Chapter 21 The systemic inflammatory response syndrome
 Access the complete reference list online at http://www.expertconsult.com
Access the complete reference list online at http://www.expertconsult.com
Definition of SIRS
The phrase ‘systemic inflammatory response syndrome (SIRS)’ was recommended by the American College of Chest Physicians/Society for Critical Care Medicine (ACCP/SCCM) consensus conference in 1992 to describe a systemic inflammatory process, independent of its cause.1 The proposal was based on clinical and experimental results indicating that a variety of conditions, both infectious and non-infectious (i.e. burns, ischemia–reperfusion injury, multiple trauma, pancreatitis), induce a similar host response. Two or more of the following conditions must be present for the diagnosis of SIRS to be made:
• body temperature >38°C or <36°C;
• respiratory rate >20/min or PaCO2 <32 mmHg;
• leukocyte count >12 000/µL, <4000/µ, or >10% immature (band) forms.
All of these pathophysiologic changes must occur as an acute alteration from baseline in the absence of other known causes. This definition is very sensitive and non-specific, and most of the SIRS criteria are also addressed in other scoring systems of injury-induced physiologic derangement, such as the Acute Physiology and Chronic Health Evaluation (APACHE), Mortality Probability Model (MPM), and Simplified Acute Physiology Severity (SAPS) systems. Several investigators have criticized the definition of SIRS as being too sensitive and encompassing the majority of ICU patients, and certainly the vast majority of patients suffering extensive thermal injury.2,3 The initial definition of SIRS also did not address the continuum of disease severity as was defined for sepsis. Criteria for the diagnosis of severe sepsis included the additional derangements of organ dysfunction, hypotension, and hypoperfusion. Evidence of hypoperfusion included, but was not limited to, the presence of lactic acidosis, oliguria, and altered mental status. Septic shock was characterized by hypotension and hypoperfusion in patients who were adequately volume resuscitated or required treatment with catecholamines or other vasoactive drugs to support cardiovascular function. Muckart and Bhagwangee,2 in an effort to define a continuum of severity for SIRS, later proposed the categories of severe SIRS and SIRS-associated shock. These conditions were defined by the same criteria as severe sepsis and septic shock in the absence of demonstrable infection. In its most severe form, SIRS can induce organ injury and subsequent multiple organ dysfunction syndrome (MODS).
Despite the limitations in the definitions of SIRS and sepsis, most clinicians and investigators generally adopted the SIRS concept. However, the initial definition and criteria were not felt to be optimized. To address these issues, a second consensus conference was assembled in 2001.4 The goal of this conference was to revisit the previously defined criteria for SIRS and sepsis as well as to determine whether revision of these criteria was indicated. The consensus was that the concepts of sepsis and SIRS are useful but the diagnostic criteria are overly sensitive and non-specific. The participants added additional criteria that defined metabolic, biochemical and functional alterations associated with SIRS and sepsis. Among these were hyperglycemia, edema, elevated plasma C-reactive protein concentration, coagulation abnormalities, thrombocytopenia, ileus, and hyperbilirubinemia. The group further proposed a staging system for sepsis and SIRS that could be used to stratify patients for prediction of outcome. This staging system, termed PIRO, defined several criteria, including the predisposition of patients to a poor outcome as determined by premorbid conditions and possible genetic factors. Other factors include the severity and type of insult, the host response to injury and the presence of organ dysfunction. The participants proposed that this model could be used to generate more specific criteria for defining the SIRS phenomenon. Although the validity of this approach remains to be established, Rubulotta recently reported that the PIRO criteria have predictive value regarding mortality in septic patients.5 Nevertheless, the validity of the staging system remains to be fully determined.
