Fig. 2.1
(a) Mammoth ivory figurine, Venus of Hohle Fels, Swabian Jura, Germany, circa 35,000 BC (Source: Silosarg). (b) Venus of Willendorf, circa 22,000 BC (Source: Don Hitchcock)
Dieting and exercise achieve significant, durable weight loss only rarely in the obese. Why is this failure rate so high? Friedman alludes to powerful biologic systems that defend body weight. These systems act at a subconscious level and are controlled by a genetic heritage that was forged, as Prentice states [2], by the selective pressure of famine present for virtually all of our evolution that acted as a constant threat to survival and reproductive fitness. From a practical standpoint, an understanding of the pathophysiology of obesity and metabolic disease will guide development of environmental and physiologic therapeutic interventions. Perhaps equally importantly, however, an understanding of the origins of the epidemic and an appreciation that obesity in most cases results not from a defect of individual willpower but rather from a physiology shaped by eons of genetic selection and thrust into a modern “obesogenic” environment provide a basis for empathy toward those afflicted with a debilitating condition.
The pathophysiology of obesity and that of its associated metabolic diseases are distinct. Part I of this chapter will address the pathophysiology of obesity itself. We will discuss the physiologic mechanisms that lead to the obesity phenotype and answer the question, “How do we become obese?” We will next discuss the genetic, evolutionary, and environmental forces that have molded these regulatory systems to create the modern epidemic. In doing so, we will answer the question, “Why do we become obese?” If obesity was simply a cosmetic condition, we might end there, but of course obesity is associated with a wide range of pathology. In Part II we will explore the pathophysiology of obesity-related metabolic disease and study the effects of nutrient excess on cellular metabolism and systemic physiology.
The Pathophysiology of Obesity
Pathophysiologic Mechanisms of Obesity
How Do We Become Obese?
Why do diets fail? Why do dieters hit a “dieter’s wall” that resists further weight loss? Why, in short, can we not simply “decide to eat less?” To answer these questions, we must explore the physiologic systems that regulate body weight. We are creatures of behavior, and so the most important of these mechanisms controls food-related behavior, which dictates how much, how frequently, and what types of food we eat. But a tenet of metabolic science is that the processes that regulate energy homeostasis are so central to our biology that they interface with virtually every aspect of physiology. We will discuss the most important of these processes, which regulate satiety and hunger, metabolic rate, thermogenesis, and adipocyte biology.
Satiety and Hunger: The Leptin Paradigm
The reason we become obese, at the most basic level, is that we eat too much. The most important regulatory mechanism for body weight in humans is the collective system that controls food intake. Hunger, which may be defined as the need or desire to eat, and satiety, which may be defined as the lack of hunger, describe fundamental aspects of eating behavior. While conceptually useful, at the cellular and molecular level these definitions are less precise, as satiety and hunger mediators utilize distinct yet intimately associated signaling pathways. Furthermore, the dichotomy between satiety and hunger is overly simplistic, as feeding behavior is highly complex and includes subtle behaviors such as eating speed, preferences, thresholds, hunger irritability, and sensory and emotional responses to food. We often consider such behaviors to be under conscious control but in fact their corresponding neural control networks reside within the hypothalamus in the midbrain, an area that, from an evolutionary perspective, long predates the frontal cortex, the seat of conscious thought and cognition. Eating behavior is regulated at a subconscious level. We can diet, but with few exceptions; such efforts are limited in magnitude and followed by weight regain. We might make the comparison to respiration, which is controlled subconsciously; we do not decide what our respiratory rate will be at any given moment. That said, we can maintain a fixed respiratory rate for a limited period of time with conscious effort in the absence of significant activity demands. Food intake is similar: We can limit our caloric intake for a period with conscious effort, but in time, the midbrain will win out, and like respiration, food intake will “snap back” to subconsciously determined levels. To understand this tightly regulated system, we must travel back two decades.
In 1994, Dr. Jeffrey Friedman and his laboratory at the Rockefeller University cloned the leptin gene [5]. Friedman’s group studied the Ob mouse—a hyperphagic obese strain, described in the 1950s, which lacked a circulating factor—that, when restored from wild-type mice in parabiosis experiments, reversed the obese phenotype (Fig. 2.2) [6]. Friedman identified that circulating factor as the 16kD protein leptin and found the leptin gene to be mutated and nonfunctional in the Ob mouse. Restoration of exogenous wild-type leptin to Ob mice reversed their obesity. Friedman’s group went on to clone the human leptin gene, and, within a few years, two obese human kindreds were identified with similar leptin mutations in whom administration of exogenous leptin also reversed obesity [7], along with equally rare kindreds with a leptin receptor mutation and a similar obese phenotype [8]. These dramatic results were met with much excitement. The hunt was on for leptin mutations in the general obese population and leptin was considered a potential treatment for obesity. It was soon found, however, that monogenic leptin mutations are a rare cause of common human obesity. Exogenous leptin, while a cure for Ob mice and Ob humans, had little therapeutic effect in obese humans in the general population. In fact, common human obesity is characterized by elevated leptin levels and resistance to leptin’s satiety effects in a situation not dissimilar from insulin resistance [9, 10].
