nodules58 have been reported. Unilateral facial involvement is another clinical variant. 59 It probably represents mosaicism.60.61.62.63. and 64.
Facial angiofibromas in tuberous sclerosis have been treated with a scanning carbon dioxide laser. 65 The benefits of therapy should be weighed against both early morbidity and the risks of long-term complications such as scarring and hypopigmentation. 65 Erbium lasers have also been used. Clinical trials using sirolimus (rapamycin), an mTOR inhibitor have shown promise.43. and 66. mTOR has a central role in the control of cell growth and proliferation.67. and 68. Its activation is influenced by heterodimers of TSC1 and TSC2. 42 It has been suggested that long-term therapy with sirolimus may increase the risk of malignant tumors in these patients. 42 The finding of increased levels of certain matrix metalloproteinases (MMPs) in lesional skin raises the possibility of antiprotease treatments in the future, or the use of retinoids, which inhibit the production of MMPs. 69
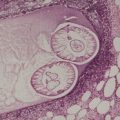
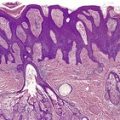
The lesions vary from rounded elevations to raised pedunculated growths (Fig. 34.1). 70 The epidermis shows some flattening of rete ridges with patchy melanocytic hyperplasia, and mild overlying hyperkeratosis. The dermal component consists of a network of collagen fibers, often oriented perpendicular to the surface in the subepidermal zone, and having an onion-skin arrangement around follicles and sometimes blood vessels (Fig. 34.2). There is an increase in ‘fibroblastic’ cells, which are plump, spindle shaped, stellate or even multinucleate. There is often a sparse inflammatory infiltrate which includes mast cells. The blood vessels are increased in number, and some are dilated with an irregular outline. 71 It has been suggested that a functional loss of tuberin may stimulate vascular growth. 72 Sporadic angiofibromas do not show loss of tuberin or hamartin. 73 Follicles may show epithelial proliferation and there may be primitive small follicles. 74 Elastic tissue is absent from the stromal fibrous tissue. The extracellular glycoproteins fibronectin and tenascin are increased in the stroma. 75 There is also overexpression of MLH-1 and psoriasin genes in the cutaneous hamartomas. 76
Adenoma sebaceum. There are pedunculated outgrowths with an angiofibromatous stroma. (H & E)
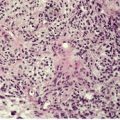
Adenoma sebaceum. Collagen is arranged around the small blood vessels in the upper dermis. Fibroblasts are increased in number, but they are not as stellate as usual. (H & E)
Staining for CD31 confirms the increased vascularity of these lesions.
Ultrastructural examination77 has shown large numbers of microvilli on the luminal surface of the endothelial cells of the vessels. The stroma contains many banded structures. No myofibroblasts have been seen.
Facial angiofibromas, both unilateral and bilateral, have already been mentioned as an important manifestation of tuberous sclerosis (see above). They have also been described in a patient with neurofibromatosis 2 (NF-2 – OMIM 101000) as a cluster of small papules on the ear. 78 Multiple facial angiofibromas are seen quite often in patients with multiple endocrine neoplasia (MEN) type 1 (OMIM 131100). 79 They tend to present in adult life. 80 Other cutaneous tumors in this syndrome include collagenomas and lipomas. 81 There may also be café-au-lait macules and confetti-like hypopigmented macules. 82 The tumors show allelic deletion of the MEN1 gene. It encodes a protein called menin, which is presumed to act as a tumor suppressor. Basic fibroblast growth factor (BFGF) is elevated in many patients and may be responsible for the formation of the cutaneous tumors. 80
Angiofibromas (often reported as perifollicular fibromas) have been reported in the Hornstein–Knickenberg syndrome, which appears to be a slightly different phenotypic expression of the Birt–Hogg–Dubé syndrome (see p. 771). Multiple facial angiofibromas have also been reported in the Birt–Hogg–Dubé syndrome (OMIM 135150). 83
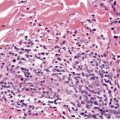
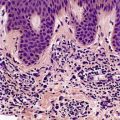
Fibrous papules of the face are usually solitary, dome-shaped papules, measuring 3–5 mm in diameter, found particularly on the nose of middle-aged adults.32.86. and 87. They are flesh colored and usually asymptomatic, although some may bleed after minor trauma. They were originally regarded as fibrosed dermal nevi,86.87. and 88. a proposition which has been disproved by electron microscopy89. and 90. and immunohistochemistry.91.92. and 93. The presence of factor XIIIa in the spindle cells and in some stellate cells suggests that fibrous papule is a proliferative reactive process consisting mainly of dermal dendritic cells.92. and 93.
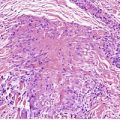
The changes are similar to those described for adenoma sebaceum. However, the vessels are sometimes more ectatic and less likely to show concentric fibrosis than in adenoma sebaceum (Fig. 34.3). Furthermore, the bizarre cells in the dermis are usually more numerous and the basal melanocytic hyperplasia more prominent in fibrous papule of the face. Rarely, the stromal cells may contain coarse cytoplasmic granules leading to a granular-cell appearance. 94 Another rare pattern involves the presence of numerous fibroblasts/histiocytes/dendrocytes with clear vacuolated cytoplasm embedded in a dense sclerotic and hyalinized stroma. 95 A few multinucleate ‘floret’-like cells may be present. Only a few cells stain for factor XIIIa and CD68. This lesion, clear cell fibrous papule, may eventually prove to be unrelated to fibrous papule, although the cases reported were all on the face, predominantly the nose (Fig. 34.4). 96 Other rare variants include hypercellular, pigmented, pleomorphic, inflammatory, and epithelioid fibrous papules. 97
(A) Fibrous papule of the nose. Note the bizarre stellate cells in the upper dermis. (B) Another case. (H & E)
Clear cell fibrous papule. This lesion from the nasal ala contains telangiectatic vessels and sheets of clear cells. (H & E)
The spindle and stellate cells in fibrous papule of the face contain vimentin and factor XIIIa (see above), but not S100 protein.92.98. and 99. α1-Antitrypsin and lysozyme were detected in one study, although this has not been confirmed subsequently. 100 A case with CD34+ cells has been reported. 101 The cells of the epithelioid fibrous papule are reactive for procollagen, and are negative for NKI/C3, unlike previously described clear cell variants.102. and 103.
Pearly penile papules are persistent asymptomatic pearly-white papules, 1–3 mm in diameter, occurring in groups or rows on the coronal margin and sulcus of the penis.31.104.105.106. and 107. Rarely they may be found on the penile shaft108 or glans. 109 They are found in 10–30% of young adult males, and are more common in black people, and in the uncircumcised.105. and 110. They may be misdiagnosed as warts, 106 but there is no causative role for HPV in their genesis. 111
Treatment with cryotherapy has been effective in some studies, but not in all. 110 Carbon dioxide laser has also proved effective. 110
There is a rich vascular network, surrounded by dense connective tissue containing an increased number of plump and stellate ‘fibroblasts’. They resemble other lesions in this group, except for the absence of pilosebaceous follicles.
Included in the acral fibrokeratoma group112 are lesions reported as acquired digital fibrokeratoma,113.114.115. and 116. acquired periungual fibrokeratoma, ‘garlic clove fibroma’, 117 and the subungual and periungual fibromas of tuberous sclerosis. 46 This unifying concept is an attempt to overcome the needless proliferation of terms, and it gives recognition not only to the common histopathological features, but also to the fact that occasional lesions have been reported in sites other than digits. 118
The lesions are usually solitary, dome-shaped or elongated thin horns, 1–3 mm in diameter and up to 15 mm in height. 48 A giant variant measuring almost 4 cm in diameter has been reported. 119 There is sometimes a history of trauma. 114 The ungual fibromas of tuberous sclerosis are often multiple, sometimes in clusters, and develop at about puberty. 120 They are found in about half the patients with tuberous sclerosis.41. and 46.
Some fibrokeratomas originate from the dermal connective tissue, whereas others appear to originate from the proximal nail fold. 121 An invaginated variant has been reported in relation to the nail apparatus. 122 This difference in the site of origin may account for the heterogeneous features observed in this entity.
Multiple acral fibromas with a myxoid but poorly vascularized stroma have been reported in a patient with familial retinoblastoma, leading to the suggestion that multiple acral benign tumors with a fibrous component might be a cutaneous marker of tumor suppressor gene germline mutation. 123 Lesions reported as familial multiple acral mucinous fibrokeratomas124 and familial myxovascular fibroma (see below) are probably further examples of this hypothesis.
The epidermal covering usually shows hyperkeratosis, and sometimes acanthosis. There is a core of thick collagen bundles which are oriented predominantly in the vertical axis (Fig. 34.5). Stellate fibroblasts are often present. There is sometimes prominent cellularity, 121 and a rich vascular supply. These latter two features have not been prominent, 115 or have been specifically excluded125 in some of the reports, suggesting that some of the lesions might best be regarded as fibromas112. and 123. rather than angiofibromas. The rare invaginated variant is characterized by a deep epithelial invagination proximal to the normal matrix. 122 A pseudo-nail plate is produced.
Acral fibrokeratoma. This lesion, which was clinically horn-like, has a core of fibrous tissue and an epidermal covering with overlying orthokeratosis. Dilated vessels are present at the tip. (H & E)
There are usually sparse elastic fibers, few inflammatory cells, and no hair follicles. Neural tissue is not present, unlike the clinically similar entity of supernumerary (rudimentary) digits.115. and 126. The stromal cells express varying amounts of factor XIIIa.
The cellular digital fibroma is composed of intersecting fascicles of thin, delicate spindle cells in the superficial reticular dermis with a fibrotic and slightly myxoid stroma. 127 It may be histogenetically distinct from other angiofibromas and digital fibrokeratomas as the constituent cells in cellular digital fibromas stain strongly for CD34, with only scattered stromal cells expressing factor XIIIa. 127 This entity needs to be distinguished from dermatofibrosarcoma protuberans. It has been suggested subsequently that cellular digital fibroma is a variant, if not the same as superficial acral fibromyxoma (see p. 844). 128
Three kindreds have been reported in which multiple verrucous papules developed on the fingers and hands.124.129. and 130. On histological examination the papules showed a fibrovascular proliferation of the papillary dermis, with variable myxoid change and overlying epidermal acanthosis and hyperkeratosis. There is some overlap with the lesions included within the concept of cutaneous myxomas (see p. 844).
This heterogeneous group of lesions forms a histological spectrum, at one end of which is the fibroma and at the other end the fibrosarcoma. In between are the ‘fibromatoses’, which have been defined as a ‘group of non-metastasizing fibrous tumors which tend to invade locally and recur after surgical excision’. 131
The ‘fibromatoses’ include entities such as palmar and plantar fibromatosis, 132 extra-abdominal desmoid,133. and 134. knuckle pads, pachydermodactyly, Peyronie’s disease of the penis, and various ‘juvenile fibromatoses’ such as juvenile aponeurotic fibroma, fibrous hamartoma of infancy, digital fibromatosis of childhood, and infantile myofibromatosis. The tumors in this latter category usually contain an admixture of fibroblasts and myofibroblasts. This is the explanation for their inclusion also as tumors of myofibroblasts (see below).
Plantar fibromatosis is a benign, but sometimes locally aggressive, proliferation of fibrous tissue involving the deep subcutis and fascia of the plantar surface of the foot. Familial cases occur. 135 In a study, from the Armed Forces Institute of Pathology (AFIP), of 56 cases of palmar-plantar fibromatosis in children and preadolescents, there was a high incidence of local recurrence of their fibromatosis. 136 Most cases had been initially managed by local excision, and in most cases, there were positive margins. 136 Most cases of plantar fibromatosis contain multinucleated giant cells but the number of these cells is quite variable. 137 They also occur in palmar fibromatosis. 138 A variant with distinct nodules occurs.132. and 136. Peyronie’s disease of the penis, nodular and diffuse fibrous proliferations of the penis and tunica vaginalis, 139 and Dupuytren’s disease of the palmar fascia are similar fibromatoses. 140 There is a high incidence of chronic liver disease in patients with Dupuytren’s disease. 141 The desmoid tumor (a clonal process) and extra-abdominal desmoid tumor are usually regarded as belonging to the domain of soft tissue tumors.142.143.144.145.146.147.148.149.150.151. and 152. They will not be considered further. Platelet-derived growth factor receptors and ligands are up-regulated in fibromatoses; they may play a role in the growth of these tumors. 153 The nuchal fibroma (see p. 320), 154 and the nuchal fibrocartilaginous tumor (see p. 375) 155 are considered elsewhere. The nuchal-type fibromas that arise in association with colonic polyps may be multiple and occur in different locations. 156 They have been called Gardner fibromas. They are discussed later in this chapter (see p. 815).
Tumors of myofibroblasts include nodular fasciitis, fibrous hamartoma of infancy, digital fibromatosis of childhood, postoperative spindle-cell nodule, dermatomyofibroma, infantile myofibromatosis, inflammatory myofibroblastic tumor, plexiform fibrohistiocytic tumor, and myofibroblastic sarcoma. Partial myofibroblastic differentiation is seen in low-grade fibromyxoid sarcoma and angiomyofibroblastoma of the vulva. Plexiform fibrohistiocytic tumor is considered with the fibrohistiocytic tumors (see p. 831) because of the dual population of cells. Congenital-infantile fibrosarcoma is usually included as a myofibroblastic lesion.24. and 27. The case with myofibroblastic proliferation confined to the skin of the neck defies classification. 157
Electron microscopy is of some value in distinguishing between the various spindle-cell tumors of the skin and soft tissues. 158 Cytogenetic analysis of tumors adds further information; some tumors have specific karyotypic aberrations. 159
Skin tags (soft fibromas, acrochordons, fibrolipomas, fibroepithelial polyps) are common cutaneous lesions that have received little attention in the dermatological literature, because hitherto they have been regarded as being of little consequence. However, there have been several reports suggesting an association between the presence of skin tags and underlying diabetes,160.161.162.163. and 164. abnormal lipid profile, 165 colonic polyps,166.167.168. and 169. or acromegaly. 170 The association with colonic polyps is controversial, and several studies have failed to confirm its existence.171.172. and 173. HPV types 6/11 were detected in a significant number of cases in one study. 174 This finding remains to be confirmed.
Skin tags have a predilection for the axilla, neck, groin, eyelids and beneath pendulous breasts. They are more common in obese females, and they may develop in pregnancy. 175 In one autopsy study they were present in 64% of individuals over the age of 50, 171 whereas in a more recent study they were present in 46% of 750 individuals examined. 176 There are three clinical types: 171 furrowed papules approximately 2 mm in width and height; filiform lesions, approximately 2 mm in width and 5 mm in height; and large bag-like protuberances, usually on the lower trunk.177. and 178. These larger lesions are very occasionally multiple.160. and 179. The term fibroepithelial polyp is sometimes used for this latter variant. Experimentally, these polyps show down-regulation or loss of tuberin and/or hamartin expression which may promote collagen formation, leading to their formation and growth. 180
Vestibular papillae of the vulva are skin tag-like smooth projections of the vestibular mucosa that appear to be normal anatomical variants.181. and 182. They are not related to human papillomavirus infection, as previously suggested.181. and 183.
Fibroepithelial polyps of the anus are relatively common lesions, some of which are thought to arise from enlargement of anal papillae. 184 They should be distinguished from the much smaller infantile perianal (perineal) pyramidal protrusion which occurs predominantly in young girls in the midline, anterior to the anus.185.186. and 187. Sometimes they are posterior to the anus. 188 Underlying constipation and anal fissures are often present. 188 A variant associated with lichen sclerosus may also occur.188. and 189. They are edematous, flesh-colored, sessile protrusions measuring 1–2 cm in length. They should not be mistaken for condylomas. 190 One case has been successfully treated with topical steroid applications. 191
The lymphedematous fibroepithelial polyp of the penis is a rare polyp of the glans penis or prepuce associated with long-term condom catheter use. 192 On rare occasions it has been associated with chronic phimosis. 193 The lesions are gray-white polyps measuring 2–7.5 cm in diameter. 192 The majority of lesions affect the ventral surface of the glans near the urethral meatus. It has been postulated that the lesions represent a reactive hyperplastic process involving the subepithelial stroma. 193 A related lesion is the polyp of the glans penis that developed in a man who practiced genital hanging kung fu. 194
The histological features vary with the clinical type. The furrowed papules show epidermal hyperplasia and sometimes horn cyst formation. These lesions, with seborrheic keratosis-like surface changes, are most common on the neck and eyelids. 195 The filiform lesions are covered by an epidermis which shows only mild acanthosis. Pagetoid dyskeratosis (see p. 272) is sometimes an incidental finding in the overlying epidermis. 196 The connective tissue stalk is usually composed of well-vascularized, loosely arranged collagen. Elastic fibers are present in normal amounts. 197 A few fat cells and, sometimes, nevus cells may be present. The larger, bag-like lesions (fibroepithelial polyps, fibrolipomas) usually have a stroma composed of loosely arranged collagen and a central core of adipose tissue. It has been suggested, and subsequently challenged, that there is little utility in submitting these lesions for histological examination.198. and 199.