Another pitfall in the designation of SIRS is the difficulty of applying the initial criteria to children. Some of the criteria, particularly those for heart rate and respiratory rate, fall within the normal physiologic range for young children. In 2002, a consensus conference was assembled to define criteria for sepsis and SIRS in children.6 The participants defined six age groups based on clinical and physiological characteristics (Table 21.1). SIRS was defined as the presence of at least two of the following four criteria, one of which must be abnormal temperature or leukocyte count:
• temperature >38.5°C or <36°C;
• tachycardia, defined as mean heart rate >2 SD above normal for age or for children <1 year,
• bradycardia, defined as heart rate <10% of normal for age;
• mean respiratory rate >2 SD above normal for age or requirement for mechanical ventilation;
Table 21.1 Pediatric age groups for SIRS criteria
The definition of septic shock is problematic in children because children can maintain blood pressure until they become severely ill. Therefore, hypotension is not a useful criterion for diagnosing shock in children. This group proposed criteria for septic shock that included the presence of tachycardia in conjunction with signs of decreased peripheral perfusion, such as decreased capillary refill, decreased peripheral pulses, decreased urine output, altered mental status and cold/mottled extremities.6
Several studies have been conducted with the goal of determining the prognostic value of the SIRS designation. In the acute setting, SIRS has been demonstrated in the majority of critically injured patients and the intensity of the response correlates directly with the severity of injury.3,7 The presence of SIRS within the first 24 hours after severe injury has not served as a reliable predictor of mortality in trauma or burn patients.3,7 However, the presence of shock is an important predictor of poor outcome, particularly when associated with MODS.2 In addition, the presence of more than two of the SIRS criteria in the setting of acute injury has correlated with increased morbidity and mortality.8,9 A study by Rangel-Frausto et al.9 showed that trauma patients who did not meet SIRS criteria had a mortality rate of 3%, compared to 6% in those with two SIRS markers. Patients with three or four SIRS criteria had mortality rates of 10% and 17%, respectively, whereas those with culture-negative shock had a 46% death rate. Haga et al.10 have shown that the persistence of SIRS for more than 3 days in surgical patients is a harbinger of complications and is associated with increased morbidity. Talmor et al.7 reported that persistence of SIRS to the second postoperative day in high-risk surgical patients correlated with an increased incidence of MODS. Additional studies have shown that persistence of SIRS criteria for more than 3 days in trauma and burn patients is associated with worse outcome.10–13 Therefore, three important factors appear to determine the effect of SIRS on the host. The first is the severity of the initial inflammatory response. This response is proportional to the severity of injury, and the presence of shock or MODS within the first 24 hours after injury carries a poor prognosis. The second determinant is the persistence of SIRS beyond the first few days of injury. Specifically, prolongation of SIRS beyond the second day after severe trauma or thermal injury is associated with an increased complication rate. Factors that appear to be important in reducing the incidence of a prolonged inflammatory state include adequate fluid resuscitation within the first 24 hours after injury, aggressive excision of necrotic tissue and enteral feeding.11–13 A third factor is the adaptive capacity of the host. Results of several studies have shown that extremes of age and the presence of coexisting disease will diminish the adaptive capacity of the host and predict a worse prognosis for any given severity of injury.14,15