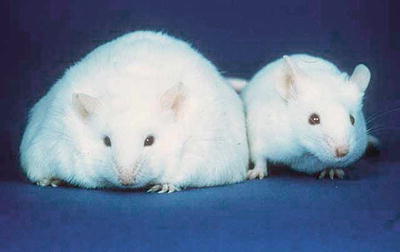
Fig. 2.2
Ob mouse (left) (Source: US Department of Energy Office of Science, Office of Biological and Environmental Research, Human Genome Program http://www.csm.ornl.gov/SC99/GENwall.html)
Despite a lack of therapeutic efficacy, the discovery of leptin nonetheless transformed our understanding of satiety and hunger. Leptin mediates a complex counter-regulatory communication between gut, adipose tissue, and brain that regulates food intake. Secreted by adipose tissue in response to a meal, leptin circulates through the bloodstream as a hormone to bind its receptor in the hypothalamus and effect satiety. When we wake, serum leptin levels are low after a nighttime fast. We breakfast, generating an adipose tissue leptin secretory response that induces satiety and limits food intake as we complete breakfast. We remain sated for a few hours, after which leptin levels wane and we find ourselves hungry by lunch and the cycle repeats. In this manner, leptin, in concert with multiple other mediators, controls daily food intake. Leptin also regulates long-term food intake. Peak postprandial leptin levels are determined in part by the adipose tissue mass available to secrete leptin, which in turn determines the magnitude and kinetics of the satiety response. When we diet and reduce adipose tissue mass, postprandial leptin levels decrease over the course of weeks. This causes us to eat progressively more at each subsequent meal until we restore adipose tissue mass and drive leptin levels higher. This mechanism provides a cogent explanation for weight regain after dieting.
The Hypothalamic Feeding Center
As early as 1940, hypothalamic ablation experiments in rats that induced hyperphagia and obesity first demonstrated the central role of the hypothalamus in weight regulation [11]. Leptin binds its receptors within the arcuate nucleus (ARCN) of the hypothalamic feeding center and activates anorexigenic (satiety-inducing), catabolic pro-opiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART)-producing first-order neurons in the dorsolateral ARCN. POMC is posttranslationally processed to yield the anorexigenic peptides α(alpha)-, β(beta)-, and γ(gamma)-MSH. Leptin simultaneously inhibits secretion of the orexigenic (hunger-inducing) mediators neuropeptide Y (NPY) and agouti-related peptide (AgRP) by the ventromedial ARCN. First-order ARCN neurons in turn project to second-order neurons, including those within the paraventricular nucleus (PVN) and lateral hypothalamic area (LHA), which in turn elaborate secondary signaling mediators. The PVN generates an anorexigenic, catabolic program via expression of CRH, TSH, and oxytocin, which increase satiety and energy expenditure via multiple endocrine and autonomic nervous system pathways. The LHA, in contrast, generates an orexigenic, anabolic program via expression of orexins A and B and melanin-concentrating hormone (MCH). Each of these pathways inhibits the other at the level of first-order neuron transmitter effects on second-order neurons (Fig. 2.3).
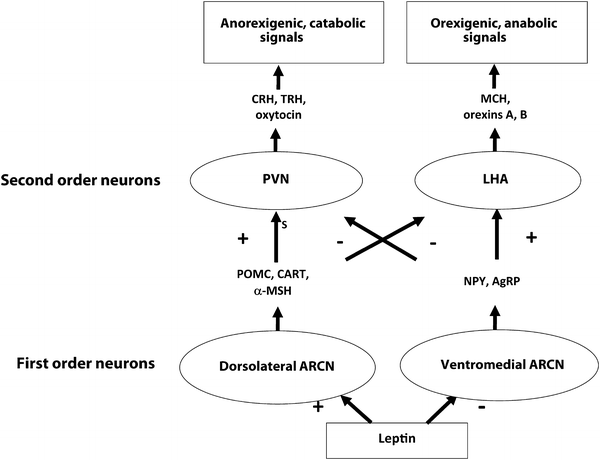
Fig. 2.3
A simplified schematic of the hypothalamic feeding center. First-order ARCN neurons communicate with second-order PVN and LHA neurons to coordinate behavioral and metabolic output. PVN signaling is primarily anorexigenic and catabolic and is enhanced by leptin and insulin, while LHA signaling is primarily orexigenic and anabolic and inhibited by leptin and insulin. Both pathways negatively regulate the other. Other peripheral and central mediators (not shown) stimulate first- and second-order neurons as well, including insulin, ghrelin, CCK, GLP-1, serotonin, endogenous cannabinoids, and norepinephrine
The hypothalamic feeding center exhibits an intrinsic “set-point” that is influenced by multiple afferent inputs from all organ systems, including prandial status, energy stores, hypothalamic leptin receptor distribution, leptin resistance, and multiple endocrine and neural stimuli. These complex inputs orchestrate diverse behavioral, metabolic, and physiologic responses to food-related stimuli via afferent and efferent projections to and from multiple brainstem and higher level central nervous system networks that regulate emotion, cognition, sensory processing, and memory. Food activates entrainable reward centers, so-called hedonic circuits that drive consumption of foods high in fat, sugar, and salt in the absence of caloric deficiency as well as dedicated memory and visual circuits. Food acquisition is such an important task in our evolutionary history that it involves all areas of the brain and all aspects of behavior and physiology.
