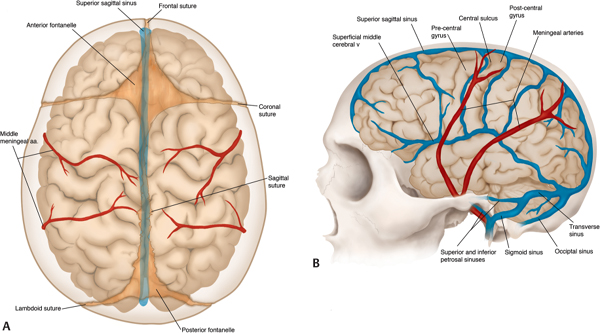
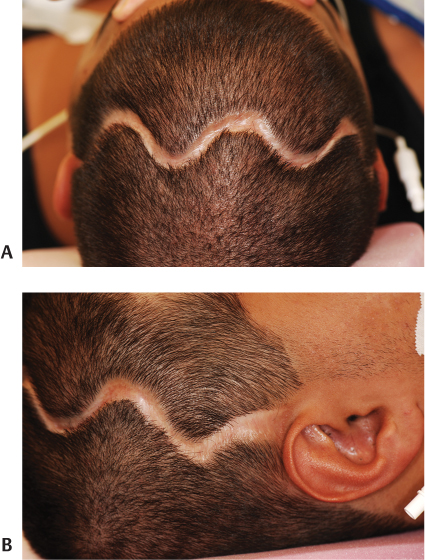
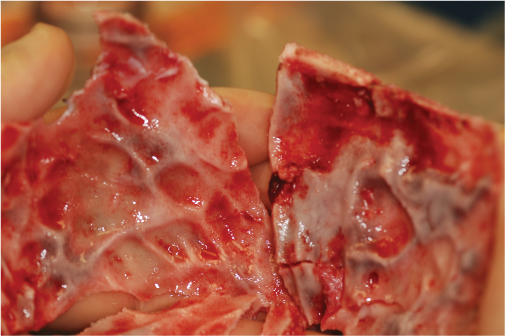
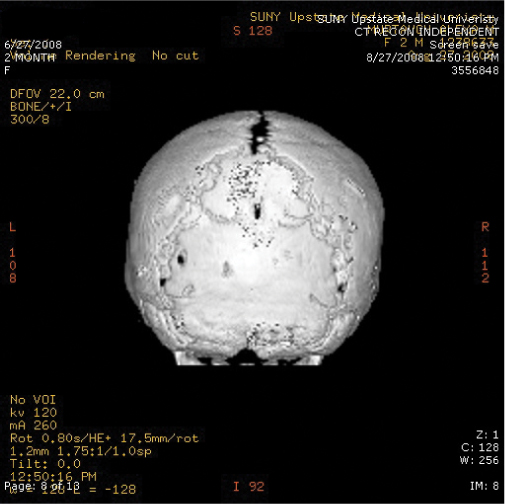
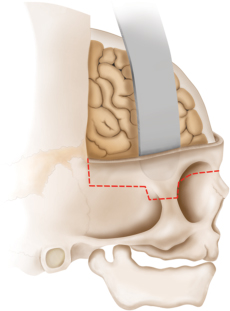
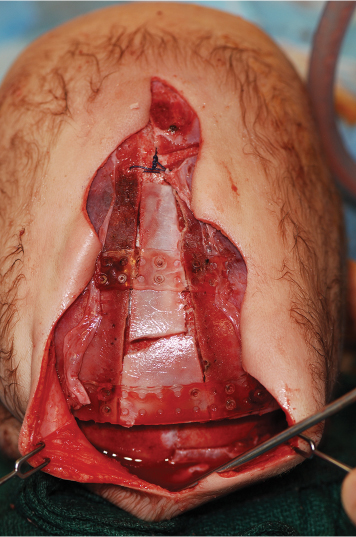
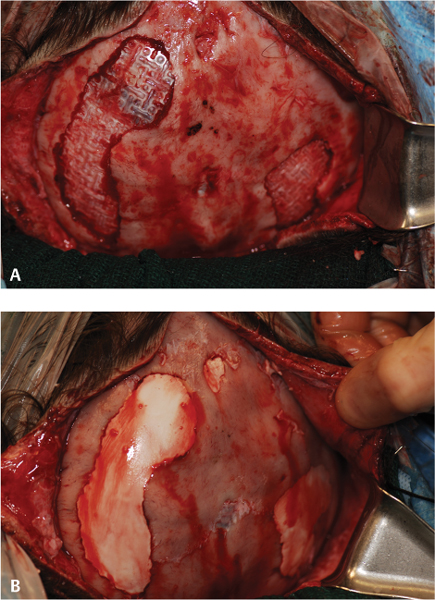
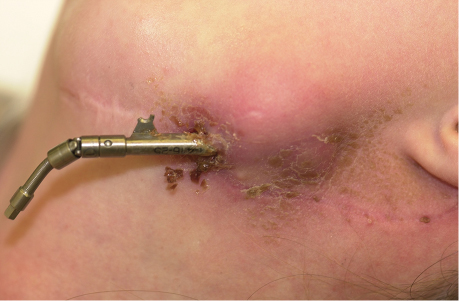
15 Surgical correction of anomalies associated with congenital malformation is both rewarding and challenging. Few areas in facial plastic surgery display such myriad presentations that require careful analysis and exact surgical planning. Many of the procedures are complex and dangerous, requiring highly specialized training and experience, so it is not surprising that surgery for congenital anomalies is associated with complications. This chapter discusses many of these complications and their management. As with all untoward results, prevention is the best practice, and attention to detail and early acknowledgment of complications are paramount to their management. Congenital craniofacial surgery refers to the correction of congenital or developmental defects, such as those resulting from premature fusion of cranial sutures or branchial arch abnormalities. The surgical treatment of craniosynostosis began in the latter half of the nineteenth century. Many of the techniques developed then are still used today in modified form. As might be imagined, any surgery involving the cutting and moving of craniofacial bones has significant risks. These include death, eye or brain damage, hemorrhage, infection, anesthesia problems, need for revision surgery, and others. Mitigating factors include patient age and size, severity of the deformity and the subsequent complexity of surgery, and comorbidities. Reported mortality rates range from zero to 3%, and morbidity rates range from 1 to 40%, with the lowest rates associated with the simplest surgeries (strip craniectomy or synostectomy) on the least significant deformities.1–4 The incidence of neurologic deficits after strip craniectomies was less than 1%. In the Shillito and Matson series, the frequency of reoperation secondary to resynostosis was 13.3% after correction of single sutures and 38% after repair of multiple sutures.1 Complication rates increase with the complexity of the surgical procedure. As more bone is exposed, there is an increased occurrence of blood loss, infection rate, risk of subgaleal collections, air embolism, dural tears associated with cerebrospinal fluid (CSF) leak, and direct injury to the brain or eye. The prevalence of obstructive sleep apnea syndrome in children with syndromic craniofacial synostosis has been reported to be as high as 40%.5,6 Airway management remains a constant and significant problem in craniofacial dysostosis. Children with craniofacial dysostoses often have airway problems related to distortion of the oropharynx by the cranial base. Midface retrusion and acute skull base angles bring the soft palate against the posterior pharyngeal