The initiating event
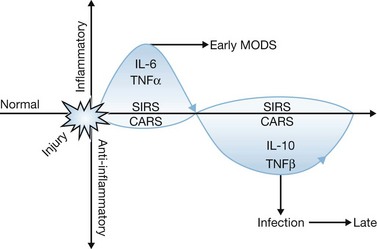
The crucial pathophysiologic event that precipitates systemic inflammation is tissue damage. This can occur both as a result of direct injury to tissues from mechanical or thermal trauma and as a result of cellular injury induced by ischemia and reperfusion. Injury results in the acute release of proinflammatory cytokines such as tumor necrosis factor-α (TNFα), interleukin (IL)-1 and IL-6. If injury is severe, such as in extensive thermal injury, a profound release of cytokines and non-cytokine mediators of inflammation occurs, resulting in the induction of a systemic inflammatory reaction (Fig. 21.1). The ability of the host to adapt to this systemic inflammatory response is dependent on the magnitude and duration of the response as well as the adaptive capacity of the host. If the insult and the host’s response to it are beyond the adaptive capacity of the host, or if adequate resuscitation is not promptly initiated, organ injury may ensue during the early post-injury period. Factors that have been implicated in the worsening or prolongation of SIRS include inadequate resuscitation during the acute phase following thermal injury, persistent or intermittent infection, ongoing tissue necrosis, and translocation of bacteria or endotoxin across the bowel.15,16
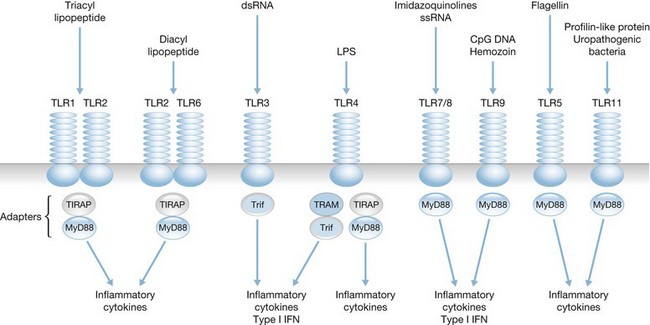
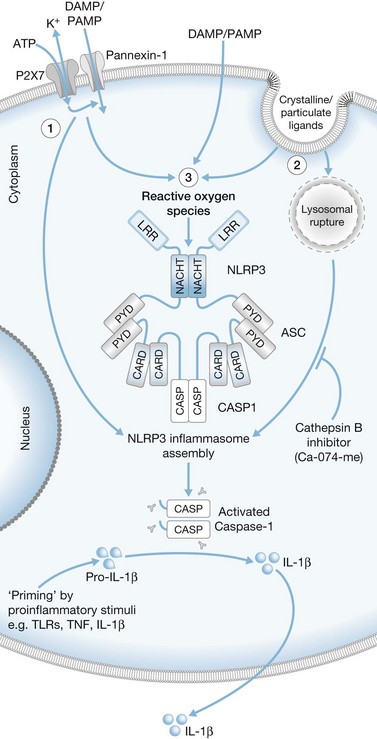
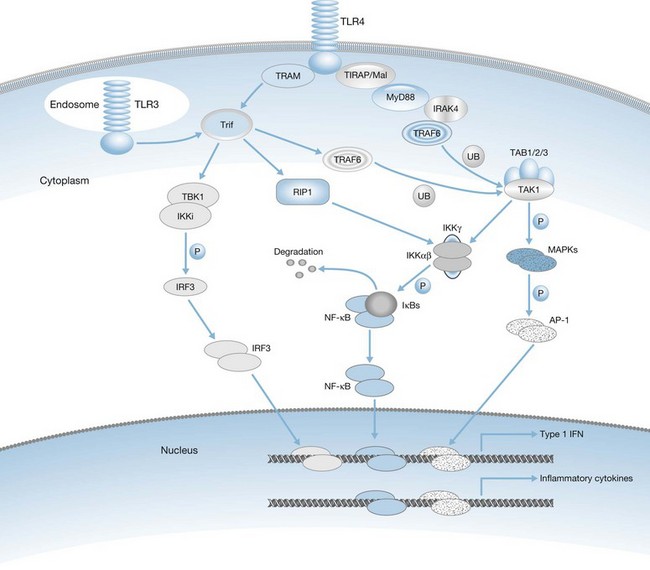
The actual mechanisms that initiate trauma-induced inflammation at the cellular and molecular levels are becoming increasingly understood. Two primary sensing systems have been identified. Recent evidence has implicated the ability of Toll-like receptors (TLR) to respond to endogenous cellular factors that are produced or released following tissue trauma (Fig. 21.2). TLR are expressed by leukocytes and some parenchymal cells. They allow the host to sense the presence of microbes and initiate an innate immune response. However, recent studies indicate that TLR have the ability to respond to endogenous ligands as well. Among the proposed endogenous TLR ligands are heat shock proteins (recognized by TLR4) and mitochondrial DNA (recognized by TLR9).17,18 A second sensing system are NOD-like receptors (NLR) that sense endogenous and exogenous ligands to cause activation of inflammasomes (Fig. 21.3).19 Inflammasome assembly ultimately results in activation of inflammatory caspases such as caspase-1. Activated caspase-1, also known as IL-1-converting enzyme, causes cleavage of pro-IL-1 and pro-IL-18 into active mature proteins. The IL-1 and IL-18 produced by this interaction are released from cells and facilitate the proinflammatory response. Proposed endogenous NLR ligands include adenosine triphosphate (ATP), uric acid and endogenous DNA.19
In some cases, SIRS may precipitate a state of immunosuppression known as the counter anti-inflammatory response syndrome (CARS). Frequently, SIRS and CARS coexist (Fig. 21.1). Current research indicates that the injured host responds to severe inflammation by activating anti-inflammatory pathways that are aimed at protecting the host from further inflammatory injury. Among these pathways are increased production of anti-inflammatory cytokines such as IL-10 and transforming growth factor-β (TGFβ) and leukocyte apoptosis.20,21 However, this anti-inflammatory state may lead to immunosuppression, which predisposes the host to opportunistic infections. If uncontrolled, local infections may escalate to severe systemic infections and sepsis. Sepsis can cause further inflammation and serve as an additional source of organ injury in severely burned patients. The pathogenesis of injury-induced immunosuppression is discussed in detail elsewhere in this book.