A Complicated Family of Mediators
Leptin opened the door to the discovery of a family of proteins that regulate food intake, broadly classified as adipokines, gut hormones, and cytokines (Table 2.1). Adipokines are secreted predominantly by adipose tissue, gut hormones by the gastrointestinal tract and viscera, and cytokines by leukocytes within adipose and other tissues. These proteins share phylogenetic and functional overlap and classification systems remain in flux. For example, leptin is structurally highly conserved and a member of the long-chain helical chain cytokine family, of which IL-6 and IL-11 are members; the term adipocytokines is therefore sometimes used to reflect the close relationship between adipokines and cytokines. In many cases, these mediators are elaborated by multiple tissues and cell types. Tumor necrosis factor-alpha (TNF-α), for example, is expressed by adipocytes and leukocytes within adipose and other tissues, although macrophages are the dominant source. Ghrelin is secreted primarily by gastric fundus cells in response to fasting but is also expressed at low levels by the placenta, kidney, pituitary, endometrium, macrophages, and hypothalamus; adipocytes are the dominant source of leptin, but small amounts are also secreted by the placenta, muscle, and stomach.
Table 2.1
Partial list of proteins involved in weight regulation: functions are complex, context dependent, and not well defined for many mediators; general effects in majority of studies listed below
Mediator | Primary source | Satiety | Glucose homeostasis | Immunity |
|---|---|---|---|---|
Adipokines | ||||
Adiponectin | Adipocytes | Minimal satiety effects | Insulinomimetic | Anti-inflammatory |
Apelin | Adipocytes, brain, heart, kidney, lung | Probably anorexigenic, data sparse | Inhibits glucose-induced insulin secretion | Anti-inflammatory |
CCL2 | Adipocytes | Unknown | Diabetogenic, likely through proinflammatory properties | Proinflammatory, macrophage homing |
Leptin | Adipocytes | Anorexigenic, hypothalamic leptin resistance in obesity | Generally insulinomimetic | Generally proinflammatory |
Lipocalin 2 | Adipocytes, monocytes, macrophages | Unknown | Diabetogenic via proinflammatory effects | Pro- and anti-inflammatory effects |
Plasminogen activator inhibitor-1 (PAI-1) | Adipocytes | Unknown | Probably diabetogenic—causality not well established | Unknown |
Resistin | Adipocytes, macrophages | Probably anorexigenic | Diabetogenic via proinflammatory effects | Proinflammatory |
Retinol-binding protein 4 (RBP-4) | Adipocytes, hepatocytes, macrophages | Unknown | Diabetogenic | Proinflammatory |
Secreted frizzled-related protein 5 (SFRP5) | Adipocytes, pancreas | Unknown | Insulinomimetic | Anti-inflammatory, suppresses Wnt signaling |
Visfatin | Adipocytes | May induce satiety, data conflicting | Insulinomimetic | Proinflammatory |
Gut hormones | ||||
Amylin | Pancreatic β(beta)-cells | Anorexigenic, increases energy expenditure | Co-released with insulin in response to food, inhibits insulin secretion | Probably proinflammatory, conflicting data |
Cholecystokinin (CCK) | Duodenal, jejunal I-cells, CNS | Anorexigenic, regulates meal termination | Probably insulinomimetic | Anti-inflammatory |
Ghrelin | Gastric fundal cells | Orexigenic | Diabetogenic | Anti-inflammatory |
Glucose-dependent insulinotropic peptide (GIP) | Duodenal, jejunal C-cells | Anorexigenic | Induces insulin secretion, conflicting data | Pro- and anti-inflammatory effects, conflicting data |
Glucagon-like peptide-1 (GLP-1) | Ileal L-cells, CNS | Anorexigenic | Insulinomimetic | Anti-inflammatory |
Glucagon | Pancreatic α(alpha)-cells | Anorexigenic, increases energy expenditure | Increases blood glucose levels, insulin secretion | Probably proinflammatory |
Insulin | Pancreatic β(beta)-cells | Anorexigenic, increases energy expenditure | “Insulinomimetic” | Generally anti-inflammatory, CNS administration may promote inflammation |
Oxyntomodulin | Ileal L-cells | Anorexigenic, increases energy expenditure | Probably insulinomimetic | Unknown |
Pancreatic polypeptide (PP) | Pancreatic F-cells | Anorexigenic, increases energy expenditure | Insulinomimetic, increases hepatic insulin sensitivity | Unknown |
Peptide tyrosine tyrosine (PYY) | Ileal L-cells | Anorexigenic | Insulinomimetic | Anti-inflammatory, inhibits NFκ(kappa)B activation |
Cytokines | ||||
IFN-γ(gamma) | T-cells, NK cells | Unknown | Generally diabetogenic | Proinflammatory, induces macrophage inflammation |
IL-1 | Macrophages | Anorexigenic | Diabetogenic | Proinflammatory |
IL-6 | Adipocytes, macrophages, lymphocytes | Unknown, likely anorexigenic in context of cachexia | Generally diabetogenic although effects are context dependent and conflicting | Generally proinflammatory |
IL-10 | Macrophages, lymphocytes | Unknown | Insulinomimetic | Anti-inflammatory |
TNF-α(alpha) | Adipocytes, macrophages, lymphocytes | Anorexigenic, mediates cachexia response | Diabetogenic | Proinflammatory |
Most but not all adipokines, gut hormones, and cytokines regulate food intake. Leptin appears to play a dominant role in regulating long-term satiety, while the gut hormones CCK, PYY, and ghrelin dominate short-term food intake, including variables such as meal size and duration. Ghrelin is secreted during fasting and binds receptors in ARCN of the hypothalamus to induce a strong orexigenic (hunger) signal. Other mediators have only modest effects on food intake. Adiponectin, an adipokine secreted by adipocytes, stimulates insulin secretion, inhibits β(beta)-cell apoptosis, and attenuates inflammation. Consistent with these beneficial effects, serum adiponectin levels are inversely correlated with obesity and metabolic disease.