Vestibular papillae of the vulva consist of connective tissue projections covered by stratified squamous epithelium. 181 Sometimes the epithelium is glycogen rich simulating koilocytes. 182
An unusual cutaneous polyp with bizarre stromal cells, thought to represent a degenerative phenomenon, has been reported as a ‘pseudosarcomatous polyp’. 200 Subsequent correspondence raised the possibility that the lesion may have been a dermal spindle-cell lipoma or ‘ancient’ (degenerative) change in a polyp. 201
The fibroepithelial polyp of the anus has a myxoid and/or collagenous stroma covered by stratified squamous epithelium which may show some swollen cells with vacuolation near the surface. 202 The stroma sometimes contains atypical cells showing fibroblastic and myofibroblastic differentiation. 184 Hyalinized vascular changes may be present near the base of the polyps. 203 There is an increase in CD34+ stromal cells. 203 The infantile perianal pyramidal protrusion reveals epidermal acanthosis, marked edema in the upper dermis and a mild inflammatory cell infiltrate. 185
The lymphedematous fibroepithelial polyp of the penis has a polypoid configuration covered by keratinizing squamous epithelium. The stroma is edematous with dilated vessels and focal proliferation of capillary-sized vessels with hyalinization. The focally myxoid stroma contains small spindle-shaped cells with ill-defined, pale eosinophilic cytoplasm. There are abundant bizarre multinucleated giant cells. A mild infiltrate of lymphocytes, plasma cells, and mast cells is present in the stroma. 193
Prepubertal vulval fibroma was the term used by Iwasa and Fletcher in 2004 for a hitherto unrecognized mesenchymal tumor of the vulva in 11 prepubertal girls. 204 They regarded them as tumors on the basis of recurrences in several cases after excision. The following year, Vargas and colleagues reported 14 cases, also from Boston, of a similar lesion that presented with enlargement of one, or occasionally both labia majora. On the basis of its occurrence at an age roughly coincident with the time of breast budding, its capacity for spontaneous regression, and its composition of elements native to the vulva, the authors concluded that the entity represented an asymmetric physiological enlargement in response to hormonal surges of pre- and early puberty. 205
The girls were aged 4 to 13 years. The lesions were ill-defined and consisted of fibro-fatty tissue ranging in size from 2 to 8 cm in greatest dimension.204. and 205. As already mentioned, several cases recurred after initial excision.
The tumors were poorly marginated, hypocellular masses in the dermis and subcutaneous tissue. They were composed of the usual constituents of vulvar soft tissue, with expansion of the fibrous component. 205 There were bland spindle-shaped cells in a variably collagenous to edematous or myxoid stroma. 204 The interconnected fibrous bands encircled lobules of fat, blood vessels, and nerves. 205 The fibroblastic cells were immunoreactive for CD34; 204 they also expressed estrogen and progesterone receptors. 205
Gardner fibroma is a benign lesion of the superficial and deep soft tissues with a predilection for childhood and adolescence and an association with familial adenomatous polyposis and desmoid fibromatosis.156. and 206. Sporadic cases also occur. The majority of cases involve the back and paraspinal region, followed by the head and neck, and the extremities. Almost 20% of patients have concurrent or subsequent desmoid tumors. 206 There is a strong association with familial adenomatous polyposis or adenomatous polyposis coli, but the proportion of sporadic cases that have the APC mutation remains to be elucidated. 206
There are some overlap features with nuchal fibroma (see p. 320) but the predilection of nuchal fibroma for middle-aged males, its location on the posterior neck, and its association in some patients with diabetes mellitus, but not familial adenomatous polyposis, usually allows a distinction to be made.
There is a bland, hypocellular proliferation of haphazardly arranged coarse collagen fibers with inconspicuous spindle cells. 206 There are small blood vessels and a sparse mast cell infiltrate. The collagen forms around adipose tissue lobules. There is no increase in small nerve bundles, as seen in nuchal-type fibroma (see p. 320).
In one study, 64% of cases tested showed nuclear reactivity for β-catenin, and 100% of cases showed nuclear reactivity for both cyclin-D1 and c-myc. 206
Pleomorphic fibroma of the skin was described by Kamino et al in 1989. 207 It presents as a slow-growing lesion, clinically indistinguishable from a polypoid skin tag. 208 A subungual lesion has been reported.209. and 210.
The lesion is usually a dome-shaped nodule with variable cellularity. The spindle-shaped cells show striking nuclear pleomorphism with rare mitotic figures (Fig. 34.6). A variant with myxoid stroma occurs.209. and 211. The cells express vimentin and CD34, but not desmin, Ki-M1p212 or S100 protein, suggesting a possible origin from dendrocytes, rather than myofibroblasts as originally thought.207. and 212. Staining for factor XIIIa has been moderate in some studies, but negative or patchy in others.209.212. and 213. The nuclear atypia is similar to the ‘degenerative’ changes seen in a number of benign mesenchymal tumors. 214
(A) Pleomorphic fibroma. (B) Some bizarre cells are present in the stroma. (H & E)
Sclerotic (‘plywood’) fibroma is an uncommon fibrocytic neoplasm that occurs sporadically as a solitary lesion, and in a multifocal form in patients with Cowden’s disease.215.216.217.218. and 219. A solitary lesion may be the presenting feature of this syndrome. 220 The terms ‘hypocellular fibroma’ and ‘circumscribed, storiform collagenoma’221 have also been used for this entity. The lesions are flesh-colored papules or nodules measuring 0.5–3 cm in diameter. Sporadic cases, unassociated with Cowden’s disease, have been reported in the oral mucosa.222.223. and 224.
Although it was initially regarded as an involutional lesion, one study has demonstrated ongoing type I collagen synthesis and deposition, suggesting that the lesion is a proliferating neoplasm.217. and 225. Local recurrence has been reported. 226
The lesions are circumscribed, unencapsulated dermal nodules, often with an attenuated overlying epidermis. 217 They are composed of thickened and homogenized eosinophilic collagen bundles arranged in a laminated fashion with intervening prominent clefts. 227 Vaguely storiform or whorled patterns of collagen may be present (Fig. 34.7). The lesions are of low cellularity. The nuclei are tapered to stellate. Elastic fibers are absent from the lesion, but there is often some stromal mucin. Similar collagenous changes have been seen as a focal phenomenon in the vicinity of inflammatory lesions, such as folliculitis, 228 and in dermatofibromas, nevi, and neurofibromas where the sclerosis may be more extensive.229. and 230. This has led to the view that sclerotic fibroma-like change may represent a common reaction pattern in the skin. 230
(A) Sclerotic fibroma. (B) There is a storiform and whorled pattern. (H & E)
The spindle cells stain for vimentin and factor XIIIa.225. and 231. CD34 positivity occurs focally, with no consistent localization.225.232. and 233. The finding of diffuse CD34 and CD99 positivity in both sclerotic fibromas and pleomorphic fibromas has led to the suggestion that these tumors may be linked in some way. 234 Both PCNA and Ki-67 immunoreactivity have been detected, as would be expected in a growing neoplasm. 217 The proliferating index (MIB-1) was very low in another series. 234
The lesion reported as a pacinian collagenoma is probably a variant. 235 It was composed of paucicellular collagen fibers arranged in concentric lamellations giving an ‘onion-skin’ appearance. The cells stained for CD34. It had some resemblance to the perineurioma (see p. 871).
Cases resembling sclerotic fibroma but with variable numbers of bizarre, multinucleated cells, often with a foamy cytoplasm, have been reported as pleomorphic sclerotic fibroma, giant cell collagenoma, and cellular storiform collagenoma.233.236.237.238.239. and 240.
Collagenous fibroma, a recently described tumor, may arise in the subcutaneous tissue or muscle.242. and 243. Dermal involvement is rare.244. and 245. The tumors are firm and non-tender. Most measure 2–3 cm in diameter but larger lesions have been reported. 246 There is a predilection for adult males. 247 Any part of the body may be involved. 247
Few cases have been subject to cytogenetic analysis. It appears that a specific breakpoint of 11q12 occurs, combined in several cases with a reciprocal translocation. 248
Surgery is the treatment of choice with no reports of recurrence. 248
Collagenous fibroma is usually a well-demarcated tumor in the subcutis, although some infiltration is often present at the periphery. It is hypocellular and composed of large, stellate, or spindle cells set in a densely collagenous or fibromyxoid stroma. There are usually no mitoses. There are inconspicuous small vessels. The cells express vimentin. They are focally positive for α-smooth muscle actin and muscle-specific actin. 245 Factor XIIIa was present in one case. S100 protein and CD34 are not expressed. Scattered cells may show a myofibroblastic immunophenotype. 249
Knuckle pads (discrete keratodermas over the knuckle and finger articulations) are well-formed skin-colored nodules overlying the interphalangeal and metacarpophalangeal joints of the hands.250.251. and 252. They are usually multiple. There are several clinical variants, including a familial group, an occupational or recreational-related group, and an acquired idiopathic group. They have followed the prolonged playing of video games. 253 An association with knuckle cracking and with pseudoxanthoma elasticum has been reported.254. and 255. Of historical interest is the prominent knuckle pad on the right thumb of Michelangelo’s statue of David. 256
Inherited knuckle pads of the feet have been described in association with leukonychia and deafness, with and without keratosis palmoplantaris. They have also been associated with acrokeratoelastoidosis. 257 They are well-defined plaques on the dorsal aspect of the feet. Acquired lesions of the feet have been associated with repetitive friction from athletic gear. 257
There appear to be at least three histological types. The usual lesions show prominent hyperkeratosis, hypergranulosis, and epidermal acanthosis. There is minimal thickening of the papillary dermis. Another type has macronodules of swollen collagen fibers surrounded by thickened elastic fibers. 258 A third variant with prominent subcutaneous fibrosis, belonging to the fibromatoses, has been documented. 256
Pachydermodactyly is characterized by fibrous thickening of the lateral aspects of the proximal interphalangeal joints of the fingers, usually in males.259.260.261.262.263. and 264. The thumbs and fifth fingers are usually not involved. More extensive lesions have been reported.262.265. and 266. It has been reported in two siblings. 267
Pachydermodactyly is regarded as a localized form of superficial fibromatosis. Trauma, such as finger rubbing, has been implicated in the etiology.268. and 269. It has also been suggested that this entity might be related to knuckle pads (see above). 270
There is thickening of the dermis, with coarse collagen bundles and a mild proliferation of fibroblasts. Increased fibroblastic activity and collagen deposition around sweat glands have been noted.267. and 271. Small deposits of mucin are sometimes present in the interstitium. 262 Elastic fibers are reduced. There is no inflammation.
The overlying epidermis shows mild hyperplasia and compact orthokeratosis.
Nodular fasciitis is a reactive proliferation of myofibroblasts with a predilection for the subcutaneous tissues of the forearm, upper arm, and thigh of young and middle-aged adults.272. and 273. Dermal and intravascular variants have been reported.274.275.276.277.278. and 279. It usually grows quite quickly to reach a median diameter of 1.5 cm. Multiple lesions have been reported. 280 Recurrences are rare, even after incomplete surgical removal, and their occurrence should lead to a reappraisal of the original histological diagnosis.281. and 282. The diagnosis can usually be made readily by fine needle aspiration. 283 Regression of the lesion has followed this procedure. 279 Regression may also occur after the use of intralesional corticosteroids. 284 Despite having high activity for proliferating cell nuclear antigen (PCNA), the cells do not express p53 or show aneuploidy.285. and 286. It is polyclonal and a reactive process. 287
Cranial fasciitis of childhood is a distinct clinical variant arising in the deep soft tissues of the scalp, with involvement of the underlying cranium.288.289. and 290. Conservative surgical excision is curative.291. and 292. Intralesional corticosteroids were used in one case with a successful outcome. 293
Nodular fasciitis is composed of a proliferation of spindle-shaped to plump fibroblasts which may be arranged in haphazard array (’tissue culture appearance’), or in bundles that form S-shaped curves (Fig. 34.8). 295 A vague storiform pattern is sometimes present focally. Mitoses are frequent, but atypical forms are rare. Cleft-like spaces may be seen between the fibroblasts. There is a variable amount of myxoid stroma and extravasated erythrocytes. Collagen is usually sparse. Capillaries with plump endothelial cells are common. Scattered lymphocytes are dispersed throughout the lesion.
Nodular fasciitis. A condensed fibrous capsule surrounds a tumor which is composed of swirling bundles of spindle-shaped cells set in a myxoid stroma. (H & E)
Intravascular fasciitis, in which the proliferation occurs within small and medium-sized arteries and veins, appears to represent an origin from myofibroblasts within vessel walls rather than extension from the extravascular component which is often present.276. and 296. It is not associated with aggressive growth or metastasis. 274 Vascular involvement results in a plexiform appearance.
Less common findings include the presence of bone and cartilage;295.297. and 298. osteoid is not uncommon in the variant known as cranial fasciitis. 288 Scattered multinucleate fibroblasts and osteoclast-like giant cells are often present and, rarely, the latter cells are quite common.281. and 295. The term ‘ossifying fasciitis’ has been used if there is a significant component of osteoid or bone. 299 A variant with numerous cells resembling ganglion cells, akin to those seen in proliferative myositis,300. and 301. may be separated off as a distinct entity – proliferative fasciitis (Fig. 34.9).302.303. and 304.
Proliferative fasciitis. Ganglion-like cells are present within a spindle-cell tumor. (H & E)
Various histological subtypes of nodular fasciitis have been proposed, based on the cellularity, the amount of myxoid stroma, and the presence of collagen or other histological features such as osteoclast-like cells, ganglion-like cells, or bone. Some of the proposed subtypes merely reflect changes in the histological composition during the evolution of the lesion. 272
Immunoperoxidase studies demonstrate HSP47 (a useful marker of skin fibroblasts), 305 smooth muscle actin, muscle-specific actin, calponin, vimentin, and KP1, but not desmin, CD34, cytokeratin, or S100 protein.275.279.306.307. and 308. A similar staining pattern is seen in proliferative fasciitis, although the ganglion cells express only vimentin. 304 The multinucleate cells in cranial fasciitis stained for CD68 in one reported case. 292
Few ultrastructural studies have been carried out but, in one study of eight cases, myofibroblasts were the predominant cell present. 309 Cells with features of fibroblasts or histiocytes are also seen. 307 Myofibroblasts and fibroblasts are also present in cranial fasciitis. 289
Atypical decubital fibroplasia (ischemic fasciitis) is a pseudosarcomatous proliferation, found in immobilized or debilitated patients, which is different from decubitus ulcers. 310 The affected area measures 1–9 cm in diameter. It involves the shoulders, ribs, sacrococcygeal region, and the tissues overlying the greater trochanter. There is involvement of the deep dermis, subcutis, and deep fascia with a zonal arrangement of fibrinoid necrosis, reactive fibrosis, neovascularization, fat necrosis, and ectatic vessels.310. and 311. Atypical fibroblasts are usually present.
A recent study of 44 cases by Liegl and Fletcher found there was an inconsistent association with immobility or debilitation. 311
Postoperative spindle-cell nodule is a rare lesion that develops soon after a surgical procedure in genital skin or the genitourinary system. 312 Lesions have now been reported at other sites. 313 It is composed of spindle-shaped cells in a pattern of interlacing fascicles. There are occasional mitotic cells. The cells express vimentin; desmin and muscle-specific actin have been present in some cases. 312 They do not express keratin. 313
Solitary fibrous tumor is a rare mesenchymal tumor that most commonly involves the pleura. It has been reported in many other sites, including the skin.314.315.316.317. and 318. It presents as a circumscribed nodule, often mistaken for a cyst. Most cases have been on the head and neck. 319 Cases have also been reported in the vagina.320. and 321. Although 10–20% of lesions in other organs have been malignant,322.323.324. and 325. all cutaneous tumors reported so far have behaved in a benign fashion. 314
The tumor cells are predominantly fibroblastic although focal myofibroblastic differentiation is often seen. 314 This tumor was originally included with the hemangiopericytomas. 326 No consistent (cyto)genetic alterations have been documented to date. 324
Treatment of cutaneous cases is surgical excision of the lesion.
Cutaneous tumors are circumscribed with alternating hypercellular and less cellular areas. There are short spindle and ovoid cells with scant cytoplasm in a fascicular, haphazard, or storiform arrangement (Fig. 34.10). Mitoses are generally rare. 327 Areas with a hemangiopericytoma-like appearance are quite common. There is a variable collagenous stroma which is focally hyalinized, and some ectatic vessels. 327 A myxoid stroma is sometimes present. 328 Lesions need to be distinguished from spindle-cell lipoma.316. and 329.
Solitary fibrous tumor. (A) There is variable cellularity. (B) The tumor is composed of spindle cells. (H & E)
Immunohistochemistry shows generalized staining for CD34, bcl-2, and vimentin. 316 Focal staining for CD99 also occurs. 317 Factor XIIIa+ dendrocytes were present in an oral lesion. 330 The cells are negative for smooth muscle and epithelial markers. The mesothelial markers calretinin and HBME-1 have not been detected in five dermal cases. 319
Fibrous hamartoma of infancy is an uncommon fibroproliferative lesion of the subcutaneous tissue that is present at birth or develops in the first 2 years of life.333.334.335.336. and 337. It most commonly occurs around the shoulder, axilla, and upper arms, but cases involving the scalp, 338 inguinal region, scrotum, 339 perianal area, 340 and lower extremities341. and 342. have been reported. There is a male predominance of 3 : 1. 334 The clinical course is benign, despite its infiltrative appearance and tendency to local recurrence. 335 It has been reported in an infant with Williams’ syndrome (see p. 348). 343 A reciprocal translocation [t(2;3)(q31;q21)] was present in one case. 344
The subcutaneous tissue of excised lesions has a glistening gray-white appearance interspersed with fatty tissue. The involved area measures 2–8 cm in maximum diameter.
This tumor should be treated by complete excision; an aggressive approach should be avoided, as the overall prognosis is excellent. 345
Fibrous hamartoma of infancy has poorly defined margins. It is centered on the subcutis. There are three different tissue components:334. and 347. interlacing trabeculae of fibrocollagenous tissue, small nests of loosely arranged mesenchymal cells, and interspersed mature fat (Fig. 34.11). The fibrous trabeculae vary in thickness and arrangement and contain spindle-shaped cells; they express vimentin but not S100 protein. 335 Desmin and smooth muscle actin are found in the fascicular-fibroblastic regions. 348 The myxoid areas are more cellular, with immature oval or stellate cells, sometimes having a whorled pattern. Sparse lymphocytes may be present in the stroma.
(A) Fibrous hamartoma of infancy. (B) There are bundles of fibrous tissue, some mesenchymal cells, and interspersed mature fat. (H & E)
The overlying skin frequently shows eccrine changes that include hyperplasia, duct dilatation, intraluminal papillary formations, and squamous syringometaplasia. 349 Primitive mesenchymal cells may replace the normal eccrine gland stroma. 350 Increased numbers of terminal hair follicles, and epidermal basaloid follicular hyperplasia have also been reported.349. and 350.