The two-hit hypothesis
Some investigators have described a phenomenon in which the injured host manifests an exaggerated inflammatory response if confronted with a secondary inflammatory stimulus during the post-injury period. This phenomenon has been termed the ‘two-hit hypothesis.’ Although the pathobiology of the two-hit hypothesis is not completely understood, monocytes and macrophages appear to play a central role in mediating the process. For example, the lymphokine α-interferon (IFN-α), produced during the initial injury, might act as the first signal and prime macrophages for a heightened inflammatory response if a second stimulus is encountered. Several changes in macrophage function, including an increase in the transcription rate of mRNA for TNFα, can be induced by exposure to IFN-α. However, TNFα protein is not produced in large amounts in response to the first inflammatory insult. If a second stimulus, such as endotoxin exposure, is provided in even a small dose, macrophages are triggered to become fully activated and to secrete large amounts of TNFα. Studies by Paterson et al.22 show that macrophages have increased responsiveness to ligands for Toll-like receptor 2 (TLR2) and TLR4 following burn injury. TLR2 and TLR4 are proteins that play an integral role as components of receptor complexes for various microbial products such as peptidoglycans, lipoproteins and lipopolysaccharide. Enhancement of TLR2 and TLR4 responses during the post-injury period may be one mechanism contributing to the two-hit phenomenon. T lymphocytes may also become hyperresponsive during the post-injury period. Zang et al.23 showed an exaggerated response to bacterial superantigens 1 day after burn injury in mice. Superantigens, such as staphylococcal enterotoxins, induce polyclonal activation of T cells and cause a shock syndrome that is similar to endotoxin shock. This mechanism may also contribute to the hyperinflammatory response seen in many patients during the post-burn period.
The exaggerated response to a secondary stimulus seen in severely injured patients appears to have functional consequences. Several studies that focused on organ injury caused by systemic inflammatory processes indicate that a phenomenon comparable to the cellular events described above can occur in severely injured patients.24 Dehring et al. found more persistent pulmonary hypertension and an exaggerated hyperdynamic response to bacteremia in sheep when a week-old thermal injury preceded systemic bacterial challenge.25 In a rat model of intestinal ischemia–reperfusion injury and endotoxemia, lung albumin leak and mortality rate increased only if both injuries occurred sequentially.26 Combined administration of low doses of endotoxin and TNFα to rats caused hypotension and metabolic effects that are commonly seen after giving a highly lethal dose of either compound alone.27 These findings are in keeping with the fact that multiple organ damage usually develops over a prolonged period during which several insults might occur. It also emphasizes why it is so important to minimize inflammatory insults such as tissue ischemia or infection, particularly in patients in whom systemic inflammation is already present.
Cytokine and non-cytokine mediators of SIRS
Several proinflammatory cytokines, chemokines, and non-cytokine inflammatory mediators play a role in the pathogenesis of SIRS. Cytokines comprise a broad group of polypeptides with varied functions within the immune response (Table 21.2). The classic mediator of systemic inflammation is TNFα. TNFα is released primarily by macrophages within minutes of local or systemic injury, and modulates a variety of immunologic and metabolic events.28 At sites of local infection or inflammation TNFα initiates an immune response that activates antimicrobial defense mechanisms and, once the infection is eradicated, tissue repair. It is a potent activator of neutrophils and mononuclear phagocytes, and also serves as a growth factor for fibroblasts and as an angiogenesis factor. However, systemic release of TNFα can precipitate a destructive cascade of events that can result in tissue injury, organ dysfunction and, potentially, death. Among the systemic effects of TNFα are the induction of fever, stimulation of acute-phase protein secretion by the liver, activation of the coagulation cascade, myocardial suppression, induction of systemic vasodilators with resultant hypotension, catabolism, and hypoglycemia.28,29 Numerous studies have shown that administration of TNFα to experimental animals will mimic the systemic inflammatory response observed in sepsis and after severe injury. Another important effect of TNFα is its ability to induce apoptosis of a variety of cell types.30 TNF-induced apoptosis may be one mechanism by which it induces tissue injury at high systemic concentrations.