In addition to regulating food intake, satiety and hunger mediators manifest multiple functions. Common to most are regulation of glucose homeostasis, lipid metabolism, and immunity. Leptin is immunostimulatory, inducing macrophage, monocyte, and T-cell proliferation and cytokine expression, with a bias toward generating Th1 responses in T-cells and inflammatory cytokine expression in macrophages [12, 13]. Leptin controls endocrine function, potentiating pituitary-hypothalamic axis activity and regulating steroid metabolism. Leptin has also been implicated in cognitive function, learning, and memory; leptin-deficient humans suffer mild cognitive deficits that are corrected with exogenous leptin replacement. The incretins are a class of gut hormones of which glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) are best described. GIP is secreted by K-cells in the duodenum and jejunum in response to a glucose load, induces insulin secretion by pancreatic β(beta)-cells, and stimulates fatty acid synthesis in adipocytes. GLP-1 is secreted by L-cells in the ileum in response to nutrient delivery and has insulinomimetic properties, decreasing peripheral insulin resistance and potentiating pancreatic insulin secretion. GLP-1 also inhibits gastric emptying and induces satiety. Altered secretion of incretins secondary to anatomic changes in the intestinal tract has been implicated in the resolution of diabetes after bariatric surgery. Similar functional diversity is characteristic of adipose tissue cytokines. TNF-α(alpha) induces insulin resistance and altered lipid metabolism and promotes anorexia in cachexia and inflammatory states [14]. In general, adipose tissue cytokines and adipokines are either proinflammatory and diabetogenic (e.g., TNF-α[alpha], IL-6, IL-8, IL-1, resistin) or anti-inflammatory and insulinomimetic (e.g., IL-10, IL-1Ra, adiponectin), with the former upregulated and the latter downregulated in obesity. Exceptions exist: The orexigen ghrelin, for example, has anti-inflammatory properties but induces insulin resistance.
Multiple other mediators regulate feeding behavior within the hypothalamus and other areas of the brain, including insulin, endogenous opioids, adrenergic agonists, and cannabinoids. Redundancy, complex regulatory control, and functional pleiotropy are characteristics of the broad family of molecules that regulate food intake. This diversity of function speaks to the intimate association between energy balance and all physiologic systems. Despite their complexity, these mediators define a conceptually simple counter-regulatory mechanism for defense of body weight that underlies the pathogenesis of obesity.
Metabolic Rate
While dominant, control of food intake is not the sole mechanism of weight regulation. Obese patients often report eating little but suffering from a “slow metabolism.” The magnitude of the effect of differences in energy expenditure on human body weight variability is debated and is less than contributions from differences in the regulation of food intake. Nonetheless, variability in metabolic rate contributes to obesity. The terms “fast burners” and “slow burners” are used to describe subpopulations at the extremes of metabolic rate.
Metabolic rate has been studied in response to weight loss in healthy volunteers using indirect calorimetry. In conditions of energy deficit, total energy expenditure (TEE), comprised of resting energy expenditure (REE, 60–70 % of TEE, defined as energy used for basic physiologic functions at rest) and non-resting energy expenditure (NREE, 30–40 % of TEE, defined as energy used for activity above and beyond REE), is decreased beyond that expected by loss of fat and fat-free mass alone. These changes are secondary to equal decreases in NREE and REE and accompanied by a corresponding decrease in voluntary physical activity. Similar changes are observed in obese subjects who achieve 10–20 % body weight loss and whose TEE decreases up to 20 % more relative to predicted values from loss of fat and fat-free mass [15]. Obese subjects who maintain weight loss have lower resting metabolic rates than lean subjects, changes that persist for years and mandate constant dietary vigilance. In contrast to diet-induced weight loss, bariatric surgery-induced weight loss is paradoxically associated with increased TEE in rodents and humans, which may provide an explanation for the durable efficacy of surgery. In the absence of surgery, however, compensatory decreases in energy expenditure counteract caloric restriction and provide yet another explanation for the plateau in weight loss and weight regain associated with dieting.