Digital fibromatosis of childhood (infantile digital fibromatosis,353. and 354. recurring digital fibrous tumor of childhood, 355 inclusion body fibromatosis356) is a rare benign tumor of myofibroblasts with characteristic cytoplasmic inclusion bodies. 336 It presents as a dome-shaped firm nodule, up to 1 cm in diameter, usually on the digits.357. and 358. The thumbs and great toes are spared. 359 Lesions are usually solitary, but a second tumor is sometimes noted at the time of presentation, or develops subsequently. 357 The tumors may be present at birth, or appear in the first year of life.360. and 361. Onset in late childhood or adult life is rare.356.362. and 363. A history of preceding trauma has been reported. 364 There is a rare syndrome consisting of recurrent digital fibroma, focal dermal hypoplasia, and limb malformations. 365
A viral etiology was originally suspected because of the characteristic inclusion bodies, but all cultures have been negative, excluding this hypothesis.366.367. and 368. The inclusions are now known to be filamentous aggregations composed largely of actin.369. and 370.
The tumor often recurs after local excision; very occasionally it may regress spontaneously, but this may be accompanied by functional impairment.371.372. and 373. Spontaneous regression increases after long-term follow-up, casting some doubt on the benefits of surgical excision with its high (up to 60%) recurrence rate. 374 Intralesional fluorouracil has also proven effective in the treatment of this entity. 373
The tumor is non-encapsulated and extends from beneath the epidermis, through the dermis and usually into the subcutis. It is composed of interlacing bundles of spindle-shaped cells and collagen bundles. There may be some vertical orientation of the cells and fibers superficially (Fig. 34.12). The appendages become incorporated within the tumor. The nuclei of the cells are oval or spindle shaped, and some stellate forms may be present. There are only occasional mitoses. The cytoplasm of the cells merges imperceptibly with the collagen. There are characteristic small eosinophilic inclusion bodies within the cytoplasm of the tumor cells, often in a paranuclear position (Fig. 34.13). A clear halo is sometimes discernible in well-stained sections. These bodies measure 2–10 µm in diameter, and may be mistaken for red blood cells. They stain red with the Masson trichrome stain, and deep purple with the PTAH method. They are PAS negative but actin positive. 376
(A) Digital fibromatosis of childhood. (B) The spindle-shaped cells and some collagen bundles have a vertical orientation within the dermis. (H & E)
Digital fibromatosis. Pale pink inclusion bodies, composed of actin, are present in the cytoplasm of the spindle-shaped cells. (H & E)
There are small capillaries and a few scattered elastic fibers in the stroma. Lymphocytes are variable in number. 377 The overlying epidermis usually shows flattening of the rete ridges. Ulceration is rare.
The immunocytochemical localization of vimentin and muscle-specific actin in the proliferating cells confirms their myofibroblastic nature. 356 The cells also express desmin. 369
The spindle cells are myofibroblasts364. and 378. and the inclusion bodies are compact masses of amorphous and granular material with some discernible microfilaments, but with no limiting membrane. Actin has been demonstrated within the myofibroblasts, and it has been suggested that the inclusions are masses of actin353 or, more likely, degradation products of it. 379 This viewpoint has subsequently been challenged. 380 Cultured tumor cells also develop inclusion bodies. 353
Fewer than 10 cases of angiofibroblastoma have been described.381. and 382. It presents as a solitary dermal nodule on the extremities. 382 It consists of stellate and spindle-shaped cells, with the phenotype of fibroblasts, embedded in a fibromyxoid to dense fibrous stroma. Numerous capillary-sized vessels, often in small groups, are scattered throughout the stroma. Staining for HHF35 is focal and pale. A small population of dendritic cells within the tumor express factor XIIIa. All other spindle-cell markers are negative. 382 It needs to be distinguished from other fibromyxoid tumors of the skin.
Surgical excision has been curative. 382
Angiomyofibroblastoma is a rare tumor of the vulva that may be confused with aggressive angiomyxoma of the pelvic soft tissues and vulval region, which appears to be a related tumor.383.384.385.386.387.388.389. and 390. Angiomyxoma will not be considered further, although rare superficial tumors have been described. 391
The morphologically similar lesion described in the male genital tract by Laskin et al as having features of both angiomyofibroblastoma and spindle-cell lipoma has now been reclassified as cellular angiofibroma (see below). Angiomyofibroblastoma is often misdiagnosed clinically as a Bartholin’s cyst. 392 The tumors are well circumscribed, measuring 0.5–12 cm in diameter. They do not recur if completely excised. Sarcomatous transformation has been reported. 393
The lesion is composed of an edematous stroma in which abundant blood vessels (predominantly of the capillary type) are irregularly distributed. Spindle cells, some of which are plump, are present in the stroma, often aggregated around the vessels. Some cells may have abundant hyaline cytoplasm. Hypercellular areas may be present. Wavy collagen bundles are scattered through the stroma. Intralesional fat is often present. A lipomatous variant with abundant fat has been reported. 395
Cellular angiofibroma is a recently described benign mesenchymal tumor, usually of the genital region, with some histological resemblance to angiomyofibroblastoma and spindle-cell lipoma. 397 It has an equal sex incidence. It occurs predominantly in middle-aged and slightly older individuals. The majority occur in the vulvovaginal region,398. and 399. and the inguinoscrotal region. 400 Most cases present as a painless mass that measures 0.5–25 cm in diameter. Lesions tend to be larger in males than in females. 397 Recurrences are rare even in cases excised with positive margins.
Two cases with deletions of the FOX1A1/13q14 region have been reported. 401 A similar finding is seen in some spindle-cell lipomas and in (extra)mammary myofibroblastoma. 401
Most cases are well-marginated tumors located primarily in the subcutaneous tissue. 397 Cellular angiofibroma is composed of bland, spindle-shaped cells, short bundles of wispy collagen and numerous thick-walled and often hyalinized vessels. Intralesional fat is present in about 20% of cases. 397 Significant cytological atypia and mitoses are uncommon.
Dermatomyofibroma is a benign tumor of fibroblasts and myofibroblasts first reported by Hügel in 1991. 404 It has a predilection for the shoulder girdle, axilla, and abdomen of young adults.405. and 406. There is a female predominance. Cases in young males have a predilection for the posterior neck.407. and 408. The lesions present as firm red-brown plaques or nodules, 1–2 cm in diameter; they may resemble a keloid. 409 A hemorrhagic form has been reported. 410 Linear and giant annular variants occur.411. and 412. A similar lesion has been reported under the term ‘myoid fibroma’.413. and 414.
Conservative surgical excision is usually carried out. There is no tendency to local recurrence. 415
The tumor is a non-encapsulated plaque-like lesion composed of fascicles of monomorphic spindle cells predominantly orientated parallel to the skin surface; some intersecting bundles are present (Fig. 34.14). 416 The cells have faintly eosinophilic cytoplasm and elongated vesicular tapering nuclei. The lesion fills the reticular dermis and sometimes extends into the upper subcutis. It spares the papillary dermis and adnexal structures. The stroma contains collagen bundles with an increase in small blood vessels. In the hemorrhagic variant (see above), there are numerous capillaries and slit-like spaces resembling the plaque stage of Kaposi’s sarcoma but there is no staining for CD34 or HHV-8. 410 Red cell extravasation is also present. Elastic fibers are preserved and some are thicker than usual. There is usually a sparse chronic inflammatory cell infiltrate around the vessels.
Dermatomyofibroma. This scar-like lesion has cells orientated parallel to the epidermis. (H & E)
Most of the tumor cells express vimentin and non-specific muscle actin.416. and 417. They are negative for smooth muscle-specific actin, desmin, S100, CD34, and factor XIIIa.416. and 417.
The entity of infantile myofibromatosis, which is regarded as a proliferative disorder of myofibroblasts, was established by Chung and Enzinger in 1981, with their report of 61 cases. 419 Previous reports had appeared under several designations including congenital fibrosarcoma, 420 congenital generalized fibromatosis,346.421. and 422. and congenital mesenchymal hamartoma. 423 Fletcher and colleagues have suggested that the spindle-cell component shows true smooth muscle differentiation rather than being of a myofibroblastic nature. 424 Requena et al have suggested an origin from myopericytes. 425 More recently, Fletcher and colleagues have described a spectrum of tumors showing perivascular myoid differentiation, which included adult cases of myofibromatosis (myofibroma). 426
The lesions are solitary in approximately 70% of cases.419. and 427. Almost half of these are situated in the deep soft tissues, and the remainder are located in the skin and/or subcutaneous tissue.336. and 428. The head, neck, and trunk are the usual sites of involvement.419. and 427. The finger is an uncommon site. 429 There is a male predominance. Most lesions are present at birth, 430 or appear in the first 2 years of life; onset in adult life has been recorded.431. and 432. The term ‘cutaneous myofibroma’ would seem to be an appropriate designation for the solitary lesions occurring in adults.433.434.435. and 436. Multiple acral myofibromas have been reported in a patient with generalized morphea. 437
In approximately 30% of cases the lesions are multicentric (congenital generalized fibromatosis, OMIM 228550) and involve the skin, soft tissues, bones and, uncommonly, the viscera.419.438. and 439. They are usually present at birth, and there is a female predominance. Spontaneous regression of soft tissue and osseous lesions sometimes occurs,419.440.441.442. and 443. but cases with visceral involvement are usually fatal.419.427. and 444. Cases with systemic involvement and a favorable outcome have been reported. 445 The various clinical types of myofibromatosis are shown in Table 34.1. Recurrence after a long period of quiescence has been documented. 446 Central nervous system abnormalities were present in one case. 447 Both autosomal recessive448 and dominant inheritance with reduced penetrance have been proposed.446.449. and 450.
• Solitary, infantile
• Congenital, multiple without visceral involvement
• Congenital, generalized with cutaneous and visceral involvement
• Solitary cutaneous myofibroma of adulthood
Macroscopically, the tumors measure 0.5–7 cm or more in diameter. They are grayish-white in color, and fibrous in consistency.
The plaque-like myofibroblastic tumor of infancy is an exceedingly rare tumor that presents in the first few months of life. 451 Histologically it resembles a dermatofibroma, the diagnosis given by the author to a similar case, seen some years ago.
The prognosis is excellent, with recurrence unlikely after excision; aggressive variants are rare. 452 The complex regional pain syndrome has followed the surgical excision of a solitary adult myofibroma. 453 There is increasing evidence that adult myofibromas show overlapping features with the myopericytoma, a tumor derived from pericytes/perivascular myoid cells. This aspect is considered further below (see p. 823).
The nodules are reasonably well circumscribed, although there may be an infiltrative border in the subcutis. There are plump to elongated spindle cells with features of myofibroblasts. They are grouped in short fascicles. Delicate bundles of collagen separate or enclose the cellular aggregates (Fig. 34.15). Mitoses are variable in number, but they are not atypical.
(A) Infantile myofibroma. (B) Short fascicles of plump, spindle-shaped cells are separated by thin bundles of collagen. (H & E)
Vascular spaces resembling those of hemangiopericytoma are often found in the center of the tumor.419. and 454. This gives most lesions a biphasic appearance, with a central hemangiopericytoma-like area and a peripheral leiomyoma-like region. A monophasic variant with a prominence of tiny capillaries has been reported. 455 Sometimes there is an intravascular pattern of growth. Necrosis, hyalinization, calcification, and focal hemorrhage may be present centrally. 424
Immunoperoxidase studies have shown that the tumor cells are positive for vimentin and α-smooth muscle actin but negative for S100 protein, myoglobin, and cytokeratin.424.431. and 454. Conflicting results have been reported for desmin,424. and 431. although it has been negative in recently reported cases.435. and 456.
The cells have the ultrastructural features of myofibroblasts. 423 Primitive vascular formations, with a pattern of irregular clefts between adjoining cells, were noted by Requena and colleagues. 425 Regressing lesions show vacuolation of the cytoplasm of spindle cells with their eventual disruption. 444
‘Perivascular myoma’ is the suggested designation by Granter, Badizadegan, and Fletcher for a histological continuum of three lesional groups of tumor – myofibromatosis in adults, glomangiopericytoma, and myopericytoma – showing perivascular myoid differentiation. 426 Hemangiopericytoma is another category of perivascular tumors, but its existence has been questioned as several other lesions may have a similar growth pattern. 362 Perivascular myomas have been reported in patients of all ages, with a predilection for the subcutis and superficial soft tissues of the extremities. 426 Some lesions are multifocal. This group of perivascular tumors has been further expanded in recent years by the addition of three further entities:
• perivascular epithelioid cell tumor (PEComa)
• clear cell myomelanocytic tumor
• dermal clear cell mesenchymal neoplasm.
It seems that these tumors are derived from a perivascular cell with different immunohistochemical and morphological features to the perivascular myoma and the glomus tumor. They will be considered next.
Myopericytoma was the term adopted by Fletcher and colleagues in 1998 for a tumor composed of a concentric perivascular arrangement of spindle cells with differentiation towards perivascular myoid cells. 426 Requena et al had used the term 2 years earlier in the text of their article on adult myofibromas to highlight the myopericytic differentiation that they perceived in this lesion. 425 This spectrum of growth patterns, ranging from myofibroma at one end to myopericytoma at the other, has been confirmed by others.457. and 458.
Myopericytomas occur predominantly on the distal extremities of middle-aged adults.457.459. and 460. There is a male predominance. Tumors are located in the dermis, subcutis, or soft tissues. 461 Despite marginal or incomplete excision, recurrence is uncommon. A malignant variant has been described. 462
Myopericytomas are characterized by thin-walled vessels and a concentric perivascular arrangement of ovoid, plump spindled to round myoid cells. The appearances range from hypocellular, fibroma-like tumors to cases resembling angioleiomyoma, myofibroma, and ‘hemangiopericytoma’ (Fig. 34.16). 459 A biphasic zonation pattern is usually present. 457 Malignant cases show increased mitotic activity. 462
(A) Myopericytoma. (B) Vessels are less conspicuous than usual. (H & E)
Immunohistochemically, all cases express α-smooth muscle actin (Fig. 34.17), and most also express h-caldesmon. 459 Desmin is focally positive in a few cases.
Myopericytoma. The tumor cells stain strongly for smooth muscle actin. (Immunoperoxidase stain)
The perivascular epithelioid cell tumor (PEComa) is a family of related mesenchymal tumors that includes angiomyolipoma, lymphangiomyomatosis, clear cell ‘sugar’ tumor of the lung, and similar lesions arising at a variety of visceral and soft tissue sites. 463 These tumors all share a distinctive cell type, the perivascular epithelioid cell, which has no known normal tissue counterpart. 463 PEComas variably express melanocytic and muscle markers, whereas S100 protein and cytokeratins are usually absent. 464
PEComas are rare tumors that occur predominantly in soft tissue, the gynecological organs, kidneys, and thorax.465.466. and 467. Cutaneous cases are exceedingly rare;468. and 469. they may be more common in the lower limb. 470 Occasional cases are associated with the tuberous sclerosis complex. 465 Chromosomal aberrations were detected in all cases in one series, and these mostly consisted of chromosomal losses. 466 The frequent deletion of 16p in which the TSC2 gene is located indicates that PEComas and angiomyolipomas are both TSC2-linked neoplasms. 466
A subset of deep tumors behaves in a malignant fashion and approximately 20% metastasize.463.465. and 471. The sclerosing variant reported by Hornick and Fletcher had a predilection for the retroperitoneum. 472 Only one case occurred on the abdominal wall.
This tumor is composed of nests and sheets of usually epithelioid but occasionally spindled cells with clear to granular cytoplasm and a focal association with blood vessel walls. 463 Multinucleate giant cells are often present. 465 High cellularity, high nuclear grade, and necrosis are present in up to a third of cases. Vascular invasion is uncommon. 465
This tumor belongs to the spectrum of perivascular epithelioid cell tumors (PEComas).464.474.475. and 476. Fewer than 20 cutaneous cases have been reported; seven of these cases were described by Mentzel et al. 464 In this series, six cases arose on the lower extremities and one case on the upper limb, all in adult females. In all cases an ill-defined dermal lesion with extension into subcutaneous tissue was noted. 464 Despite incomplete/marginal excision in three of the cases, none has recurred locally so far. 464 In a recent report from Fletcher and colleagues this tumor is now regarded by them as a cutaneous PEComa. 469
The tumors are composed of numerous blood vessels with a lace-like pattern and slightly thickened walls, surrounded by epithelioid cells containing clear or focally granular pale eosinophilic cytoplasm. 464 The nuclei are round and vesicular.
Immunohistochemically, tumor cells express HMB-45, microphthalmia transcription factor (MITF), and NKI/C3. 464 Perivascular expression of α-smooth muscle actin is uncommon. 476 S100 protein and pancytokeratin are not expressed. 464 Melan-A was positive in most cases in one series, 469 but negative in the other report. 464 Scattered cells express desmin and h-caldesmon.469. and 475. Focal calponin staining is sometimes present. 464
This term was applied by Lazar and Fletcher to five cases of a tumor arising on the lower limbs of adults. 477 The tumors were composed of large clear cells with vesicular nuclei centered on the reticular dermis. They usually extended into the subcutis but spared the papillary dermis.
All cases expressed NKI/C3 and 2/5 expressed CD68. They were negative for S100, HMB-45, melan-A, and muscle markers. 477 A further case that expressed NKI/C3 has been reported. 478
McAlhany and LeBoit reported a further five cases in abstract form, stating that these lesions were the same as those reported as clear cell myomelanocytic tumor (see above). 479 The cytoplasm contained glycogen.