wall, leading to breathing problems and sleep apnea. Small nasal airways contribute to the problem as well. In addition, laryngotracheal anomalies may be associated with craniofacial problems. Signs associated with increased respiratory difficulties include swallowing or gagging problems, increased reflexes and spasticity, and sleep apnea (often described by the parent as a worsening in the child’s snoring). Preoperative endoscopic evaluation is essential. In some cases, a tracheostomy must be established before surgery. Of children with syndromes involving bony deformities of the face and skull, 20% unambiguously required tracheostomy in a study of 251 patients by Sculerati et al.7 Tracheostomy also reduces the risk of postoperative problems, such as airway edema, aspiration pneumonia, or pneumocephalus after monobloc advancement. In some cases, Chiari malformations must be decompressed to reduce brainstem dysfunction. The main issues regarding timing of surgery are skull thickness and malleability, patient size and blood volume, brain growth potential, and intracranial constraint. In the first year of life, the human brain grows from roughly 335 g to 925 g, almost tripling in mass. Fused sutures may restrict the skull growth required to accommodate this brain growth. Before 1965, surgical correction was undertaken later rather than earlier (i.e., after an age of 1 year) because of anesthesia and blood volume issues. During the 1970s and into the 1980s, arguments were advanced for surgery from one to six months of age to leverage the growth potential and reduce elevated ICP. Scheduling surgery at four to nine months of age allows for relative safety due to patient size, takes advantage of rapid brain growth to support and enhance the correction, and provides for malleable skull bone that is thick enough to hold fixation materials without being too thick and stiffto allow easy remodeling. The osteogenic dural stimulus in the infant (age less than 12 months) allows for filling in of bony gaps after repositioning, so bony defects are rare at this age. If ICP is elevated, then an early operation is necessary to prevent brain damage. Excessive blood loss is still the most worrisome complication.8 This problem results from constant slow ooze throughout cases requiring ten hours or more as well as from acute bleeding from vascular injury. Dissection over dural sinuses, particularly more posteriorly, risks massive hemorrhage from dural sinus injury, especially in the region of the torcular herophili (Fig. 15.1A,B). Knowledge of the locations of these vessels relative to the skull surface anatomy is critical. Sagittal sinus ligation to control hemorrhage can result in serious neurologic injury or death if it is done posterior to the anterior one-third of the sinus. Most deaths and serious neurologic sequelae associated with craniosynostosis surgery reflect an inability to maintain adequate blood volume perioperatively. Hypovolemia also increases the risk of air