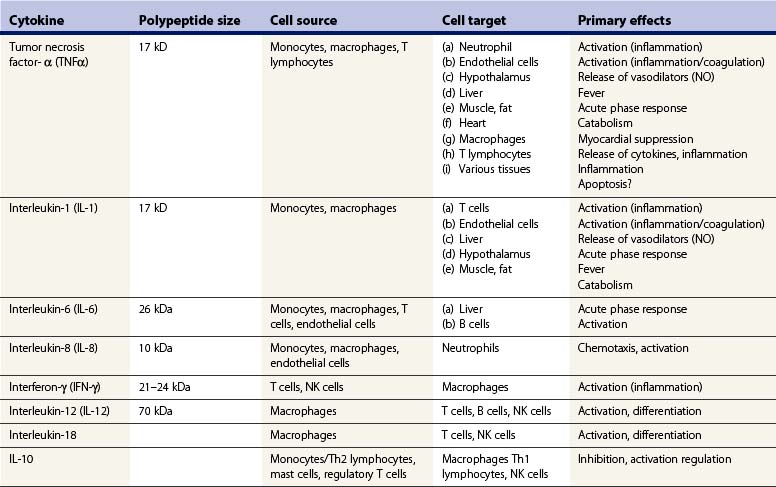
TNFα is also a potent stimulus for the release of other proinflammatory mediators, particularly IL-1 and IL-6. IL-1 is released primarily by mononuclear phagocytes and its physiologic effects are essentially identical to those of TNFα.31 However, important differences exist between the functions of IL-1 and TNFα. Most notably, IL-1 does not induce tissue injury or apoptotic cell death by itself, but can potentiate the injurious effects of TNFα. The IL-1 family of proteins, including IL-18, are the only group of cytokines for which known natural antagonists have been identified. The IL-1 receptor antagonists (IL-1ra) bind to the IL-1 receptor but do not induce receptor activation.31 These proteins appear to function as competitive inhibitors of IL-1 action. As noted earlier, the systemic release of IL-1 is dependent on the activation of inflammasomes and caspase-1.19
IL-6 is another protein that is commonly increased in the circulation of patients with SIRS.31 Macrophages, endothelial cells, and fibroblasts secrete this protein. IL-6 itself does not induce tissue injury but its presence in the circulation has been associated with poor outcome in trauma patients, probably because it is a marker of ongoing inflammation. The primary effect of IL-6 is to induce secretion of acute-phase proteins from the liver as well as to serve as a growth and differentiation factor for B lymphocytes.
Interferon-γ (IFN-γ) is a cytokine that facilitates the amplification of the acute inflammatory response, particularly the stimulation of cytokine secretion, phagocytosis, and respiratory burst activity by macrophages. IFN-γ is secreted primarily by T lymphocytes and natural killer (NK) cells in response to antigen presentation as well as macrophage-derived cytokines such as IL-12 and IL-18. The primary effect of IFN-γ is to amplify the inflammatory response of macrophages. In response to IFN-γ, the phagocytic and respiratory burst activities of macrophages are increased, secretion of inflammatory mediators such as TNFα and IL-1 is enhanced, and antigen presentation is potentiated by upregulation of class II major histocompatibility complex. Blockade of IFN-γ production or function has been shown to markedly reduce the deleterious inflammatory effects induced by bacterial endotoxin.32 Therefore, IFN-γ is believed to be an important factor in the amplification of SIRS.