The immunohistochemical findings in their cases were as follows: strong positivity for NKI/C3 and nuclear MITF in all cases, variable positivity for CD68 and S100, 4/5 with focal positivity for HMB-45, and no staining for melan-A, or muscle markers. 479 In their recent report on primary cutaneous PEComas, 469 Fletcher and colleagues acknowledged that this entity was morphologically similar to cutaneous PEComa, but they did not accept that they were synonymous on the basis of the negative melanocytic markers of the distinctive dermal clear cell neoplasm. 469
Inflammatory myofibroblastic tumor, also known as inflammatory fibrosarcoma, occurs predominantly in the lungs and mesentery, although the soft tissues of the head and neck, or the extremities, may be involved.480.481. and 482. The mean age at presentation is 13 years. 481 Systemic symptoms are often present. It appears to represent a spectrum of myofibroblastic proliferations, some of which have been included in the past as inflammatory pseudotumor, a heterogeneous ‘entity’.24.483.484. and 485. Helmut Kerl and colleagues believe that inflammatory pseudotumor includes tumors with detectable myofibroblasts that represent inflammatory myofibroblastic tumor and a group best called plasma cell granuloma, that represents a reaction pattern found in disorders, such as spirochete-induced fibroid nodules and localized chronic fibrosing vasculitis.484.486. and 487. HHV-8 specific DNA has been detected in one cutaneous case. 488
This tumor has a potential for recurrence and persistent local growth. 489 Metastases have been recorded but no reliable morphological parameters have been identified that predict prognosis.480. and 490. A recent report of 59 cases by Coffin, Hornick, and Fletcher noted that metastases were confined to ALK-negative cases. 481 Approximately 50% of cases recurred locally. 481 It has been suggested that inflammatory myofibroblastic tumor and inflammatory fibrosarcoma are synonymous or closely related entities. 491 They have been regarded as synonymous here. Recently it has been suggested that this tumor is derived from fibroblastic reticulum cells (myoid cells), and not from myofibroblasts. 492
The neoplastic nature of this lesion has been confirmed by the finding of recurrent clonal aberrations involving chromosome 2p23 similar to the ones found in anaplastic large cell lymphoma. 396 The fusion genes found are ALK-TPM4 and TPM3-ALK. 159 Approximately 50–70% of all tumors harbor ALK gene rearrangements. 481
Surgery remains the preferred treatment option for this tumor. Re-excision can be effective in managing recurrences. 481 Adjunctive therapy such as radiation and chemotherapy have been used, but there have been insufficient cases for meaningful analysis. 481 Steroids and NSAIDS have also been used. The use of imatinib has been reported to be effective. The development of kinase inhibitors of ALK is awaited.
The lesions are characterized by an admixture of myofibroblasts and fibroblasts, usually arranged in short interwoven fascicles (Fig. 34.18). 493In addition there is a polymorphic inflammatory cell component, consisting principally of lymphocytes and plasma cells. 493 Xanthoma cells are sometimes prominent. Myxoid areas and focal stromal hyalinization are usually present.
(A) Inflammatory myofibroblastic tumor. (B) There is an intimate admixture of inflammatory cells and spindle-shaped cells. (H & E)
Immunoperoxidase stains for vimentin, muscle-specific actin, smooth muscle actin, and cytokeratin are usually positive, indicative of a myofibroblastic tumor with admixed inflammatory cells.489. and 494. ALK-1 staining is present in approximately 50% of tumors.396. and 495. The staining pattern is usually cytoplasmic. Less consistent positivity is seen with desmin, and the epithelial markers AE1/AE3 and CAM5.2. 396 Skeletal muscle markers are negative.
There has been one report of a benign fibroblastic tumor, with a myxoid matrix, that does not fit into one of the recognized entities. 496 The well-circumscribed tumor was confined to the dermis and composed of stellate and spindle-shaped cells arranged loosely in a fascicular pattern resembling ‘tissue cultures of fibroblasts’. There was abundant stromal mucin. The cells expressed vimentin only and had the ultrastructural characteristics of fibroblasts. 496 A further case has since been reported. 497 It was on the nose of a 27-year-old woman. 497
Low-grade fibromyxoid sarcoma is a rare tumor characterized by bland histological features and a paradoxically aggressive clinical course.498.499.500.501. and 502. The hyalinizing spindle-cell tumor with giant rosettes (low-grade fibrosarcoma with palisaded granuloma-like bodies) is a variant of low-grade fibromyxoid sarcoma.502.503. and 504. They share the same cytogenetic abnormality (see below). 505 Low-grade fibromyxoid sarcoma has a tendency to develop in the deep soft tissues of young adults, whereas the myxofibrosarcoma occurs in older patients. 506 A variant of low-grade fibromyxoid sarcoma occurring in the superficial soft tissues has been described. It appears to affect children at a higher rate than the usual deep variant; its prognosis is also better than the deep tumors. 507 Local recurrence is common; distant metastasis, particularly to the lungs, occurs in up to 50% of cases.
The majority of low-grade fibromyxoid sarcomas bear either the t(7,16)(q32–34;p11) or t(11,16)(p11;p11) translocations, resulting in FUS-CREB3L2 or FUS-CREB3L1 fusions, respectively. 504 The former translocation is the more common. 504
The immunophenotype and ultrastructure suggest a fibroblastic tumor possibly with focal myofibroblastic differentiation. A recent study suggested a relationship to sclerosing epithelioid fibrosarcoma. 504
The tumor consists of bland spindle cells, showing a mainly whorled or focally linear arrangement, set in areas with an alternating fibrous or myxoid stroma. 499 Cellularity is low to moderate, and mitotic figures uncommon. 498 The tumor cells are small, spindle to stellate, with pale, poorly defined cytoplasm.
In a comparative study with myofibroblastic sarcoma, low-grade fibromyxoid sarcoma was found to be composed of bland spindle cells arranged in a whorled pattern with alternating myxoid and fibrous stroma, and few curvilinear vessels, whereas myofibroblastic sarcoma had prominent elongated curvilinear vessels and pseudolipoblasts, accompanied by abundant myxoid matrix. 506
The hyalinizing spindle-cell tumor with giant rosettes, a variant of low-grade fibromyxoid sarcoma (low-grade fibrosarcoma with palisaded granuloma-like bodies) is characterized by a collagenized fibroma-like component, cellular patches of hyperchromatic spindle cells, and hyalinized bodies ringed by nuclei giving the appearance of a rosette or palisaded granuloma.502. and 508.
Formerly regarded as a myxoid variant of malignant fibrous histiocytoma (MFH), myxofibrosarcoma is now considered to be a distinct entity on the basis of its reproducible cytoarchitecture and consistent clinicopathological characteristics.509.510.511. and 512. Myxofibrosarcoma is one of the most common fibroblastic sarcomas of older individuals.513.514.515. and 516. It is often located in the subcutaneous tissue of the limbs. Dermal involvement has also been reported but in such cases the bulk of the tumor is usually in the subcutis in the form of ill-defined nodules.517. and 518.
Myxofibrosarcoma is believed to be derived from fibroblasts. A large number of immature dendritic cells are present in the stroma. 519 The tumor is characterized by complex chromosomal aberrations, none of which is specific for this entity. 511
Treatment is wide surgical excision, with at least a 2 cm clearance. Radiotherapy has also been used for cases with infiltrative morphology, although there is no evidence that this is of any value. 517 Superficial tumors less than 5 cm in diameter have an excellent prognosis while disease-specific mortality rates are significantly related to tumor necrosis, large size (>5 cm in diameter), and decreased myxoid areas. 520
There is a spectrum of histological changes ranging from hypocellular, low-grade myxoid tumors to high-grade pleomorphic sarcomas. 513 When dermal involvement is present, it is usually predominantly myxoid in appearance, leading to confusion with benign myxoid neoplasms in small biopsy specimens.513. and 521. Low-grade areas are composed of abundant vacuolated, myxoid matrix with scattered spindled fibroblast-like cells and stellate cells. Intracytoplasmic mucin is present in some of these cells giving a signet-ring appearance. 513 The stroma contains curvilinear blood vessels. Intermediate-grade areas are more cellular with more nuclear hyperchromatism and pleomorphism. High-grade tumors resemble what was called pleomorphic MFH in the past. Cases mimicking pleomorphic hyalinizing angiectatic tumor have been described.522. and 523.
Nascimento, Bertoni, and Fletcher have reported 17 cases of an epithelioid variant of myxofibrosarcoma which accounted for less than 3% of myxofibrosarcomas in Fletcher’s consultation material. 511 It appears to be a more aggressive variant with approximately 70% local recurrence and 50% metastasis. 511 The cases were characterized by alternating hypercellular and hypocellular myxoid areas, the latter having prominent curvilinear blood vessels. The cellular areas had many epithelioid cells with round nuclei, vesicular chromatin, prominent nucleoli, and moderate amounts of eosinophilic cytoplasm. 511 Immunostains were negative for all markers studied.
In the more usual variant, strong staining with vimentin has been recorded, while scattered cells show myoblastic or histiocytic differentiation. 513 CD34 positivity has been recorded in some cells. 517 There is no staining with desmin, S100, EMA, or a keratin cocktail. The extracellular matrix has a heterogeneous composition that includes glycosaminoglycans and albumin. 524
The myxoinflammatory fibroblastic sarcoma is a rare, low-grade sarcoma of the hands and feet.510.525. and 526. It has been described at other sites.527. and 528. It is thought to be of fibroblastic origin, but a synovial origin has not been excluded. Its low-grade nature has been questioned, as metastases have been recorded. 529
Chromosomal abnormalities (translocations and supernumerary ring chromosomes) have been reported. 530
There are some histological similarities with myxofibrosarcoma, but with the addition of numerous inflammatory cells. 510 Three types of tumor cell occur in this lesion: spindle cells, large bizarre ganglion-like cells (virocyte or Reed–Sternberg-like cells), and a variable number of multivacuolated cells resembling lipoblasts and seen mainly within the myxoid areas. 529 In most cases CD34, epidermal growth factor receptor (EGFR) and CD163 are diffusely positive. 531
Enzinger and Weiss have defined a fibrosarcoma as a malignant tumor of fibroblasts that shows no evidence of other cellular differentiation. 532 There has been a marked decline in the diagnosis of fibrosarcoma in the last 20–25 years as a result of the delineation of the fibromatoses as a diagnostic entity and the recognition of low-grade fibromyxoid sarcoma and its variants hyalinizing spindle-cell tumor with giant (collagen) rosettes, and sclerosing epithelioid fibrosarcoma (see below). Furthermore, other spindle-cell tumors, which in the past were sometimes misdiagnosed as fibrosarcoma, such as spindle-cell melanoma and spindle-cell squamous carcinoma, as well as malignant peripheral nerve sheath tumors, can now be more confidently diagnosed with the assistance of various monoclonal antibodies and immunoperoxidase techniques. Malignant fibrous histiocytoma, while extant, also included cases that had previously been diagnosed as fibrosarcoma.
Very little has been written on fibrosarcoma in recent years. 533 However, as it is primarily a tumor of the deep soft tissues which only rarely develops in the skin and superficial subcutis, only brief mention will be made of it. Cutaneous fibrosarcomas may follow thermal burns and radiation therapy, or result from extension of a tumor arising in deeper tissues. The tumor affects predominantly middle-aged adults, although there is an uncommon clinical subset involving neonates and young children (congenital-infantile fibrosarcoma).534.535.536. and 537.
Congenital-infantile fibrosarcoma is much less aggressive than adult-type fibrosarcoma despite a similar histological appearance. It accounts for 20–50% of malignant soft tissue tumors in infants and neonates. Up to one-half may be present at birth, with the remainder appearing in the first 2 years of life. 538 Approximately 70% develop on the extremities, with the remaining 30% occurring on the head and neck. Axial lesions are uncommon. The lesions may be highly vascular and masquerade clinically as a hemangioma. 539 Recently, a novel chromosomal translocation t(12;15)(p13;q25) has been identified in the congenital-infantile group. 540 This translocation gives rise to an ETV6–NTRK3 gene fusion.159. and 540. It can be detected in paraffin-embedded tissue. 541
The term ‘fibrosarcoma, low-grade fibroblastic type’ has been used as a diagnosis of exclusion, for tumors not encompassed by the entities of low-grade fibromyxoid sarcoma and its histological variants. 542 It remains to be seen whether this represents a distinct fibrosarcoma type. 542 The cases reported as fibroblastic/myofibroblastic sarcoma of the skin are difficult to categorize. 148 There was a quantitative predominance of fibroblasts over myofibroblasts. All five cases were free of recurrence although the follow-up period for some cases was short. 543
Typically, fibrosarcomas are composed of interlacing fascicles of spindle cells forming a so-called ‘herringbone’ pattern. Mitoses are common. There is a variable meshwork of collagen and reticulin between the individual cells, the amount depending on the differentiation of the tumor.
The sclerosing epithelioid fibrosarcoma is composed of small to medium-sized cells with a clear or pale cytoplasm and arranged in cords and strands.545. and 546. The cells are surrounded by a prominent collagenous stroma. It appears to be related to low-grade fibromyxoid sarcoma.
The inflammatory fibrosarcoma appears to be synonymous with the inflammatory myofibroblastic tumor (see above).
The congenital infantile variant is usually more cellular and composed of smaller cells with prominent mitotic activity. Foci of necrosis may be present. Unlike the adult variant, the interlacing fascicular (herringbone) pattern is not always seen. 538
The tumor cells express vimentin but not as a rule, desmin, S100 protein, or smooth muscle actin. 537
Malignant tumors composed of myofibroblasts are increasingly being recognized, although their existence remains controversial. 27 They have a spectrum of histological grades, and this has led to a proliferation of terminology. Furthermore, some tumors contain a mixture of cells that mark for fibroblasts and myofibroblasts. In a review of myofibroblastic malignancies in 2004, Fisher classified myofibroblastic sarcomas as follows: 27
• Low-grade
Myofibrosarcoma (myofibroblastic sarcoma)
Inflammatory myofibroblastic tumor
Infantile fibrosarcoma
• Intermediate-grade
Myofibrosarcoma
• High-grade
Pleomorphic myofibrosarcoma (histologically identical to MFH).
Most cases of myofibrosarcoma occur in the oral cavity, deep soft tissues, and bone, but about 10% or more involve the subcutaneous tissue. Low-grade lesions are usually indolent and often cured by complete excision, whereas some intermediate-grade myofibrosarcomas may recur and even metastasize. Pleomorphic myofibrosarcomas are usually regarded as being the same as pleomorphic malignant fibrous histiocytomas (see p. 838). 27
Low-grade myofibrosarcomas (myofibroblastic sarcomas, low grade) are distinctive spindle-cell tumors showing myofibroblastic differentiation. 547 Of the 18 cases reported by Mentzel et al in 1998, only two involved the subcutis. 547 Recurrences and metastases were recorded for some of the cases, but there was no follow-up for the subcutaneous cases. 547 Fisher has subsequently suggested that at least one of these cases is likely to have been of intermediate-grade malignancy. Other series have been reported that have included low-grade and intermediate-grade tumors. 548 Unlike inflammatory myofibroblastic tumor, low-grade myofibrosarcoma (myofibroblastic sarcoma) is not a member of the family of ALK-positive tumors. 549
Myofibrosarcomas are composed of stellate or spindled cells with tapered or ovoid nuclei with small nucleoli and scanty or moderate amounts of eosinophilic cytoplasm with variably distinct cell boundaries. 27 The cells are arranged in fascicles, sheets, or storiform whorls, with variable collagenous or myxoid stroma. There is focal nuclear atypia. Abnormal mitotic figures are rare and necrosis occurs in a minority of cases. 27
The majority of low-grade myofibrosarcomas are positive for smooth muscle actin, and fewer than half express desmin, usually focally. 27Calponin is usually diffusely positive, whereas h-caldesmon is focally expressed in an occasional case. 27
The term ‘fibrous histiocytoma’ was introduced for a group of tumors that share certain morphological features, such as the presence of fibroblast-like spindle cells and presumptive histiocytes, cells that were assumed to arise from a tissue histiocyte that could function as, or transform into, a fibroblast. Although these tumors appear to be histogenetically heterogeneous, it is still convenient to retain the term ‘fibrohistiocytic’ in a morphologically descriptive sense for tumors which show features of fibroblastic and histiocytic differentiation. Some of these tumors appear to be derived from the dermal dendrocyte. Others are of uncertain lineage. 551
The large number of terms that have been used for dermatofibroma (histiocytoma,552. and 553. fibrous histiocytoma,554.555. and 556. sclerosing hemangioma, 557 nodular subepidermal fibrosis558. and 559.) reflects the remarkable variation in its histological features, and continuing controversy regarding its histogenesis. 560 However, the presence of transitional patterns and different morphological components in the same lesion is strong evidence in favor of its basically common nature and histogenesis. 554 There has been a tendency to use the term ‘dermatofibroma’ for the usual fibrocollagenous and storiform variant and ‘fibrous histiocytoma’ for some of the unusual variants.