Studies of overfeeding and weight gain in obese subjects demonstrate converse changes with notable differences. First, overfeeding leads to a compensatory increase in TEE primarily due to increased NREE, in contrast to weight loss, in which equal decreases in NREE and REE are observed [15]. Second, voluntary overfeeding studies in twin cohorts demonstrate a significant genetic component to variability in weight gain in response to overfeeding [16]. Third and importantly, in general, increases in TEE in response to overfeeding are transient, lasting months then reverting to baseline, unlike sustained weight loss for which such changes tend to be long lasting. Furthermore, while compensatory increases in TEE oppose weight gain, this effect is weaker than that of decreased TEE with weight loss. Taken together, these observations suggest that regulation of metabolic rate is more robust in conditions of negative rather than positive energy balance: The system is better designed to prevent leanness than avoid adiposity, a common theme.
The mechanisms underlying the regulation of energy expenditure and metabolic rate are multiple, but the sympathetic nervous system and hormonal system are dominant. Voluntary choice certainly controls NREE to some extent, but much like food intake, we consciously control our activity levels to a lesser degree than we imagine. Differences in autonomic nervous system activity have been implicated in determining metabolic rate: Overfeeding increases and underfeeding decreases sympathetic nervous system activity in rodents, and variability in sympathetic nervous system activity is observed in humans, with higher levels predicting successful weight loss. Obese subjects also manifest alterations in thyroid hormone and catecholamine balance that are associated with decreased metabolic rates. At the cellular level, these variables control metabolic rate and energy expenditure by regulating thermogenesis.
Thermogenesis
In 1783, Laplace and Lavoisier invented the first calorimeter and measured energy balance in animals. They found that biologic systems obey the first law of thermodynamics, taking in nutrients and storing and expending energy (as activity or heat) and demonstrating that throughout these processes, energy is conserved. As Lavoisier famously concluded, “Life is combustion.”
All cellular biochemical reactions are less than 100 % efficient and thus generate heat. Thermogenesis occurs in all cells, but is most active in brown adipose tissue (BAT) and skeletal muscle. Skeletal muscle thermogenesis occurs during exercise, during movement not associated with exercise (non–exercise-induced thermogenesis, NEAT), and during cold–induced shivering. Skeletal muscle energy utilization efficiency during exercise is increased in weight loss subjects and decreased in subjects who gain weight, and lower levels of NEAT have been demonstrated in obese humans [17]. These observations suggest that differences in skeletal muscle thermogenesis contribute to the pathogenesis of obesity. BAT is a dominant site of thermogenesis, in which it underlies non–shivering thermogenesis, a critical thermoregulatory mechanism in rodents and neonatal humans. BAT, relative to white adipose tissue (WAT), is associated with higher thermogenic potential, increased mitochondrial number and function, decreased lipid storage, and improved metabolism. Diet–induced thermogenesis (DIT), the increase in metabolic rate in response to a meal observed in rodents and humans, is an important component of the thermogenic response. At the cellular level, DIT increases mitochondrial heat production and decreases energy extraction, limiting exposure to postprandial increases in nutrient flux. Humans demonstrate significant variability in the magnitude of DIT, with obese humans having lower DIT responses [18].
While all biochemical reactions generate heat, membrane ion leaks that occur in all cells and triglyceride-fatty acid futile cycling within skeletal muscle are examples of processes that involve significant thermogenesis. Uncoupling of electron transport from oxidative phosphorylation within mitochondria is also a dominant-regulated thermogenic process and plays an important role in determining metabolic rate. Mitochondria, evolved from intracellular parasites to become endosymbiotic cell organelles, utilize oxidative phosphorylation coupled to electron transport along their inner membrane to generate ATP from glycolytic energy substrates. The efficiency of this process is regulated by uncoupling proteins (UCP). These proteins uncouple oxidative phosphorylation from electron transport, creating a proton leak that effectively runs the mitochondrial engine in “neutral,” generating heat rather than ATP. Control of UCP activity adjusts mitochondrial efficiency and acts as a thermoregulatory mechanism. This control mechanism extends across animal and plant phyla: Uncoupling occurs in potatoes in response to cold weather. Three dominant human UCP isoforms have been described with two other candidates also identified. UCP-1 expression is restricted to BAT; UCP-2 is widely expressed in multiple tissues, while UCP-3 is expressed in BAT, skeletal muscle, and brain.
The role of UCPs in the pathogenesis of obesity is an area of active study. In humans, the correlation between UCP function and thermogenesis, specifically, and metabolic rate, in general, is imperfect. Skeletal muscle UCP expression is paradoxically upregulated in response to starvation, for example, confusing the role of uncoupling in response to nutrient deficit. The type and quantity of dietary constituents, along with variables such as environmental temperature and photoperiod, also influence UCP-1 expression, adding to complexity. Finally, the magnitude, duration, and direction of changes in UCP-1 expression in BAT in response to overfeeding in rodents are heterogeneous and strain dependent, reinforcing the importance of genetic background on the regulation of metabolic rate. Nonetheless, a transgenic mouse overexpressing UCP-1 under control of the adipose tissue-specific AP2 promoter is resistant to obesity, and polymorphisms in UCP genes correlate with obesity and metabolic disease in humans, supporting a role for UCPs in obesity pathogenesis.