Chemokines are a family of proteins that function primarily as chemotactic factors for leukocytes and, when produced inappropriately or in excess, can contribute to damaging systemic or local inflammation (Table 21.3). IL-8 is the most widely studied chemokine in the setting of sepsis and SIRS; it is a potent chemoattractant for neutrophils and is a major factor in recruiting neutrophils to inflammatory foci. Several studies have shown that IL-8 plays a role in mediating tissue injury in the setting of trauma and burn injury, particularly in the lung.33,34
Table 21.3 Classification of chemokines
| Chemokine type | Target cell |
|---|---|
| CXC Chemokines | |
| CXCL8 (IL-8, mouse MIP-2) | Neutrophils |
| CXCL1 (GROα, mouse KC) | Neutrophils |
| CXCL2 (GROβ, mouse KC) | Neutrophils |
| CXCL3 (GROγ, mouse KC) | Neutrophils |
| CXCL5 (ENA-78) | Neutrophils |
| CXCL6 (GCP-2) | Neutrophils |
| CXCL4 (PF4) | Fibroblasts, stem cells |
| CXCL9 (Mig) | T and NK cells |
| CXCL10 (IP-10) | T and NK cells |
| CXCL11 (I-TAC) | T and NK cells |
| CXCL12 (SDF-1α/β) | T lymphocytes |
| CC Chemokines | |
| CCL3 (MIP-1α) | Monocyte/macrophages, T and B cells, NK cells, basophils |
| CCL4 (MIP-1β) | same as above |
| CCL22 (MDC) | Monocyte, T lymphocytes |
| CCL25 (TECK) | Macrophages, T lymphocytes |
| CCL17 (TARC) | T lymphocytes |
| CCL5 (RANTES) | Monocyte/macrophages, T/NK cells |
| Basophils | |
| CCL14 (HCC-1) | Monocytes |
| CCL16 (HCC-4) | Monocytes, lymphocytes |
| CCL18 (DC-CK-1) | T lymphocytes |
| CCL20 (MIP-3α) | T lymphocytes |
| CCL19 (MIP-3β) | T lymphocytes |
| CCL2 (MCP-1) | T lymphocytes, monocytes, |
| CCL8 (MCP-2) | same |
| CCL7 (MCP-3) | same |
| CCL13 (MCP-4) | same |
| CCL11 (Eotaxin) | Eosinophils |
| Other Chemokines | |
| XCL1 (Lymphotactin) | T lymphocytes, NK cells |
| CX3CL1 (Fractalkine) | T lymphocytes, monocytes, |
The current conventional names of chemokines are presented and original names are provided in parentheses for reference.
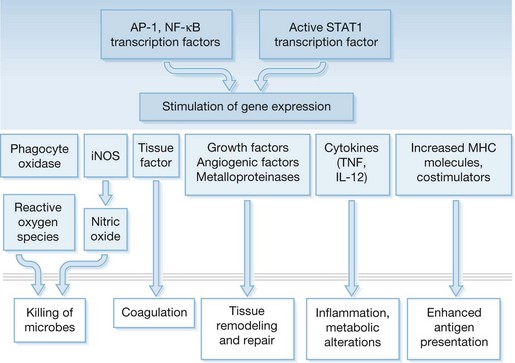
Production of most soluble mediators of inflammation is regulated at the transcriptional level. Some of the key transcription factors that control proinflammatory gene expression include nuclear factor-κB (NF-κB), AP-1 and IRF-3 (Fig. 21.4). NF-κB is composed of a family of proteins including p50 (NF-κB1), p65 (RelA), C-Rel, and p52 (NF-κB2) that combine to form homo- or heterodimers and ultimately function to regulate the transcription of a variety of cytokine, chemokine, adhesion molecule, and enzyme genes involved in SIRS.35 Increased translocation of NF-κB has been associated with a poor outcome in some studies. Activation of NF-κB in peripheral blood monocytes correlates with increased mortality in septic patients, and alveolar macrophages from patients with adult respiratory distress syndrome (ARDS) exhibited higher nuclear NF-κB levels than critically ill patients without ARDS.36,37 The AP-1 complex is activated through activation of MAP kinases by stimuli that are similar to those required for NF-κB mobilization. IRF-3 is mobilized through activation of the Trif-associated signaling pathway and results primarily in transcription of type I interferon (IFN-α) genes. The STAT1 pathway is induced by activation of type I (IFN-α) and type II (IFN-γ) receptors. Together, these transcription factors mediate the transcription of numerous factors involved in inflammation and tissue repair (Fig. 21.5).