Dermatofibromas are common, accounting for almost 3% of specimens received by one dermatopathology laboratory. 561 There is a predilection for the extremities, particularly the lower, of young adults.552. and 561. There is a female preponderance. 561 Rare sites of involvement have included the fingers,558. and 562. palms and soles, 563 the scalp, 558 the face, 564 a vaccination scar, 565 and within a tattoo. 566
Dermatofibromas are round or ovoid, firm dermal nodules, usually less than 1 cm in diameter. A ‘giant’ variant has been recorded.567.568.569. and 570. It preferentially develops on the lower limbs.571. and 572. Satellitosis is a rare finding at the edge of a giant dermatofibroma. 573 Polypoid, flat, atrophic, and depressed configurations also occur.574. and 575. Both the Meyerson phenomenon and a halo of asteatotic eczema have been reported around a dermatofibroma. 576 Lesions with a preponderance of histiocytes are often larger, and the aneurysmal (angiomatoid) variants may measure up to 10 cm in diameter.577. and 578. Dermatofibromas are usually a dusky brown in color but aneurysmal variants may be red, and tumors with abundant lipid can be cream/yellow, particularly on the cut surface of the excised lesion. They often show a characteristic central white, scar-like patch on dermatoscopic examination,579. and 580. and a delicate pigment network at the periphery. Other findings have been recorded,581.582. and 583. including homogeneous blue pigmentation simulating a blue nevus. 584 Dermatofibromas are most often solitary, but 2–5 lesions are present in 10% or so of individuals. 552 Multiple lesions have been reported,585.586.587. and 588. usually as a rare complication of immunosuppressive therapy563.589.590. and 591. or the acquired immunodeficiency syndrome.592.593.594.595.596. and 597. Eruptive lesions have appeared after the commencement of highly active anti-retroviral therapy (HAART), 598 and in other clinical circumstances.599.600.601.602.603.604.605.606. and 607. Familial eruptive dermatofibromas have also been reported. 605 There are only a few reports of patients with large numbers of tumors;556.608.609.610.611.612. and 613. one such case was reported as ‘disseminated dermal dendrocytomas’. This case may have been a different entity – progressive nodular histiocytosis (see p. 955). Clinical variants include the aneurysmal type, already referred to, 577 and the rare annular hemosiderotic histiocytoma in which multiple brown papules in annular configurations were present on the buttocks. 614 Some authors regard the hemosiderotic dermatofibroma as an early stage in the development of the aneurysmal variant. 615 Some aneurysmal lesions may recur. 616 Spreading satellitosis has been reported in one case. 617
Although regarded as a benign tumor or reactive inflammatory process that may uncommonly recur locally (see below), several cases of metastasizing cellular dermatofibroma have been recorded.618.619.620. and 621. Clonality has since been found in some cases of dermatofibroma but no consistent karyotypic aberration has been found.622. and 623. Mentzel and colleagues have drawn attention to the aggressive nature of dermatofibromas arising on the face. 624 They often involve deeper structures, and have an increased rate of local recurrence. They need to be excised with wider margins than the classic type. 624
The deep fibrous histiocytoma, initially reported by Fletcher in 1990, occurs in the subcutaneous or deep tissues. 625 A larger series of 69 cases from Fletcher’s consultation files was reported in 2008. 626 The series included 41 males and 28 females, ranging in age from 6 to 84 years. More than half were located on the extremities, but about 10% occurred in the deep soft tissues of the peritoneum, mediastinum, or pelvis. 626 The non-visceral lesions were subcutaneous in location. They were well-circumscribed tumors that ranged in size from 0.5 to 25 cm (median, 3.0 cm). Of those cases with available follow-up, 8 (22%) had a local recurrence, but in all 8 cases, the tumor had been marginally or incompletely excised. 626 Metastases occurred in two patients; both ultimately died of the disease. Apart from their large size, the metastasizing tumors were otherwise identical to the other tumors. 626 A case associated with a melanoma has been reported. 627
The atypical fibrous histiocytoma (dermatofibroma with monster cells) 628 is another uncommon variant with a low recurrence rate and the rare development of distant metastases. 629
Fibroblasts, non-lysozyme containing histiocytes, 559 and endothelial cells have all been regarded at some time as the cell of origin. 552 Dermatofibromas have been regarded by some authors as a benign tumor, and by others as a fibrosing inflammatory or reactive process.628.630. and 631. In support of the latter concept is the history of trauma, blunt or piercing, recorded in up to 20% of cases. 558 An insect bite has preceded the development of a dermatofibroma. 552 HHV-8 has not been detected. 632 On the basis of the immunoreactivity of 30–70% of the constituent cells with factor XIIIa, an origin from the ‘dermal dendrocyte’ has been proposed.633.634. and 635. The dendritic cells appear to be derived from blood monocytes or a stem cell common to both. 636 Recent studies have found HSP47-positive fibroblasts in dermatofibromas.305. and 637. They were the major constituent cells in the fibrocollagenous type. The authors postulated two cell lineages – fibroblastic, and bone-marrow derived monocyte/macrophages (dermal dendrocytes). 637 A recent study concluded that a dermatofibroma is composed predominantly of cells of macrophage phenotype with a variable amount of myofibroblastic elements. 638 It has been suggested that fibroblastic tumor cell-derived stem cell factor is responsible for the epidermal hyperpigmentation that overlies dermatofibromas. 639
Surgical excision (including shave excision) is the usual method of treatment. Recurrence is uncommon, even in cases with positive margins. Two exceptions to this statement exist. The cellular variant, which often extends into the subcutis, and dermatofibromas of the face need to be excised with wider margins than the classic type to prevent recurrence. 624
Dermatofibromas are poorly demarcated tumors centered on the dermis. There is sometimes extension into the superficial subcutis which may take the form of septal extension or a well-demarcated bulge.640. and 641. A ‘pure’ subcutaneous form is exceedingly rare. 642 A grenz zone of variable thickness is present in about 70% of cases. 552 Sometimes this contains dilated vessels. 552 Dermatofibromas are composed of a variable admixture of fibroblast-like cells, histiocytes, some of which may be xanthomatous or multinucleate, and blood vessels. The terms ‘nodular subepidermal fibrosis’, ‘histiocytoma’, and ‘sclerosing hemangioma’ were used in the past for variants where one of these three components predominated. Nowadays it is usual practice to refer to fibrocollagenous, histiocytic, and angiomatous variants to reflect these differences in composition.554. and 643. Storiform, aneurysmal (angiomatoid), and fibrous histiocytoma variants are also recognized (see below). Numerous other variants have been described in recent years. Many are histological curiosities. They are listed in Table 34.2.
• Fibrocollagenous
• Storiform
• Cellular
• Histiocytoma
• Lipidized
• Angiomatous
• Aneurysmal
• Clear cell
• Granular cell
• Halo
• ‘Monster’ cell
• Osteoclastic
• Myofibroblastic
• Myxoid
• Keloidal
• Palisading
• Atrophic
• Subcutaneous
• Combined
The fibrocollagenous type has a predominance of collagen and fibroblast-like cells in an irregular or whorled arrangement (Fig. 34.19). 554 Sometimes there is striking cellularity of the lesion beneath the epidermis, with less cellular areas deeply. 554 The lichenoid dermatofibroma has a dense cellular infiltrate impinging on the undersurface of the epidermis producing basal cell damage that often leads to clefting. 644 Mitoses are rare. Uncommonly there is a high mitotic rate but this does not necessarily indicate the likelihood of recurrence. 645 Histiocytes are usually imperceptible, but there is a variable vascular component. Hemosiderin is sometimes present in the cellular areas, but is uncommon elsewhere. Bone646 and calcification558 are rare constituents of the stroma. Another variant of the fibrocollagenous type has nuclear palisading and prominent Verocay-like bodies in part of the lesion, usually the center. 643 This palisading variant is positive for vimentin and factor XIIIa. 647 A rare variant with sclerotic areas resembling a sclerotic fibroma has been reported. 648 The author has seen several such cases.
Dermatofibroma. (A) The epidermis overlying the dermal tumor is acanthotic and papillomatous. There is a central ‘dell’. (B) The cells have a storiform arrangement. (H & E)
Those cases with a prominent storiform pattern are sometimes termed ‘fibrous histiocytoma’ to distinguish them from the more usual fibrocollagenous type.554. and 649. Included within this group is the so-called ‘cellular benign fibrous histiocytoma’.621.650. and 651. These tumors often extend into the subcutis; they sometimes recur locally after incomplete surgical removal. 652 Normal mitotic figures are common in the cellular variant, and 12% of cases show central necrosis. Up to 60% of cases show focal positivity for smooth muscle actin in a minority of the cells. The cellular variant differs from dermatofibrosarcoma protuberans by its smaller size, fewer mitoses, and less extensive involvement of the subcutis. 649 It is negative for CD34, in contrast to dermatofibrosarcoma protuberans. 650
Another variant of fibrous histiocytoma occurs in the subcutis and deep soft tissues.625. and 626. This deep fibrous histiocytoma consists of bland ovoid to spindle-shaped cells arranged in a storiform pattern with admixed lymphocytes. 626 Just less than half have been monomorphic while the others have had multinucleate giant cells, osteoclastic giant cells, and/or foam cells. 626 In a significant number of cases, a hemangiopericytoma-like growth pattern has been present.626. and 653. Stromal hyalinization has sometimes been present. Expression of CD34 was present in 4% of cases, while smooth muscle actin was positive in 38% of cases. 626
The cerebriform collagenoma occurring in patients with Proteus syndrome may have a distinctive storiform pattern, that may even resemble a dermatofibrosarcoma protuberans. 654
The histiocytic variant (‘histiocytoma’) has nests and sheets of histiocytes in a poorly cellular collagenous stroma (Fig. 34.20).552. and 554. There are often many foam cells and giant cells, which may be of foreign body or Touton type. 552 Hemosiderin and lipid are commonly present. 558 Foam cells and cholesterol clefts may be prominent in dermatofibromas if there is underlying hyperlipoproteinemia. 655 These foam cells are different from the clear cells seen in the rare clear cell dermatofibroma (Fig. 34.21), in which the intrinsic spindle cells have pale cytoplasm.656.657.658. and 659. The balloon cell dermatofibroma also has a clear cell ‘phenotype’ (Fig. 34.22). 660 The granular cell dermatofibroma has epithelioid cells with a granular cytoplasm (Fig. 34.23).661.662.663.664. and 665. More traditional-appearing areas are usually present.
(A) Dermatofibroma (histiocytic variant). (B) Sheets of ‘histiocytes’, some with vacuolated cytoplasm, are admixed with spindle-shaped cells. (H & E)
Dermatofibroma of clear cell type. (H & E)
Dermatofibroma of balloon cell type. (H & E)
Dermatofibroma of granular cell type. (H & E)
The lipidized variant, which tends to be large and situated around the ankle region, is composed of numerous foam cells, smaller numbers of siderophages, and stromal hyalinization, which is typically ‘wiry’, keloid-like or osteoid-like. 666 These stromal changes set it apart from the more usual histiocytoma variant. There is no abnormality in serum lipids in this group. 667
In the angiomatous (vascular) variant there are numerous small branching vessels in a variable collagenous stroma. Focal hypercellularity resembling that seen in the fibrocollagenous type is often present. Hemosiderin is present in about half the cases.
The aneurysmal (angiomatoid) variant is distinct, with blood-filled spaces occupying up to one-half of the lesion.577.668.669.670. and 671. These vary from narrow clefts to large cavernous cysts (Fig. 34.24). The vascular channels are surrounded by histiocytes which contain hemosiderin, and by foam cells and fibroblasts. 577 Squamous-lined cysts were present in the stroma of two cases.616. and 672. Solid areas with the more usual appearance of a dermatofibroma are almost invariably present. 673 This feature, together with the presence of foam cells, allows a distinction to be made from Kaposi’s sarcoma. 668 The aneurysmal variant of malignant fibrous histiocytoma is much more pleomorphic and is usually centered on the deeper soft tissues.
(A) Dermatofibroma (aneurysmal variant). (B) There are large vascular spaces and abundant hemosiderin in the surrounding stroma. (H & E)
Large, bizarre cells with abundant foamy cytoplasm and hyperchromatic nuclei have been reported in some dermatofibromas.628.650.674.675. and 676. These ‘monster cells’ are sometimes binucleate or multinucleate.570.628. and 677. Pleomorphism varies from focal and minimal, to moderate or marked. 629 Atypical mitoses are found in one-third of cases. 629 Numerous cells express Ki-M1p.212. and 678. The term ‘atypical fibrous histiocytoma’ has also been used for this variant; it is the preferred designation (Fig. 34.25). 629In the review of 59 cases of atypical fibrous histiocytoma of the skin reported by Kaddu, McMenamin, and Fletcher in 2002, they noted multinucleated giant cells, sometimes hemosiderin-rich, and atypical mitoses in nearly a third of cases. 629 A variant of dermatofibroma with osteoclast-like giant cells (osteoclastic) has also been reported.679. and 680. In the combined dermatofibroma two or more variant patterns coexist in a single lesion. 681 A combined lesion with islands of indeterminate cells and eosinophils set in a spindle-cell tumor with the features of a dermatofibroma has been reported. 682
(A) Atypical fibrous histiocytoma. (B) Giant (‘monster’) cells can be seen. (H & E)
Two other rare variants are the myxoid dermatofibroma with abundant stromal mucin mimicking a cutaneous myxoma604. and 683. and the keloidal dermatofibroma with areas of thick eosinophilic collagen. 684 There is marked thinning of the dermis in the atrophic variant. 685 The proliferating cells in this variant appear to phagocytose the elastic fibers within the lesion. 686 Ulcerated and erosive variants have also been described. 644
Other histological changes that can be seen in dermatofibromas include the presence of lymphoid nodules.552. and 687. They are an uncommon finding. The lymphoid tissue is usually in the subjacent fat, or at the periphery of the lesion. 687 Germinal center formation is faint but definite in many instances. 687 Mast cells and a sparse infiltrate of chronic inflammatory cells are sometimes present. A diffuse stromal infiltrate of eosinophils was present in one case. 688 Another uncommon finding is the presence of smooth muscle bundles in the adjacent dermis. 689Myofibroblastic differentiation may occur within the tumor cells. 690 Rarely it is a prominent feature.691. and 692. The cells express smooth muscle actin and HHF35. 691
A morphologically related entity is the epithelioid cell histiocytoma (see below), which is composed of large angulated epithelioid cells that have some resemblance to the cells of a Spitz nevus. 693 It is considered separately because of its distinctive appearance.
Various changes may be present in the overlying epidermis in dermatofibromas. 694 Acanthosis, sometimes with basal hyperpigmentation, is present in approximately 70% of lesions.695. and 696. Other changes include a seborrheic keratosis-like pattern, 696 epidermal atrophy696 or, rarely, intraepidermal carcinoma697 or pseudoepitheliomatous hyperplasia and focal acantholytic dyskeratosis. 698 In 5–8% of cases there is a spectrum of distinctive epidermal changes ranging from basaloid hyperplasia, often with rudimentary pilar structures, to undoubted basal cell carcinoma.553.694.699.700. and 701. Sebaceous structures and sebaceous hyperplasia are uncommonly present (Fig. 34.26).553.702. and 703. Up-regulation of the EGF-EGFR and the HH-Patched signaling pathways may be involved in this sebaceous hyperplasia. 704 Unequivocal basal cell carcinoma is uncommon,646. and 705. and some of the cases reported as this are examples of exuberant basaloid hyperplasia; 706 the distinction is not always easy. Recently, a significant increase in CK20-stained Merkel cells has been demonstrated in these basaloid proliferations as compared to basal cell carcinoma. 707 It has been suggested that the hair follicle-like structures represent regressive changes in pre-existing follicles and not the induction of new pilar units. 561 Epidermal growth factor or some other factor produced by the underlying tumor cells may be responsible for these proliferative changes.708. and 709. Loss of heterozygosity in the patch gene is often present in the basal cell carcinoma-like changes in the epidermis. 710
Dermatofibroma with overlying sebaceous glands. (H & E)
A small percentage of cells, usually at the periphery, are S100 positive. 643α1-Antitrypsin and macrophage markers such as CD68 and HAM-56 have been reported in some of the tumor cells.711. and 712. Vimentin and α-smooth muscle actin are often present in the spindle-shaped cells.713. and 714. The finding of factor XIIIa in many dermatofibromas provides a marker for this tumor, 633 although it is also present in acral angiofibromas, scars, keloids, and some atypical fibroxanthomas, but not in dermatofibrosarcoma protuberans (DFSP). 634 The staining in dermatofibromas is often restricted to the periphery of the lesions, leading some authorities to question its specificity for dermatofibromas. 713 It is usually absent in the aneurysmal variant. 671 It was initially thought that the use of CD34 and factor XIIIa staining would differentiate dermatofibroma from DFSP. Unfortunately some overlap occurs,715. and 716. including the occurrence of indeterminate fibrohistiocytic lesions with a dual population of CD34 and factor XIIIa-positive cells (see below). In a recent study of the immunohistochemical properties of dermatofibroma and DFSP, CD34 was strongly expressed in 25% of dermatofibromas and 80% of DFSP, whereas factor XIIIa was strongly expressed in 95% of dermatofibromas and 15% of DFSP. 717 This same study demonstrated the strong expression of tenascin (an extracellular matrix glycoprotein) at the dermal–epidermal junction in all cases of dermatofibroma but not in any DFSP. 717 Staining within the lesion was of no help in differentiating these two lesions. Similar results were recorded in an earlier study. 718 Matrix metalloproteinases are expressed in dermatofibromas but are not up-regulated in DFSP. 719 Procollagen 1 and syndecan-1 are also found in dermatofibromas.720. and 721. The expression of metallothionein has been reported in dermatofibromas but not DFSP. 641 A high proliferative index detected by MIB-1 staining excludes the possibility of a common dermatofibroma, 722 but not the cellular or deep variant. 723 CD10, a marker for atypical fibroxanthoma and positive in many other tumors, is also expressed in dermatofibromas. 724
Ultrastructural studies have given conflicting results, with the cells variously reported as resembling fibroblasts, histiocytes, and even myofibroblasts.725. and 726. Endothelial cells with Weibel–Palade bodies were the prominent cell noted in one study. 557 Vessels are conspicuous in the angiomatoid variant. 669 There is a need for a reappraisal of the ultrastructural findings in the light of the immunoperoxidase studies implicating the dermal dendrocyte as the cell of origin (see above).
Epithelioid cell histiocytoma is regarded as a variant of fibrous histiocytoma (dermatofibroma). 693 It is considered separately from dermatofibroma because of its distinctive histological appearance, which resembles to some extent the intradermal variant of Spitz nevus. 727 The lesion usually presents as an elevated nodule on the extremities of adults, although other sites have been recorded. 728 In one patient a lesion on the arm was followed a month later by another lesion on the nostril. 729 Although a dendrocyte origin is usually favored, an endothelial cell origin has also been proposed on the basis of the ultrastructural findings in one case. 730 On the basis of immunohistochemistry, an origin from microvascular fibroblasts and dendritic cells has been proposed (see below). 731 The term ‘solitary epithelioid histiocytoma’ has recently been proposed as a group term for tumors previously called reticulohistiocytoma and multicentric reticulohistiocytosis (see p. 956). 732 This is bound to lead to confusion.