Adipocyte Biology and Other Metabolic Processes
Humans demonstrate differences in kinetics of adipocyte hypertrophy, proliferation, differentiation, and metabolism. Much data are derived from in vitro analysis, so caution must be applied in extrapolation to in vivo function. Nonetheless, obese humans manifest a higher adipocyte proliferative capacity than lean subjects. It was once thought that adipocytes did not proliferate or increase in number in adults, but recent data suggest otherwise, and obese subjects, especially those with early-onset childhood obesity, have a greater number of adipocytes than lean subjects as well as preadipocyte precursors with different and metabolically adverse phenotypic potentials [19]. While as of yet poorly understood, these differences contribute to obesity pathogenesis.
Homeostasis, Allostasis, and Homeorhesis
The regulatory systems described above comprise only a partial list of the many physiologic processes that occur within adipose and other tissues that control energy balance, all of which are regulated by homeostatic systems with differing amplitudes and kinetics that contribute to variability in human body weight. Despite the power of weight regulatory systems, we have a degree of control over our body weight, and it is an individual responsibility to strive to optimize our health within our defined limits. As such, personal choice plays a role in the pathogenesis of obesity. But I have argued that the magnitude of the effect of personal choice on weight is less than we believe and food intake is instead subject to tightly regulated subconscious mechanisms. The limits imposed on personal choice by these systems are powerful and in fact represent a fundamental characteristic of all biologic systems.
All of biology has but one goal: the maintenance of homeostasis. Perfect homeostasis is of course elusive: Our heart rate is not a constant 72 beats per minute for our entire lives; rather, we are constantly buffeted by external forces that drive us away from homeostasis. In the case of energy homeostasis, this buffeting comes in the form of periodic alterations in food resources, physical activity, illness, temperature, and innumerable other environmental and physiologic variables. The term allostasis was proposed by Drs. Sterling and Eyer at the University of Pennsylvania in 1988 to describe the process by which biologic systems evolved to achieve stability not through strict adherence to a homeostatic mean but rather through a dynamic response to external stimuli, the allostatic load, that achieves controlled variability around a homeostatic mean within a defined allostatic range. The robustness of biologic systems derives not from adherence to homeostasis but rather from the capacity for dynamic allostasis. Biologic systems function well within their allostatic range, deviation from which is highly disruptive. Furthermore, most humans, most strains of laboratory rodents, and many wild animals gradually gain weight over the course of their lives [4], suggesting not only a predisposition to excess weight but also toward gradual lifelong increase. Biologist Conrad Hal Waddington proposed the term homeorhesis, in contrast to homeostasis, to describe regulation of a system along such a trajectory [20] (Fig. 2.4).
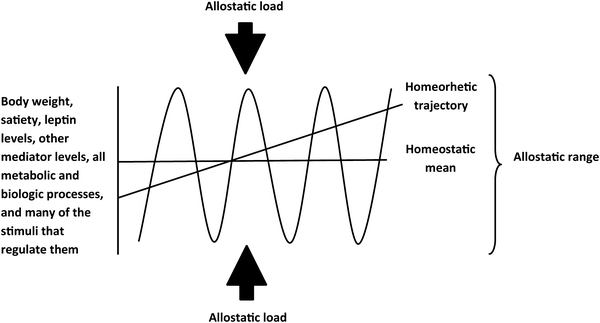
Fig. 2.4
Homeostasis, allostasis, and homeorhesis. Levels of leptin and other mediators, the outcome measures they regulate (food intake, thermogenesis, and all other metabolic and physiologic processes), as well as physical activity and many environmental and physiologic stimuli that regulate these processes all cycle in an allostatic manner around a homeostatic mean or, alternatively in some cases, along a homeorhetic trajectory (e.g., body weight over the course of a lifetime). This cycling is limited by multiple respective overlapping interacting allostatic ranges in response to diverse stimuli, which comprise the allostatic load for any given mediator, outcome, or system. Biologic systems are confined within their allostatic ranges, which are dynamically regulated and differ among individuals
Leptin represents a paradigm for allostatic behavior but is by no means unique; all aspects of metabolism, indeed all biologic processes, are governed by allostasis. In fact, it has been suggested that body weight is not the primary variable regulated by metabolism—rather, the regulated variables are the sum total of all the metabolic processes that we have discussed, and body weight is simply the resulting secondary phenotype [20]. When we ask obese patients to lose significant weight with diet and exercise, we are asking them to step outside their allostatic range, an impossible task from a biologic perspective. This is why efforts to address the obesity crisis at the individual level fall short. This is why diets fail. To impact on the obesity epidemic, we must understand the genetic, evolutionary, and environmental influences that define metabolic allostatic range and how these factors conspire so vigorously to defend body weight.
Evolutionary and Genetic Contributors to the Pathogenesis of Obesity
Why Do We Become Obese?