Several non-cytokine factors have been implicated in the pathogenesis of SIRS. Platelet-activating factor (PAF) is a phospholipid autocoid released by endothelial cells that regulates the release of cytokines and amplifies the proinflammatory response. It appears to be an important factor in the adhesion of neutrophils to endothelial cells. The prolonged presence of PAF in the serum of patients with SIRS has correlated with poor outcome.38 Eicosanoids are arachidonic acid metabolites that regulate many aspects of the immune response. Leukotrienes (LTC4-LTE4) induce contraction of endothelial cells and encourage capillary leakage.39 Thromboxane A2, a macrophage and platelet-derived factor, promotes platelet aggregation, vasoconstriction and, potentially, tissue thrombosis.40
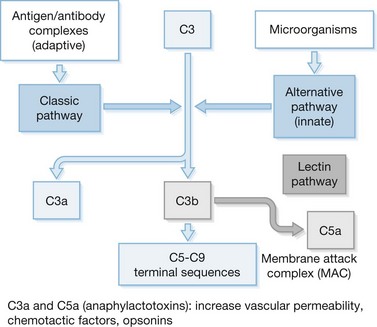
The complement cascade is composed of more than 30 proteins that interact in a complex fashion to mediate inflammation and direct lysis of microbes and other cells (Fig. 21.6). However, in SIRS, excessive complement activation appears to cause significant cellular injury in the host. Products of the complement cascade, most notably C3a and C5a, are potent activators of inflammation and leukocyte chemotaxis.41 C3a and C5a also directly activate neutrophils and promote release of reactive oxygen intermediates and proteases. Excessive release of these factors can result in significant tissue injury. The membrane attack complex (MAC) is the terminal component of the complement cascade. MAC results from the aggregation of the complement components C5–C9 on biological membranes. The accumulation of MAC on cell surfaces can result in significant tissue and cellular injury and may be a major factor in the pathogenesis of MODS. Suber and colleagues42 have reported that complement-mediated responses to self-antigens exacerbate tissue injury after burn injury.
Circulating cytokines as markers of SIRS and predictors of outcome
Numerous studies have been undertaken with the goal of using plasma cytokine levels as diagnostic and prognostic markers in patients with SIRS or sepsis. This approach seems logical, based on the observation that circulating cytokines have been observed in several clinical studies of trauma, sepsis, and thermal injury. Given its central role in the activation and regulation of the proinflammatory response, TNFα has been studied extensively as a plasma marker of SIRS. The results have been inconsistent. Martin et al.43 showed that TNFα levels were markedly elevated in patients with septic shock and correlated with fatal outcome. However, their results also showed that trauma patients did not exhibit the same marked elevations in circulating TNFα, nor did circulating TNFα concentrations correlate with increased mortality in trauma patients. In some published studies, measurement of circulating TNFα levels in burn patients has not provided a useful marker of outcome.44,45 Overall, plasma TNFα levels have been variable and inconsistent and have not correlated with mortality or the development of MODS. However, a study by Zhang et al.46 in 25 patients with >30% total body surface area (TBSA) burns demonstrated marked increases in plasma TNFα levels and a significant correlation between TNFα concentration and shock, MODS, and death. These findings support the results of Marano et al.,47 who showed a significant correlation between circulating TNFα concentration and mortality in burned patients. Therefore, taken together, these results show that TNFα could serve as a useful marker of ongoing inflammation as well as an indicator of morbidity and mortality in the setting of burn injury, but controversy remains.
TNFα interacts with two known cell surface receptors designated tumor necrosis factor receptor (TNFR)-I and TNFR-II. TNFR-I, also known as TNF-R55 or p55, is expressed on a variety of cells and its activation mediates most of the activities of TNFα, including induction of apoptosis. Activation of TNFR-II (TNF-R75 or p75) results in cellular proliferation and activation. During inflammatory states, TNFR are released from cells and may serve as antagonists of TNFα. Several investigators have characterized surface-bound and soluble TNFR (sTNFR) in sepsis and trauma.46,48,49 Hubl et al.48 showed that surface TNFR-I were upregulated, whereas TNFR-II were downregulated in patients with SIRS. Increased TNFR-I correlated with increased body temperature but not with survival. SIRS patients with decreased surface TNFR-II had a trend toward increased mortality. A study by Zhang et al.46 showed a higher incidence of shock, MODS, and mortality in burn patients with increased plasma sTNFR-I and sTNFR-II levels. Presterl et al.49 showed a correlation between sTNFR and APACHE III scores, as well as the incidence of shock and mortality in septic patients. Sikora and colleagues50,51 have reported increased concentrations of sTNFR-I and sTNFR-II in the plasma of burned children. sTNR concentrations correlated with burn surface area and decreased with adequate treatment. Children with hypovolemic shock had higher plasma concentrations of sTNFR. Overall, in the studies published to date, the presence of high levels of circulating sTNFR correlate with ongoing inflammation and may serve as an indicator of poor prognosis.
Another family of proteins that has been extensively analyzed as markers of SIRS is IL-1 and IL-1 receptor antagonist (IL-1ra). In burn patients, low IL-1ra levels correlated with mortality in two independent studies.52,
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