The polypoid lesions often have an epidermal collarette. 728 There is relatively sharp circumscription. 733 All lesions are characterized by sheets of angulated epithelioid cells with abundant eosinophilic cytoplasm. Scattered mitoses are usually present. There are many small blood vessels, and there is often a perivascular mosaic pattern of epithelioid and plump spindle cells. Sometimes this results in hemangiopericytoma-like features. 734 Conspicuous cellular whorls of spindle cells were present between the epithelioid cells in one case. 735 Xanthoma and/or giant cells are often present in small numbers. 736 The stroma contains a light mononuclear inflammatory cell infiltrate. 728 A cellular variant, with one or more dermal nodules composed of large epithelioid cells, has been reported. 737 It usually extends into the deep dermis. A granular cell variant has also been described. 738 Rare cases occur with combined features of dermatofibroma and epithelioid cell histiocytoma (Fig. 34.27).
(A) Epithelioid cell histiocytoma. (B) This case has both epithelioid cells and overlap features with dermatofibroma. (H & E)
Factor XIIIa is expressed by 50–70% of the cells, whereas S100 and HAM-56 may be expressed by up to 5% of cells. HMB-45 is negative. 737 The granular cell variant expresses CD68. 738 The vascularity is confirmed by a CD31 or CD34 preparation. 739 Early lesions, but not late ones, contain up to 40% of CD34+ cells, leading to the suggestion that the constituent cells appear to arise from the activation of resident microvascular CD34+ dermal fibroblasts and the accumulation of factor XIIIa+ dendritic stromal-assembly histiocytes. 731 Vimentin positivity was recorded in one study. 740
Plexiform fibrohistiocytic tumor is a rare tumor that involves predominantly the upper limb of infants and children.741. and 742. It presents as a slow-growing, painless mass. Over 100 cases have now been reported. 742 It has an immunophenotype suggestive of myofibroblastic differentiation.743. and 744. Grossly, the tumor involves the superficial soft tissue, often arising at the dermal–subcutaneous junction. 745 Dermal variants have been described. 746 The lesion is poorly demarcated, lobulated or micronodular. It is white-gray in color, and averages 1–3 cm in size. 747
Local recurrence is common, usually because of incomplete removal. Metastasis has been recorded, 748 but not in infants. 747 It is included here with the fibrohistiocytic lesions, rather than the myofibroblastic tumors, because of the dual cell population.
Wide local excision is the recommended treatment. 747
The appearances vary, depending on the relative proportion of the two major components – fascicles of fibroblastic cells, and aggregates or discrete nodules of histiocyte-like cells. The fascicles often ramify, giving a plexiform appearance. 747 The ‘histiocytic’ nodules are composed of eosinophilic epithelioid cells. Osteoclast-like giant cells with 3–10 nuclei are often present (Fig. 34.28). 749 Hemosiderin is often present in the stroma. 747 Granular cell change is a rare finding. 750 A myxoid variant has also been reported. 751 The cases reported as plexiform xanthomatous tumor had some areas resembling this lesion with the addition of numerous xanthoma cells. 752
Plexiform fibrohistiocytic tumor. (A) This subcutaneous tumor has fibroblastic fascicles and histiocytic nodules. (B) Osteoclast-like giant cells are present in the nodules. (H & E)
The tumor cells express vimentin, smooth muscle actin but not factor XIIIa. They are positive with HHF35.742. and 746. A minority of the cells in the histiocytic nodules stain for CD68 but not CD45 or Mac387.745. and 748. The osteoclast-like giant cells are positive with KP1. 746
Ultrastructurally, the tumor cells have features of myofibroblasts and histiocyte-like cells. 742
Giant cell fibroblastoma is a rare soft tissue tumor occurring predominantly in childhood. 753 The back, thigh, and chest are the favored sites. The vulva is a rare site of involvement. 754 There is a male predominance. Macroscopically, the tumor appears gray-pink with a partly gelatinous consistency. 755 Cases recurring as, or transforming to, dermatofibrosarcoma protuberans (DFSP) have been reported.756.757.758. and 759. Foci resembling giant cell fibroblastoma have also been seen within a DFSP (hybrid lesions),760. and 761. as has the phenomenon of a DFSP recurring as a giant cell fibroblastoma.762. and 763. Furthermore, both possess a similar cytogenetic abnormality (see below).761. and 764. These findings have led to the suggestion that giant cell fibroblastoma is a juvenile variant of DFSP. Giant cell fibroblastoma has been misdiagnosed as dermatofibroma. 765
Wide local excision is the recommended treatment. 761 Local recurrence is common, following incomplete surgical removal. 766
The dermis and subcutis are involved by an infiltrating spindle-cell tumor in which the tumor cells are embedded in a loose connective tissue matrix showing areas of mucinous change. Scattered through the tumor are uninucleate and multinucleate giant cells. A characteristic feature is the presence of branching sinusoidal spaces lined by cytoplasmic extensions of the spindle and giant cells.767. and 771. These cells are negative for S100 protein, desmin, muscle-specific actin, and factor VIII, but show scattered positivity for α1-antitrypsin.767.768. and 772. Both the spindle cells and the giant cells stain consistently for vimentin. 770 Although earlier reports gave conflicting results on the expression of CD34 by this tumor, 773 recent reports have found positivity in a significant number of cases. 774 The giant cells are almost invariably positive.773. and 775.
Dermatofibrosarcoma protuberans (DFSP) is a slow-growing, locally aggressive tumor of intermediate malignancy, and of disputed histogenesis, with a marked tendency to local recurrence, but which rarely metastasizes.777.778.779. and 780. Its annual incidence in the United States is 4.2 per million; 781 it is slightly less in one area of France. 782 It occurs in all races.783. and 784. It has a predilection for the trunk and proximal extremities of young and middle-aged adults,785.786. and 787. but other sites are rarely involved.775.788.789.790. and 791. Congenital and familial cases, and onset in childhood are all rare.792.793.794.795.796.797.798.799.800.801. and 802. Congenital cases may be misdiagnosed as benign lesions because of their deceptive appearance;803.804. and 805. they may be mistaken for a vascular birthmark. 806 In the United States, DFSP is more common in blacks than in whites, and there is a higher incidence in women than men. 781 The lesions are solitary or multiple polypoid nodules, often arising in an indurated plaque of tumor.807.808.809. and 810. Approximately 40% of cases are initially non-protuberant. 811 Atrophic variants are uncommon; they may be difficult to diagnose clinically.792.799.812.813.814. and 815. The involved area of skin measures from 0.5 to 10 cm or more in greatest diameter. A violaceous or red color is sometimes present, but at other times the nodules are flesh colored. 808 A history of previous trauma at the site is sometimes given.788. and 816. Accelerated growth has been reported during pregnancy; 817 metastases followed pregnancy in one case. 818 Previous arsenic exposure was present in one case. 819 It has also been reported in a patient with X-linked agammaglobulinemia, 820 in a renal transplant recipient, 821 and at the site of prior leishmanization, and in a smallpox vaccination scar.822. and 823. DFSP has also been reported in association with a nuchal-type fibroma, 824 and with multiple spindle-cell lipomas. 825 Rarely, DFSP may masquerade as a skin tag, 826 a cyst, 827 or as a primary breast lesion when located in the skin of the breast. 828
Local recurrence occurs in up to one-third of all cases.808. and 829. Lower recurrence rates have been reported using Mohs micrographic surgery.830. and 831. Hematogenous metastases, although rare, 832 are more common than those to regional lymph nodes.833.834.835. and 836. In a recent study, the relative 5-year survival was estimated to be 99.2%. 781 Progression of a recurrent DFSP to a malignant fibrous histiocytoma, 837 and the development of fibrosarcomatous, neurofibrosarcoma, and myxofibrosarcomatous areas in DFSPs is well documented, occurring in 10–15% of cases.838.839.840.841. and 842. This progression to fibrosarcoma is associated with microsatellite instability and p53 mutations.843. and 844. The fibrosarcomatous variant of DFSP is said to be a much more aggressive lesion with metastatic potential;845. and 846. however, a study of 17 cases showed that only four cases recurred and none developed metastasis within the 5-year follow-up period. 847 The lesions were treated initially with wide local excision with negative margins. 847 In a recent study from the Mayo Clinic on the prognostic significance of fibrosarcomatous transformation, four patients (10%) had metastases and two patients died of disease. 844 Giant cell fibroblastoma has a close relationship with DFSP; either tumor may recur with features of the other (see above).
The pigmented storiform neurofibroma of Bednar (Bednar tumor) is now regarded as a pigmented variant of DFSP.848.849.850.851.852.853.854. and 855. It is an exceedingly rare tumor which accounts for only 1–5% of all cases of DFSP. 853 It occurs in a similar clinical setting to DFSP but, as yet, no metastases have been recorded. It is thought to represent the colonization of a DFSP by melanocytes. 856 Dermal melanocytosis has been reported in one case raising the possibility of neuroectodermal multidirectional differentiation for both this and the DFSP components. 857 A case has been reported that arose at the site of a previous immunization. 858 A congenital variant has also been reported. 859
The myxoid DFSP is uncommon. In a review of 23 cases from his consultation files, Fletcher found that the majority of cases were found on the extremities, and the head and neck. Three cases were from the anogenital region. 860 Clinical follow-up in eight cases revealed local recurrence in two, but there was no evidence of metastasis. 860
Although DFSP is traditionally regarded as a fibrohistiocytic tumor, several ultrastructural and immunohistochemical studies have questioned this viewpoint; 861 a neuroectodermal origin has also been suggested, but the immunophenotype (see below) does not support this concept. Fletcher and colleagues have commented that ‘the most pragmatic solution at this time may be to regard these tumors as a heterogeneous group, in line with the pluripotentiality of mesenchyme’. 862 A fibroblastic origin seems most likely on the basis of the immunophenotype and ultrastructure of the cells. 863 Platelet-derived growth factor has been identified in DFSP and other fibrohistiocytic lesions. 864
Cytogenetically, DFSP is characterized by a reciprocal translocation, t(17;22)(q22;q13), and a supernumerary ring chromosome derived from the translocation r(17;22).791. and 865. Other rare abnormalities have been found. Cloning studies reveal that the translocations result in the fusion of two genes, COL1A1 and PDGFB, related respectively to the formation of type I collagen and platelet-derived growth factor.865.866.867. and 868. A recent study has shown that COL1A1–PDGFB fusion can be identified in virtually all cases of DFSP when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in-situ hybridization assays. 869 Cases without a detected cytogenetic abnormality are in the literature from earlier days. 870 This fusion product has also been demonstrated in the granular cell and myxoid variants, in the Bednar tumor, and in giant cell fibroblastoma.761.871. and 872. A recent study has suggested that genomic gains of COL1A1–PDGFP are associated with the histological evolution of giant cell fibroblastoma into DFSP. 873
Macroscopically, the cut surface of the tumor nodules appears firm, and gray-white in color. Recurrent lesions with abundant mucin may have a more glistening appearance. The pigmented variant may appear slate gray or black if sufficient melanin is present within the tumor. 853
Aggressive surgical treatment with margins up to 5 cm has been recommended for this tumor. 874 In one French study, the local recurrence rate was 47% when margins less than 3 cm were used compared to a recurrence rate of only 7% when margins ranging from 3–5 cm were used. 782 The recurrence rate is higher on the head and neck than on the trunk because of limitations in the clearances in the former site. Adjuvant radiotherapy reduces the local recurrence rate when wide local excision is impossible because of anatomical constraints or functional and cosmetic concerns.782. and 842. Mohs surgery can be used in these cases,802. and 875. as well as in recurrent disease. 876 Preoperative MRI may be used in children to delineate the size and extent of the lesion prior to surgery. 877
Dermatofibrosarcoma protuberans is a dermal tumor which almost invariably extends into the subcutis, where it infiltrates around small groups of fat cells in a characteristic manner (Fig. 34.29 and Fig. 34.30). 807 A variant localized to the subcutis has been reported.879. and 880. Deeper extension to underlying muscle is rare. 881 A superficial grenz zone is often present, but extension to the epidermis, with ulceration, is sometimes seen. 862 Dermal appendages are surrounded but not invaded.
Dermatofibrosarcoma protuberans. This deep biopsy shows a tumor involving dermis and subcutis. (H & E)
Dermatofibrosarcoma protuberans. Tumor cells insinuate between fat cells in the subcutis and extend into the underlying muscle. (H & E)
DFSP is a spindle-cell tumor composed of interwoven bundles of rather uniform, small spindle cells with plump nuclei. There are small amounts of intermingled collagen. At points of intersection of the fascicles there may be an acellular collagenous focus from which the fascicles appear to radiate (Fig. 34.31). 832 This is referred to as a storiform or cartwheel pattern. Scattered mitoses are present, but atypical forms are rare. Increased cellularity of the tumor, and more than eight mitoses per 10 high-power fields, appear to be associated with a predisposition to metastasis. 832 Other histological features include an occasional histiocyte and multinucleate giant cell. 861 Thin-walled capillaries are randomly distributed. Fibrosarcomatous change, seen particularly in recurrences, represents a form of tumor progression.777. and 882. The proportion of this element varies markedly from case to case. 774 The fibrosarcomatous areas may show focal myxoid change,805. and 839. keloid-like hyalinization, giant rosettes, 883 pigmented melanocytes, 884 and myoid nodules and bundles.885.886.887. and 888. The leiomyomatous nodules appear to be, at least in part, of vascular origin and reactive in nature. 889 The fibrosarcomatous areas express CD34 in less than 50% of cases. 884 The expression of fusion transcripts in both conventional and fibrosarcomatous areas supports a common histogenesis of the two components. 865
Dermatofibrosarcoma protuberans. The spindle-shaped cells have a storiform or cartwheel arrangement. (H & E)
Myxoid DFSP is a distinct variant with greater than 50% myxoid stroma. 860 It is often found for the first time in recurrent lesions. 872 There are large myxoid areas which are paucicellular, surrounded by more typical areas with storiform features. 890 Pigment is sometimes present. 860Atrophic and granular cell variants of DFSP have also been proposed.891.892. and 893. Some cases of DFSP have areas resembling giant cell fibroblastoma (see above). 774 Furthermore, either tumor may, rarely, recur as the other.756.757.758.760.762. and 773. The related giant cell angiofibroma may also occur within a DFSP. 332
The pigmented variant has melanin-containing dendritic cells (melanocytes) scattered throughout the tumor. 853 The amount of melanin pigment is quite variable (Fig. 34.32). Although Bednar reported three cases in which this pigmented variant was present in the core of a nevocellular nevus, 849 no further examples of this phenomenon have been reported. There is one report of a recurrent DFSP transforming into the pigmented form. 894 Rare cases occur with histological overlap between a pigmented dermatofibroma and a pigmented DFSP. 895 In two recently reported cases, both showed focal positivity for CD34. They did not express factor XIIIa. 895 Both patients were disease free 2 years later. The various types of DFSP are listed in Table 34.3.
Dermatofibrosarcoma protuberans with melanin pigment. This variant is known as a Bednar tumor. (H & E)
• Fibrosarcomatous
• Fibrosarcomatous with myoid/myofibroblastic change
• Myxoid
• Granular cell
• Atrophic
• Sclerosing
• Palisaded
• Giant cell fibroblastoma
• Combined
• Indeterminate
Although usually inconspicuous, the stromal collagen in a DFSP will stain with the trichrome method, but unlike the collagen in a dermatofibroma it is not polarizable. 896 Sclerotic changes with localized areas of abundant collagen have been reported.897. and 898. Meningothelial-like whorls have been reported in a Bednar tumor. 899 Unlike dermatofibromas, hemosiderin is rarely present in a DFSP. 777 Furthermore, a dermatofibroma is usually smaller and less cellular, with fewer mitoses than DFSP.
Lesions with overlap features between dermatofibroma and DFSP have been reported as indeterminate fibrohistiocytic lesions. 900 The tumors all had keloidal collagen, infiltration of the subcutis in a honeycomb pattern, low mitotic counts, and a dual population of CD34+ and factor XIIIa cells in varying proportions from tumor to tumor. A single recurrence was noted. 900 The continuation of this diagnosis does not seem tenable in the light of cytogenetic studies.
Immunohistochemical studies have shown that the tumor cells in DFSP contain vimentin,863. and 901. but they are negative for S100 protein. A few scattered cells may stain for lysozyme and α1-antichymotrypsin. 862 About 80% of cases express CD99. 902 The most diagnostic marker is the human progenitor cell antigen, CD34, which stains 50–100% of the cells in DFSP.903.904.905.906.907. and 908. Conflicting results have been reported in the giant cell fibroblastoma component that is sometimes present.760. and 773. In contrast, CD34 stains only focal areas, in a small percentage of cases, in dermatofibroma909 and some neural tumors. 910 However, a recent study reported strong, diffuse staining in 20% of cases of dermatofibroma. 911 Factor XIIIa, which stains most dermatofibromas, is only focally positive in a small number of cases of DFSP. MIB-1 is not a useful marker in distinguishing DFSP from dermatofibroma; 912 nor is CD117 which is negative in both DFSP and cellular dermatofibromas. 913 Low-affinity nerve growth factor receptor (p75) is present in 95% of cases of DFSP but not in dermatofibromas. 914 Whereas stromelysin-3 is expressed in all cases of dermatofibroma it is present in less than 10% of DFSPs. 915 It was absent from all cases of DFSP in one study. 916 CD163, a hemoglobin scavenger receptor, was present in 17/19 dermatofibromas, 23/23 cellular fibrous histiocytomas, but only 3/18 DFSPs. 917 CD44 was expressed strongly in all cases of dermatofibroma in one study, but only weakly or not at all in DFSP. 918 Whereas 96% of dermatofibromas expressed HMGA1 and HMGA2 only a small number of cases of DFSP expressed these proteins. 919 Transforming growth factor-β receptors are expressed much less in DFSP than in dermatofibroma. 920 APO-D staining is strong in DFSP and neural tumors but negative in dermatofibromas. 921 Cyclooxygenase-2 is expressed in both dermatofibromas and DFSP. 922 The pigment-laden cells in the Bednar tumor are positive for S100 protein and vimentin, whereas the non-pigmented cells are usually positive only for vimentin. 856 The expression of CD34 is not present in all cases of pigmented DFSP.855.858. and 923.