Rare reports document humans with a paucity of adipose tissue secondary to congenital and acquired lipodystrophy syndromes and genetic errors of metabolism. These patients have voracious appetites, require high caloric intake to maintain lean body weight, and, like the obese, suffer from diabetes and steatosis, stressing the central role of adipose tissue in metabolism and demonstrating that its absence is as detrimental as its excess. Why are such patients rare while overweight and obesity are so prevalent? We have discussed the physiologic systems that defend body weight, but why are these systems predisposed to excess weight? Why, in other words, is there not instead an epidemic of wasting and leanness? The explanation for why humans are prone to excess adiposity but stringently protected from underweight conditions is rooted in genetics and influenced by powerful environmental and evolutionary selective pressures.
Why Do We Overeat?
Despite knowing that it is unhealthy, humans, along with many animals not capable of such knowledge, will eat to excess if surplus food is available. Hedonic circuits, mentioned above, and metabolic thrift, discussed below, are examples of physiologic and evolutionary mechanisms underlying this phenomenon. But why did evolution select for such behavior if it is detrimental to health? The reason is that evolution did not mold us to optimize long-term health or longevity. Rather evolution selects for physiology and behavior that optimizes reproductive fitness, a goal that conflicts to some extent with that of longevity. Despite its detrimental long-term effects, surplus nutrition, to a point, optimizes reproductive fitness in youth, as evidenced by increased fertility rates and younger age of onset of puberty in Western societies. Reproduction is energy intensive and evolution selected for a propensity to maximize energy resources to ensure adequate reproductive potential. Long-term health and longevity are secondary (or absent) “goals” of evolution. Humans, unlike other animals, have the ability to self-reflect and, to an extent, “decide” to eat less, but as has been discussed, this ability is constrained by allostatically governed mechanisms. The distinct and separate goals of reproductive fitness and long-term health and longevity—the former hardwired by evolution, the latter unique to human cognition—lead to our conflict with overeating.
Metabolic Thrift
Geneticist James Neel at the University of Michigan proposed the “thrifty genotype” hypothesis in 1962 [21]. Neel postulated that throughout evolution, the constant pressure of famine led to selection of genes that regulated metabolism in a highly “thrifty” manner: Polymorphisms in metabolic genes were strongly selected for if they imparted metabolic thrift, or a tendency toward overweight, obesity, and energy conservation, while polymorphisms in genes that imparted a tendency toward a less thrifty metabolism were strongly selected against. Metabolic thrift provides a robust reproductive advantage in food-sparse environments, but leads to a blossoming of the obesity phenotype in “obesogenic” environments in which food is plentiful, exactly what we observe today. Polymorphisms resulting from random Darwinian mutation that predisposed to overweight, obesity, and thrift accumulated in the human genome, while those predisposing to leanness and a lack of thrift became rare. The thrifty gene hypothesis has evolved and remains debated. Thrifty gene candidates are sparse and proof of causality elusive. Alternative hypotheses such as predation release and genetic drift may also contribute to obesity. Debate also exists regarding the magnitude of food scarcity during human evolution, although strong historical data support that, until recently, famine acted as a constant selective pressure [2]. Despite these controversies, the thrifty genotype hypothesis provides a coherent and accepted explanation for the human predisposition toward overweight and obesity and its dramatic recent increase.
Why Adipose Tissue?
Hummingbirds do not accumulate significant adipose tissue but rather maintain a caloric intake per unit body weight 50 times that of humans and undergo dramatic nocturnal downregulation of metabolic rate. Other animals conserve energy by hibernating, in some cases for the majority of their lives. Rabbits have very low adipose tissue mass and are metabolically unthrifty but compensate with a high reproductive rate during periods of feast. Such diverse strategies for metabolic thrift abound in living creatures. Among these strategies, storage of calories for future use is common. Storage may be ectopic: Hamsters store up to 50 % of their body weight in their cheek pouches, providing food for a rainy day but with the price of a host of pouch pathologies. Migratory geese store excess calories in the liver, the source of foie gras. Humans in contrast, along with many other species, have evolved to store energy in the form of adipose tissue as a dominant mechanism of metabolic thrift.
Adipose tissue is present across phyla, including yeast and insects [19], but is particularly well developed in vertebrates. Why did humans settle on adipose tissue? Adipose tissue is particularly energy dense—humans can store less than 1 day’s worth of calories in skeletal muscle in the form of glycogen but over 2 months worth of calories in fat within adipose tissue. Unlike rabbits, humans invest significant resources into a single offspring with each pregnancy. Furthermore, human development is characterized by a prolonged period of rapid postnatal growth and brain development that requires a constant supply of calories. Nutritional deficits during this period have widespread detrimental effects and exerted a strong reproductive disadvantage during evolution. Adipose tissue provides a constant source of calories during this critical period, reflected by the fact that human body fat percentage is highest during the rapid neonatal growth phase. Adipose tissue thus protects our fragile, singleton, energy-hungry offspring. Finally and additionally, humans can be considered to some extent a migratory species: Much of our evolution was shaped by the out-of-Africa emigration that led to pan-global colonization 50,000–75,000 years ago. Adipose tissue allowed us to weather variability in food supplies during famines and treacherous migrations to different environments. For these reasons, adipose tissue is a particularly good strategy for metabolic thrift in humans.