The spindle-shaped cells usually have an indented or deeply lobulated nucleus. In some studies, including one concerning the Bednar tumor, 853 a basal lamina suggesting a perineurial origin was present.924. and 925. In other studies the predominant cell has been interpreted as a modified fibroblast926. and 927. or histiocyte. 928 Of interest are the rare reports in which the metastases of a DFSP have assumed a histiocytic appearance. 836 The primary lesion in such cases may have been a malignant fibrous histiocytoma incorrectly diagnosed as a DFSP. 862
‘Atypical fibroxanthoma’ (AFX) is the term coined by Helwig for a cutaneous tumor with marked cellular pleomorphism, yet with a course that is usually benign. 929 As such it has been regarded as one of the so-called ‘pseudomalignancies’ of the skin. 930 This view must be modified somewhat in the light of rare reports of metastasis (see below). Similar cases were reported about the same time as those of Helwig, under several different names.931. and 932. The tumors usually occur as a solitary, gray to pink or red nodule, often dome-shaped, on the head or neck of the elderly.933. and 934. There is a rare plaque form. 935 In some series there has been a predominance in males.935. and 936. They may develop quickly, but they are usually less than 2 cm in diameter. Another clinical variant occurs in younger patients, and these lesions may be larger, more slowly growing, and with a predilection for the trunk and extremities.934.937. and 938. AFX has been reported in children with xeroderma pigmentosum939. and 940. and, rarely, in children without this condition. 941 In both groups, the lesions may be ulcerated and bleed. It has also arisen in an area of syringocystadenoma papilliferum associated with nevus sebaceus (organoid nevus). 942
The nature of AFX is speculative. It has been regarded as a proliferative mesenchymal, possibly fibrohistiocytic, response to a variety of cutaneous injuries. 943 The diversity of immunoperoxidase markers raises the possibility of heterogeneous, bimodal ‘fibrohistiocytic’ and ‘myofibroblastic’ phenotypes. 944 In older patients there is often a history of prolonged actinic exposure, but lesions may also develop at sites of irradiation and even trauma. 945 This tumor may also develop in renal transplant recipients.946.947. and 948. It has been suggested that there is a close relationship to malignant fibrous histiocytoma (MFH), and that it is merely the small size and dermal location of most cases of atypical fibroxanthoma that lead to its benign behavior.949. and 950. A more likely explanation for these differences in behavior is the absence of ras-oncogene mutations in AFX, whereas MFH has both H- and K-ras gene mutations.951. and 952. Although the large atypical cells in AFX are aneuploid, as they are in MFH, the smaller spindle-shaped cells in AFX are diploid.953. and 954. These theories will need reconsideration in the light of the ‘break-up’ of MFH as an entity.
Recurrences develop in approximately 5% of cases, but in most instances this is due to incomplete removal. Extension into subcutaneous fat is another marker for local recurrence. 955 Lesions may even regress spontaneously or after incomplete removal. The diagnosis of AFX has been questioned in those cases that have behaved aggressively. 956 There is no doubt that AFX, as reported in the past, was a heterogeneous entity that included cases of malignant melanoma and the spindle-cell variant of squamous cell carcinoma.957. and 958. The advent of immunoperoxidase markers, and the wider use of electron microscopy, should allow more accurate assessment and diagnosis of these tumors in the future. More than 20 cases of AFX with metastases have now been reported, and several of these have been studied with immunoperoxidase or electron microscopy to exclude melanoma and squamous cell carcinoma.943.959.960. and 961. The cases reported have tended to metastasize to regional lymph nodes, 943 but more widespread systemic spread has been reported. 962
The author’s personal (anecdotal) experience with over 300 cases may be of interest. The tumor is usually histologically distinctive with its bizarre cells, numerous mitoses, level 4 extension (except for the rare plaque form), frequent ‘collarette’, and usual occurrence on the ear, forehead, or bald scalp. Experience has shown that it is prudent to perform immunohistochemistry for S100 protein and keratin, as melanoma and squamous cell carcinoma can produce morphological simulants. The author is not aware of any metastasis in these cases but at least 10 have recurred. In our published series of 89 cases of AFX, no recurrences or metastases were found on follow-up, despite encountering recurrences in earlier cases. 935 All these recurrent lesions were excised with positive or narrow margins and there was usually subcutaneous extension. It should be noted that most lesions were completely excised, as shave excisions have not been used widely in Australia until recently.
It has been suggested that atypical fibroxanthoma is a ‘myth’, and yet another variant of squamous cell carcinoma. 963 This statement has been strongly refuted by others.935. and 964. The majority of cases do not have adjacent actinic keratosis change, which might be expected if AFX were truly a variant of squamous cell carcinoma. 935 Most authors with experience of numerous cases of AFX believe it is a distinct clinicopathological entity with established clinical, histological, and immunohistochemical features. 955
Atypical fibroxanthomas should be treated by wide local excision, even though several cases in our published series did not recur, despite the presence of positive margins in the original excision specimen. Mohs technique may be of benefit in cases on the head and neck.
Atypical fibroxanthoma is usually a well-circumscribed, non-encapsulated, highly cellular tumor centered on the dermis (Fig. 34.33). It is contiguous with the epidermis, or separated from it by a thin zone of collagen. Sometimes cells appear to stream out from the basal layer, and in these areas the dermoepidermal junction is not always distinct. 956 In one series, nearly half the tumors extended into the subcutaneous fat, 957 but most observers would regard subcutaneous extension as uncommon, and an indicator of possible recurrence. The epidermis is often thin or even ulcerated, but there may be peripheral acanthosis and, occasionally, a peripheral ‘collarette’. 935
(A) Atypical fibroxanthoma. (B) This cellular tumor is composed of spindle-shaped cells and some bizarre cells with hyperchromatic nuclei. Several mitotic figures are present. (H & E)
There is florid pleomorphism and polymorphism, with many atypical and often bizarre cells. There are usually admixtures of three cell types, although a spindle-cell variant without the other two cell types has been recorded.650. and 965. The predominant cell is a plump, spindle-shaped cell in poorly arranged fascicles. Although it may occur in an atypical fibroxanthoma, 933 a storiform arrangement should raise the possibility that the correct diagnosis is in fact an MFH or dermatofibrosarcoma protuberans. The spindle cells have a prominent nucleus, which is often vesicular. There is also a haphazard arrangement of large polyhedral cells in the tumor. Some have vacuolated, lipid-containing cytoplasm. A clear cell variant has been described (Fig. 34.34).966.967.968.969. and 970. They do not contain glycogen. 971 True xanthoma cells are uncommon. The third cell type is a giant cell, which may be mononucleate, multinucleate, or osteoclast-like. 972 These cells have hyperchromatic nuclei, and mitoses, including bizarre forms, are common. The cytoplasm of these cells is often partly vacuolated. Lipid stains on frozen sections will show variable amounts of lipid, usually in the polyhedral and giant cells, with only sparse amounts in the spindle cells. Hemosiderin may also be present.
(A) Atypical fibroxanthoma of clear cell type. (B) A mitotic figure is present. (H & E)
Necrosis is uncommon and is usually limited to the ulcerated surface. Large areas of necrosis should raise doubts about the diagnosis of atypical fibroxanthoma. The adnexae may be distorted by the growth of the tumor, but they are not usually destroyed.
There is usually only a very small amount of interspersed collagen, although variants with prominent fibrosis and sclerosis have been reported.934. and 973. Foci of stromal osteoid974 or chondroid975 differentiation, and the presence of numerous osteoclast-like giant cells975.976.977.978. and 979. are rare findings. Pigmented and granular cell variants have also been reported.980.981.982.983.984. and 985. The various forms are listed in Table 34.4. A ‘carcinosarcoma’ has been reported in which a basal cell carcinoma and a sarcomatoid AFX were present. 986 Some tumors are richly vascular. The adjacent uninvolved dermis may show solar elastosis. Any inflammatory infiltrate is usually sparse and peripheral, although neutrophils may be abundant near an ulcerated surface.
• Spindle cell
• Clear cell
• Osteoid
• Osteoclastic
• Chondroid
• Pigmented
• Granular cell
Features that may portend a more aggressive behavior include vascular invasion (the most reliable feature), deep tissue invasion, tumor necrosis, 943 and possibly impaired host immunity. 959 Perineural and intraneural invasion have been reported; no recurrence occurred in these two cases, but follow-up was extremely short in one case. 955
Immunoperoxidase studies for α1-antitrypsin and α1-antichymotrypsin are positive in more than half of the cases.937. and 987. Although some S100-positive cells may be found, these constitute only a small percentage of the cells present.944.988. and 989. A case has been reported in which the giant (multinucleated) cells expressed HMB-45 and MART-1. 990 However, S100A6 is expressed in many cases. 991 These cells may represent part of the inflammatory response. The tumor cells are reactive for vimentin and, in a few cases, scattered cells stain for factor XIIIa634. and 992. but not usually for cytokeratin or epithelial membrane antigen. 993 Weak cytokeratin expression has been reported. 994 In one study, 41% of cases stained for muscle-specific actin or smooth muscle actin, 57% expressed CD68 (a monocyte–macrophage marker), but no case expressed CD34 (other than stromal blood vessels). 944 CD31 sometimes stains tumor cells, as well as the stromal blood vessels. 942 Weak positivity with CD74 has been reported in AFX, in contrast to the strong staining for this marker in MFH. 995 Positivity for CD99 has been reported in two-thirds or more of the cases tested.996. and 997. Staining is less frequent in MFH. 998 It has been stated recently that CD99 expression is not specific for AFX and it should have no role in its diagnosis. 999 CD117 was detected in 15 of the 16 cases studied in one series. 1000 It also highlights stromal mast cells.1001. and 1002. It is also positive in melanoma in sun-exposed areas of the body, reducing the usefulness of this marker in AFX. Melanoma may simulate AFX in other ways, and when negative for S100 protein such melanomas can pose a diagnostic dilemma. 1003 Gadd45, a multifunctional protein involved in the nucleotide excision repair pathways and cell-cycle checkpoint control, is expressed in some cases of AFX, MFH, and leiomyosarcomas. 952 It has little use in diagnosis.
Strong staining with CD10 is found in most cases of AFX.935.1004. and 1005. It is also positive in most cases of MFH, and dermatofibroma, and also in some cases of leiomyosarcoma. Weak staining has been reported in some melanomas and squamous cell carcinomas, 1004 but in the author’s experience CD10 remains a useful antibody, when used in a panel of antibodies for spindle and pleomorphic tumors of the skin (Fig. 34.35).
Atypical fibroxanthoma. The cells stain strongly with CD10. (Immunoperoxidase stain)
Procollagen 1 (PC1) is expressed in most cases of AFX. 1006 The combination of CD10 and PC1 is a useful adjunct in an immunoperoxidase panel for the diagnosis of AFX. 1007
The spindle cells have abundant rough endoplasmic reticulum, small vesicles and cytoplasmic filaments, and surface indentations in the nuclei. 1008 These features suggest a fibroblastic or myofibroblastic origin. The large cells have a histiocytic appearance with lipid vacuoles. 1009 Transition forms between these two cell types have been noted, as have histiocytic cells with Langerhans granules.988. and 1010.
Angiomatoid fibrous histiocytoma (AFH) is a rare soft tissue neoplasm first described by Enzinger in 1979. 1011 It occurs predominantly on the extremities of children and young adults. 1012 It was included for many years as a variant of malignant fibrous histiocytoma despite its more superficial location, the younger age of the patients, and its favorable prognosis.1013.1014. and 1015. It is now recognized as a distinct entity.
The lesions are generally small, measuring 2–4 cm in largest diameter. Most cases behave in a relatively indolent manner although rare metastases have been recorded. 1016 It is regarded as a tumor of intermediate biologic behavior. 1013
Two specific translocations have been demonstrated – t(12:16)(q13:p11) and t(12:22)(q13:q12). The first mentioned involves fusion of the FUS and ATF-1 genes and the second the fusion of the EWSR1 gene with ATF-1.1013. and 1017. Sometimes the fusion transcript is not identified. 1018 The first mentioned translocation is also seen in myxoid liposarcoma although it involves the FUS and DDIT3 (CHOP) genes. 1013 The EWSR1/ATF-1 fusion product is also seen in clear cell sarcoma.1019.1020.1021. and 1022. The histogenesis of this tumor is uncertain. An origin from fibroblastic reticulum cells has been suggested. 1013
This tumor is usually well demarcated with a thick fibrous capsule that may contain inflammatory cells. It tends to involve the subcutis. 1011 There are large blood-filled cystic spaces and areas of hemorrhage in addition to solid nests of spindle and ovoid cells.1011. and 1023. Xanthoma cells, siderophages, and giant cells are often present. A peripheral lymphoplasmacytic infiltrate is present to some degree in about 80% of cases. 1024 The cells show intermediate staining for CD34.1025. and 1026. Approximately 50% of cases show positivity for desmin and CD99. 1024
Malignant fibrous histiocytoma (MFH), formerly the most common soft tissue sarcoma of late adult life, is a controversial entity that is currently undergoing a systematic break-up into several entities, leaving behind the pleomorphic variant as a wastebasket of undifferentiated sarcomas.1027. and 1028. In the 40 years that the author has been practicing pathology, there have been rotating wastebaskets into which the diagnostic dilemmas of soft tissue tumors have been placed for a decade or more. This is not a criticism of those pathologists working in the area, who have kept us well informed, but rather an acknowledgement of the complex histogenesis of many of these tumors, which have remained an enigma despite advances in electron microscopy, and then in immunohistochemistry. Some of these tumors are becoming transparent at last, under the surveillance of cytogenetics.
The angiomatoid variant of MFH is now classified as angiomatoid fibrous histiocytoma (see above) on the basis of its better prognosis, its more superficial location, and its occurrence in younger individuals than those with pleomorphic MFH.
The myxoid variant of MFH has been reclassified as myxofibrosarcoma (see p. 825) on the basis of its distinctive and consistent clinicopathological features, most notably the presence of prominent myxoid foci comprising nearly 50% of the tumor.509.511. and 514.
The giant cell variant, which was not consistently included as a variant of MFH, is now regarded as a distinct entity – the giant cell tumor of soft tissues (see p. 839). It is exceedingly rare and has morphological similarities to the bone tumor of the same name.
The inflammatory variant remains, for the present, in the category of MFH. Some cases probably represent undifferentiated liposarcomas. Cytogenetic studies will undoubtedly lead to a reclassification of some of the tumors in this group.
The pleomorphic MFH, also called pleomorphic undifferentiated sarcoma, remains largely ‘unscathed’. It is a surprisingly large group, if the findings of the Scandinavian Sarcoma Pathology Review Group are in any way representative of the situation in other countries. 1029 Fletcher, on the other hand, believes this entity will eventually be dropped. 1027 The account which follows refers to the pleomorphic variant, although some of the references given include cases now reclassified into other groups.
Pleomorphic MFH usually involves the deep soft tissues and striated muscles of the proximal part of the extremities, particularly the lower. 1030 The retroperitoneum is another favored site.861. and 1031. Up to 20% of cases may arise in the subcutaneous tissues, although less than 10% are confined to the subcutis without underlying fascial involvement.1031.1032.1033. and 1034. Cutaneous tumors are even rarer.1035. and 1036. They usually extend into subcutis and deeper structures.1037. and 1038. Adults between the ages of 50 and 70 years are most often affected. There are usually no predisposing factors but, rarely, cases have followed radiotherapy,1032. and 1039. or developed at the site of a chronic ulcer, 1040 osteomyelitis, 1041 vaccination scar, 1042 or burn scar. 1043 MFH has also developed in HIV-positive patients, 1044 and in a renal transplant recipient. 1038
The tumors are multilobulated, often circumscribed, gray-white fleshy masses. 1032 The majority are 5 cm or more in diameter. Focal areas of hemorrhage and necrosis are quite common. The inflammatory variant may be yellow in color.
The prognosis is generally poor, with 5-year survivals ranging from 15% to 30%; 1045 recent studies have recorded much-improved survival rates. 1046 Tumors that are small and superficially located have a better prognosis than large, deep ones.1031.1047. and 1048. Tumor size, local tumor recurrence, and necrosis are associated with a greater risk of metastasis. 1029 Proliferative activity (MIB-1 index) is not an independent prognostic parameter, despite being so in other soft tissue sarcomas. 1049 Tumor vascular endothelial growth factor (VEGF) expression correlates with stage, grade, and prognosis in soft tissue sarcomas in general. 1050 Proximal deep tumors and those in the retroperitoneum have a poor prognosis. Local recurrence and metastases are common, with the lungs and regional lymph nodes most often affected. 1031
The histogenesis of this tumor has been controversial. The tumor cells show partial fibroblastic and histiocytic differentiation, as reflected by collagen production and the presence of cells which may be immunoreactive for the histiocytic markers, as well as showing occasional phagocytosis. 1051 Others believe that there are no histiocytic features. It may be derived from a poorly defined mesenchymal cell, which may differentiate along histiocytic and fibroblastic lines.1052.1053. and 1054.
Multiple structural and numerical aberrations in many chromosomes have been found in this tumor. 1055 It appears that genes involved in the RB1- and TP53-associated cell-cycle regulatory pathways may play a prominent role in the development of malignant fibrous histiocytoma. 1056
Wide surgical resection is usually recommended for this tumor.1037. and 1046. Radiotherapy and chemotherapy have generally been used as adjunctive or palliative measures but chemotherapy appears to have marginal efficacy. 1057 There is increasing use of preoperative neoadjuvant treatments such as chemotherapy, radiotherapy, hyperthermia, or a combination of these methods for high-grade soft tissue sarcomas. While some tumors show a good response to these combined-modality treatments, the prognosis is still poor. 1058
All tumors usually have an infiltrative margin. They may show areas of hemorrhage and necrosis, particularly the larger tumors. Chronic inflammatory cells are usually present throughout the tumor, particularly at the periphery. The pleomorphic tumors are now synonymous with MFH, if the inflammatory lesions are excluded.