The Genetic Basis of Obesity
Obesity is a genetic phenomenon. In addition to leptin mutations, over 20 rare inborn genetic syndromes are associated with human obesity. In virtually all, obesity is one of a number of phenotypic abnormalities, emphasizing the interaction of weight regulation with all aspects of physiology. Causative genes have yet to be identified for Prader-Willi, Bardet-Biedl, Cohen, and Alstrom syndromes, while in other syndromes, specific genes have been implicated, all associated with feeding behavior, stressing the dominance of these systems in the pathogenesis of obesity. Some of these disorders are monogenic and include mutations in genes encoding POMC, PCSK1, and MC4R, among others [22]. But in contrast to monogenic obesity syndromes and similar to many chronic diseases, common human obesity is a complex polygenic phenotype, significantly complicating analysis.
Despite inherent challenges, epidemiologic and statistical methods based on Mendelian genetics have accurately quantified the cumulative genetic contribution to obesity and demonstrate a high level of hereditability and a strong genetic basis. Quantitative genetic analysis of twin cohorts is a powerful tool that is widely used to quantify genetic contribution to complex polygenic diseases and clinical phenotypes. Twin studies rely on comparison of thousands of monozygotic and dizygotic twin pairs to quantify shared genetic material based on Mendelian genetics, followed by statistical analysis that calculates of the contribution of genetic influences to phenotypic and clinical traits with a high degree of analytic power. Further detail can be gleaned from adoption studies of twin pairs separated at birth. When applied to obesity, multiple twin studies demonstrate that approximately 70 % of the tendency toward a particular body habitus, lean or obese, is determined by genetic contributors and 30 % by nongenetic, presumably environmental factors, values that have been reproduced consistently [16, 23]. Twin studies also reveal enlightening aspects of food-related behavior. For example, differences in suckling time, eating speed, food preferences, and crying frequency manifest at birth and correlate strongly with genetics, suggesting that such behaviors are hardwired before the onset of environmental influences.
While twin studies demonstrate a strong genetic contribution to obesity, as do similar tools such as genetic linkage and familial aggregation analyses, other methodologies are required to identify specific genetic mutations in thrifty gene candidates. Genome-wide association mapping and sequencing techniques rely on broad DNA sequencing of the genome in thousands of humans to identify single-nucleotide polymorphisms (SNPs) followed by correlation of SNP prevalence with phenotypic and clinical characteristics. These methods permit correlation of specific DNA polymorphisms with clinical disease-related traits. While powerful, a number of issues currently limit the utility of these techniques. First is the aforementioned polygenic nature of obesity. More than 200 specific genetic mutations have been associated with obesity in mice and more than 20 candidate genes have been identified in humans, likely only a minority of the total genes involved. These polymorphisms are found throughout the genome in and near genes associated with multiple physiologic processes, but the individual effect size of each locus is weak, contributing less than 1–3 % to the tendency toward obesity and confounding analysis of the functional role of any single gene on global phenotype. Tens of thousands of subjects are required to adequately power studies to identify SNPs with such low effect magnitudes. In addition, multiple loci interact in concert, further increasing complexity and confounding assignment of specific functions. Finally, most SNPs lie in noncoding regulatory regions of the genome, areas that are currently poorly understood and present significant challenges with respect to interrogation and functional analysis. Despite these limitations, however, genome-wide analysis has begun to identify thrifty gene candidates. Among these emerging candidate genes, MC4R represents a singularly important locus. More than 90 polymorphisms have been identified within the human MC4R gene locus, which together account for up to 6 % of cases of common human obesity, and are transmitted in monogenic Mendelian fashion with incomplete penetrance [24
 Duodenal Switch: Technique and Outcomes
The Rationale for Metabolic Surgery
Liability Reduction, Patient Safety, and Economic Success in Bariatric Surgery
Duodenal Switch: Technique and Outcomes
The Rationale for Metabolic Surgery
Liability Reduction, Patient Safety, and Economic Success in Bariatric Surgery
 Operative Outcomes of Bariatric Surgery in Patients with a Low Body Mass Index (30–35 kg/m2)
Operative Outcomes of Bariatric Surgery in Patients with a Low Body Mass Index (30–35 kg/m2)
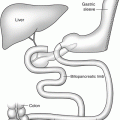 Gastrointestinal Obstruction After Bariatric Surgery
Gastrointestinal Obstruction After Bariatric Surgery
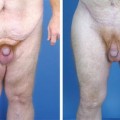 Body Contouring After Massive Weight Loss
Body Contouring After Massive Weight Loss




Related posts:
Duodenal Switch: Technique and Outcomes
The Rationale for Metabolic Surgery
Liability Reduction, Patient Safety, and Economic Success in Bariatric Surgery
Operative Outcomes of Bariatric Surgery in Patients with a Low Body Mass Index (30–35 kg/m2)
Gastrointestinal Obstruction After Bariatric Surgery
Body Contouring After Massive Weight Loss
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