They are composed of an admixture of plump spindle-shaped cells, clusters or sheets of histiocytes, and scattered pleomorphic multinucleate giant cells (Fig. 34.36).1031.1032. and 1059. Mitoses are common. The spindle cells may be arranged in whorls or have a storiform appearance. There is usually a delicate collagenous stroma, which in some areas may be more prominent. Focal myxoid change is quite common. Rarely, metaplastic osteoid or chondroid material is formed. 1060 Xanthoma cells and siderophages are sometimes present. Similar appearances can be seen in pleomorphic sarcomas presumed to be of other cell lineages. 1054 For this reason, pleomorphic MFH should now be regarded as an undifferentiated pleomorphic sarcoma.
Malignant fibrous histiocytoma composed of spindle-shaped and polygonal cells and scattered pleomorphic giant cells, some of which are multinucleate. (H & E)
The rare inflammatory variant, which is most frequently retroperitoneal, has a grave prognosis. Cutaneous lesions have been reported. 1061 A diffuse, and at times intense, neutrophilic infiltrate, unassociated with tissue necrosis, is present not only in the primary tumor, but also in recurrences and metastases.1062.1063.1064. and 1065. Xanthoma cells, both bland and anaplastic, are also present. A storiform fibrous pattern is usually seen in some areas of the tumor. 1062 As stated at the beginning of this section, this tumor is probably a mixture of undifferentiated sarcomas, particularly liposarcoma.
The tumor cells contain vimentin;1066. and 1067. cytokeratin has also been present in a few tumors. 1068 Strong staining for CD74 has been reported. 995 Various histiocyte markers can be demonstrated, using immunoperoxidase techniques, in from 60% to 80% of malignant fibrous histiocytomas. Those used have included α1-antitrypsin and α1-antichymotrypsin. 711 CD68 and CD10 are frequently positive, whereas CD34 is negative.1014.1015. and 1069. Rare cases with S100 expression have been reported. 1070
Primary giant cell tumor (osteoclastoma) of soft tissues is a rare tumor that may arise in both the superficial and deep soft tissues.1027.1074.1075.1076. and 1077. It is found predominantly on the thighs, trunk, and upper extremities. It occurs in a broad age range. 1078 There is no sex predilection. Morphologically, it has some resemblance to giant cell tumor of bone. Cases involving the dermis, but extending into the subcutaneous fat also, have been reported.1075. and 1079. Although no recurrences had occurred at the time of publication of these cases, the follow-up period was brief. Metastases have been rare in cases having a deeper location. 1078
There are round to spindle-shaped cells forming the stroma for uniformly scattered, osteoclast-like multinucleated giant cells (Fig. 34.37). Mitoses are not uncommon and average 2–3 per 10 high power fields. Mitotic ‘hot spots’ may be present. Foci of hemorrhage, hemosiderin deposition, and foamy macrophages are usually present. Aneurysmal bone cyst-like areas may also be present. There is some fibrosis, usually at the edge. 1075 The tumor cells express vimentin and CD68. The latter marker is usually strongly expressed in giant cells; it is focal in mononuclear cells.
Giant cell tumor of soft tissue composed of numerous osteoclast-like giant cells. (H & E)
This tumor must be distinguished from the malignant diffuse-type tenosynovial giant cell tumor, the malignant variant of tenosynovial giant cell tumor. Like its benign counterpart, it tends to occur within or near the large joints of the extremities. 1080
Chung and Enzinger formally documented fibroma of tendon sheath in 1979, with a report of 138 cases. 1081 It is a solitary, slow-growing subcutaneous tumor with a predilection for the fingers, hands, and wrists of middle-aged adults, particularly males.1082. and 1083. Recurrences occur in approximately 20% of cases.
The fibromas are well-circumscribed, often lobulated, gray to white tumors that measure 1–2 cm in diameter. They are usually attached to a tendon sheath.
A (2;11) translocation has been found in some of the tumor cells in one case, suggesting that this lesion is neoplastic and not a reactive process. 1084
These tumors are situated in the subcutaneous tissue, sometimes with dermal extension. There are relatively sparse spindle or stellate cells embedded in a dense fibrocollagenous stroma. Cellular areas are present, sometimes toward the periphery. The stroma shows variable hyalinization and sometimes has a whorled pattern with associated artifactual clefting. Myxoid degeneration and, rarely, focal calcification of the stroma may be present. A characteristic feature is the presence of dilated or slit-like vascular channels. A sparse mononuclear cell infiltrate is sometimes present at the periphery.
A rare pleomorphic variant (pleomorphic fibroma of tendon sheath) has been reported. There are scattered large cells with pleomorphic, hyperchromatic nuclei, but no mitoses. 1086 A further variant of pleomorphic fibroma exists: it has overlap features with the giant cell tumor of tendon sheath (see below). This has given rise to the suggestion that fibroma of tendon sheath is the end and sclerosing stage of giant cell tumor, 1087 although this has been strongly disputed. 1088
Giant cell tumor of tendon sheath is a not uncommon benign tumor with a predilection for the dorsal surface of the fingers, in the vicinity of the distal interphalangeal joint.1091. and 1092. Periungual lesions are rare. 1093 It occurs particularly in young and middle-aged adults. It is a slow-growing, usually asymptomatic lesion, which may measure up to 3 cm in diameter at the time of removal. Multicentric lesions are rare. 1094 Local recurrence, usually a result of incomplete removal, occurs in 15% of cases. 1091
Giant cell tumors are lobulated and gray-brown in color, with yellowish areas. They are usually attached to a tendon sheath or joint capsule, but cutaneous involvement can occur.1095. and 1096. The histogenesis of giant cell tumors has been controversial. They have been regarded as a variant of fibrous histiocytoma, and as a tumor derived from mesenchymal cells. 1097 One study concluded that the cells are of monocyte–macrophage lineage, closely resembling osteoclasts. 1098 The immunophenotype (see below) is more suggestive of a synovial cell origin. 1088 It appears to be a polyclonal proliferation. 1099
This tumor must be distinguished from the giant cell tumor of soft tissue, a rare tumor resembling the lesion of bone (see p. 839).
The lesion has an eosinophilic collagenous stroma with variable cellularity. In the sparsely cellular areas the cells are plump and spindle shaped, set in a partly hyalinized stroma, whereas in the more cellular areas the cells are usually polygonal. Small clusters of lipid-laden histiocytes are often present. Multinucleate giant cells with up to 60 or more nuclei are a characteristic feature (Fig. 34.38). They are variable in number and haphazardly distributed. Hemosiderin is invariably present, and sometimes there are cholesterol clefts.
(A) Giant cell tumor of tendon sheath. (B) This subcutaneous tumor is composed of polygonal cells and collections of multinucleate giant cells. (H & E)
The proliferating mononuclear cells stain positively for CD68, HAM-56, and vimentin, but not for S100, cytokeratins, EMA, CD45, CD34, desmin, or smooth muscle actin. 1088 The giant cells stain for CD68, vimentin, and CD45. 1088
Ultrastructural studies have usually supported a synovial or fibrohistiocytic origin, 1101 although another report documented a pleomorphic cell population in which the giant cells had some similarity to osteoclasts and the stromal cells similarities to primitive mesenchymal cells, osteoblasts, fibroblasts, and histiocytes. 1097
Epithelioid sarcoma, first delineated by Enzinger, 1102 is a rare aggressive tumor that usually involves the deep subcutis and underlying soft tissues. Infrequently, it arises in the dermis and superficial subcutis, where it may be confused both clinically1103 and histologically with various benign cutaneous diseases, including granuloma annulare.1104. and 1105. It has a predilection for the extremities, particularly the hands, of young adult males.1106.1107. and 1108. Childhood cases occur. 1109 Lesions of the face, 1110 vulva, 1111 and penis1112 have also been described. A history of trauma is sometimes given. 1106 A case has been reported in a patient with neurofibromatosis type 2. 1113
It presents as one or more slow-growing tan-white nodules with an indistinct infiltrating margin. Ulceration develops late, if at all. Local recurrence occurs in nearly 80% of cases, and metastases in 30–45%.1106.1114.1115. and 1116. The regional lymph nodes and lungs are the most common initial sites of metastases. The skin of the scalp represents another common site of distant metastasis. 1117 The tongue is one of several rare metastatic destinations. 1118 Adverse prognostic features are older age, a proximal or axial location, tumor size greater than 5 cm, deep extension, necrosis, rhabdoid cytomorphology, vascular invasion, incomplete excision, metastases, and numerous mitotic figures.1106.1119. and 1120.
Epithelioid sarcoma is of disputed histogenesis. A synovial origin, which was favored at one time,1121. and 1122. now seems unlikely. 1120 A histiocytic, fibroblastic, 1123 myofibroblastic, 1124 perineurial, 1125 and mesenchymal reserve cell1126 origin have all been suggested. One tumor produced granulocyte colony-stimulating factor, but it is difficult to draw any histogenetic conclusions. 1127 An N-ras oncogene mutation has been demonstrated in the tumor cells in one study. 1115 In another, allelic loss on chromosome 22q was present in 60% of cases. 1128 Subsequent studies have shown chromosomal gains at 22q and other sites, 1129 but there are no consistent or specific chromosomal rearrangements or changes. 1130
Fletcher and colleagues have drawn attention to an aggressive tumor with epithelioid and rhabdoid features, which they have called ‘proximal-type’ epithelioid sarcoma. 1131 Most of the cases involved the deep soft tissues in axial proximal locations but several, in the region of the vulva, involved the subcutis. It usually occurs in middle-aged or older adults, 1130 but cases in children have been reported. 1132 It recurs more frequently and metastasizes earlier than the classic type. 1133
Treatment of epithelioid sarcoma of both types consists of radical surgical excision of the tumor and, if indicated, therapeutic lymph node dissection. 1134 In patients who have large tumors, isolated limb perfusion with tumor necrosis factor and melphalan may be useful. 1134 The role of postoperative radiotherapy after wide surgical excision needs further research. Its use is usually confined to cases with positive surgical margins. 1135 Sentinel lymph node biopsy has also been suggested as a useful procedure. 1134
The low-power appearance often resembles a necrobiotic or granulomatous process, or an epithelial tumor. Epithelioid sarcoma is an ill-defined cellular lesion forming vague nodules in the dermis and subcutis. There is subtle extension into the contiguous fascial and tendinous structures. Perineural invasion is present in approximately 20% of cases. 1119 The tumor is composed of oval to polygonal cells with abundant, often eosinophilic, cytoplasm, and a gradual transition to plump spindle cells (Fig. 34.39).1136. and 1137. Rarely, a spindle-cell pattern predominates. 1138 Nuclear pleomorphism and scattered mitoses are present. Multinucleate giant cells are rare or absent. Sometimes the pattern can look deceptively bland. 1139
(A) Epithelioid cell sarcoma. (B) There are sheets of epithelioid and polygonal cells. (H & E)
A characteristic feature is the presence of central necrosis (geographic necrosis) or fibrosis. Collagen also extends between the cells. Calcification is present in approximately 20% of cases, and osseous metaplasia in 10%. 1106 Hemosiderin is often present. A case with angiomatoid features has been reported. 1140 A perinodular inflammatory cell infiltrate of lymphocytes and histiocytes is a usual feature. 1141
The ‘proximal-type’ variant grows in a multinodular pattern and shows prominent epithelioid and rhabdoid features. It lacks a granuloma-like pattern. There is marked cytological atypia. The cells are positive for vimentin, cytokeratin, and EMA. Some tumors express desmin and CD34. 1131 This variant of epithelioid sarcoma needs to be distinguished from extrarenal malignant rhabdoid tumor (see p. 863), an ‘entity’ which undoubtedly includes many cases of proximal-type epithelioid sarcoma. 1132
Immunoperoxidase studies of the usual (classic) type show that many tumor cells contain cytokeratin, epithelial membrane antigen (EMA) and vimentin, but not CD45, S100 protein, desmin, FLI-1, and CK20.1120.1142.1143.1144.1145. and 1146. The combination of cytokeratin, EMA, and vimentin positivity is a characteristic feature of this tumor, although exceptions occur (Fig. 34.40). 1147 CAM5.2 is a useful marker for detecting the cytokeratins that are usually present. 1146 Cytokeratin 5/6 is found in only rare cells in epithelioid sarcoma, in contrast to spindled squamous cell carcinoma in which it is diffusely positive. 1148 Nearly all epithelioid sarcomas express cytokeratin 8 (CK8), whereas about 75% express CK19. 1149 Patchy membrane staining for CD34 and more diffuse staining for muscle-specific actin1149 occur in more than half of the cases. 1147 Most cells express vascular-endothelial cadherin. 1150 Histiocytic markers have usually been negative. 1144
(A) Epithelioid sarcoma. (B) The tumor cells are quite pleomorphic. The inset shows positive staining for EMA. (H & E and immunoperoxidase inset)
Light and dark cells are present within the tumor cell population. There are many filopodia-like surface extensions of cytoplasm1121 and there are bundles of intermediate filaments in the cytoplasm. 1144 Occasional pseudoglandular spaces have been present in some tumors. 1122 In summary, there are epithelial and mesenchymal features, including myofibroblastic differentiation. 1120
Only brief mention will be made of this soft tissue sarcoma, which has a predilection for the extremities of young and middle-aged adults. It exhibits a wide spectrum of biological behavior. The high-risk group includes older patients, 1151 tumor size greater than 5 cm, and poor differentiation.1152. and 1153.
Only one case of superficial synovial sarcoma involving the dermis has been reported. 1154 It involved the skin overlying the knee in a young woman. It recurred six times over a 24-year period. 1154
A study of 21 minute synovial sarcomas of the soft tissues of the hands and feet was reported from the AFIP in 2006. 1155 All lesions were less than 1 cm in diameter. The median age of the affected patients was 29 years (range, 8–60 years). 1155 SYT–SSX fusion transcripts were demonstrated in five cases. Minute tumors appear to have a favorable outcome if completely excised. 1155 Genomic deletion of p16INK4A (CDKN2A) is a frequent genetic event in synovial sarcomas. 1156
The chromosomal translocation t(X;18) has been described in 90% of synovial sarcomas, and the presence of SYT–SSX fusion products is quite characteristic.1157. and 1158. Usually the proximal portion of the SYT gene at chromosome 18q11 is fused to the distal portion of one of several duplicated SSX genes at chromosome Xp11. 1159SYT/SSX1 translocations are nearly three times more frequent than SYT/SSX2 translocations, and this latter pattern is usually associated with monophasic tumors. 1159
In general, aggressive treatment has been advocated for synovial sarcomas, and in earlier times, this often meant amputations. Postoperative radiation is usually used after conservative excisions, but its efficacy has been questioned. 1155 Chemotherapy is used for large, inoperable, and recurrent tumors, but it does not seem necessary for the minute variants. 1155 It is often used in children with synovial sarcoma. 1160 As some tumors express epidermal growth factor receptors and HER-2/neu protein, treatment with tyrosine kinase inhibitors has been suggested as a future alternative treatment modality. 1159
Synovial sarcoma occurs in two predominant patterns: a monophasic form composed of spindle cells, sometimes in a fascicular pattern, and a biphasic form with spindle cells admixed with glandular structures of varying size. The minute lesions (see above) were biphasic in 7 cases, and monophasic in 14. Microscopic calcification was present in 8 cases. 1155 The biphasic lesions usually stain for cytokeratin and EMA while about 70% of the monophasic form stain for at least one of the cytokeratins or EMA.1152. and 1161. Focal staining for S100 protein is seen in about 25% of cases. 1162 Matrix metalloproteinase-2 expression correlates with epithelial differentiation in synovial sarcomas. 1163
The miscellaneous group of disorders includes several very rare entities in which fibrous tissue forms a significant component, and another group with a myxoid stroma. Collagenous papules of the ear are no longer considered in this chapter. They result from the deposition of amyloid in the papillary dermis (see p. 381). 1164.1165. and 1166.
Fibro-osseous pseudotumor is a rare tumor of the subcutaneous and soft tissues of the digits, usually in young adults.1167. and 1168. There is a close histological resemblance to myositis ossificans, with osteoid formation and a background stroma of fibroblasts, collagen and myxoid material.1167. and 1169. Unlike myositis ossificans, the lesion usually involves the subcutis and has an irregular multinodular growth pattern. 1167
Ossifying fibromyxoid tumor is a rare tumor of soft tissues of intermediate malignant potential, that sometimes involves the subcutaneous tissue.1170.1171.1172. and 1173. Dermal involvement is exceedingly rare. 1174 It occurs preferentially on the upper and lower extremities and, rarely, in the head and neck region. It has also presented as a scalp cyst. 1175 There is a male predominance. The tumors measure 1–14 cm in diameter (median 4 cm). 1176
Ossifying fibromyxoid tumor was initially thought to be of neural origin, but it seems more likely that it is derived from mesenchymal stem cells that also express neural differentiation, which is often weak, but occasionally quite prominent. 1174 Genomic analysis of malignant tumors has shown a clonal pattern, but no consistent chromosomal abnormality. 1175
In the vast majority of cases, complete surgical excision is curative, but local recurrences are common unless an adequate clearance is obtained. Mohs surgical technique has been used for this purpose. 1175 Metastases were recorded in 16% of patients in a series of consultation cases. 1176 There were no metastases in the series of 104 cases reported from the AFIP. 1177
The tumor is circumscribed, with a characteristic fibrous capsule and an incomplete peripheral shell of new bone. 1171 Bone is not present in all cases, and its absence, particularly in a small biopsy, can lead to difficulties in diagnosis. 1175 Sometimes the bone is central rather than peripheral in location. 1178 There are cords, nests, and sheets of oval to round and spindle-shaped cells with a well-vascularized stroma. 1170 The stroma may be mucoid, fibromyxoid, or fibrous. The stroma stains with the colloidal iron method, and with Alcian blue (pH 1.0–2.5). The presence of high cellularity, high nuclear grade, or >2 mitotic figures/50 high power fields may be associated with local recurrence and/or metastatic disease, while an infiltrative growth pattern leads to an increased risk of recurrence. 1176 Tumor cells stain for vimentin and show strong focal staining for S100 protein. Some cases have shown staining for desmin, smooth muscle actin, pancytokeratin, glial fibrillary acidic protein, and neuron-specific enolase.1176. and 1177.