embolism. Blood should be available from the beginning of the procedure, and the patient’s hematocrit should be monitored routinely throughout the procedure. To reduce blood loss, some centers maintain systemic hypotension (a mean blood pressure of 50 mm Hg) during the operation. Epinephrine is injected into the skin before the incision is made. Hemostasis at the incision site is achieved with Raney clips or with cauterizing instruments, such as the Shaw scalpel or the Colorado needle. Topical agents like thrombin and collagen reduce blood loss as well. Systemic aminocaproic acid also reduces blood loss in select cases. Many centers now routinely arrange for donation of pedigree blood from the family before surgery. Pneumocephalus is expected after such dissections, but tension pneumocephalus is a life-threatening emergency and must be managed with decompression. Subdural and epidural fluid collections are likewise expected, but hematomas exerting a mass effect should also be decompressed. Brain contusion or hemorrhage may occur as a result of excessive retraction, and it will be visible on postoperative computed tomography (CT) imaging. The contusions and hemorrhage generally are self-limiting and resolve, but neurologic sequelae may result. The best way to manage complications is to avoid them with gentle retraction on the dura. The temporal branch of the facial nerve can be damaged sharply during scalp elevation or injured by cautery or traction. Again, avoidance is the best management. Widening of the scalp scar (Fig. 15.2A,B) can result from excessive tension on the closure or damage to the adjacent hair follicles. Careful tissue handling and wound closure generally avoid these problems. In particularly long cases, strangulating scalp clips or sutures should be used with caution. Fig. 15.1 Overlay of skull surface anatomy with dural and brain anatomy: (A) vertex view, (B) lateral view. (Illustration by Robert Brown.) Hydrocephalus and increased intracranial pressure, although not synonymous, are closely related. They may exist together or separately. Findings include neurologic dysfunction, irritability, and visual disturbance. Complete neurologic, ophthalmologic, and CT radiographic examinations are necessary if these conditions are suspected. Hydrocephalus is uncommon in children with isolated single-suture synostosis but becomes more frequent in multisuture synostosis and in cases involving the skull base (i.e., craniofacial dysostosis such as Crouzon and Apert syndromes). Various series have reported hydrocephalus in less than 50% of cases.1,9,10 Renier et al. performed intraoperative ICP monitoring on an extensive series of patients and found that the risk of increased ICP in children with Kleeblattschädel or multiple suture closures can be as high as 95%. Many patients present with surgical emergencies because of increased ICP.11 The decision of when to place a ventriculoperitoneal (VP) shunt for cerebrospinal fluid (CSF) diversion can be complex. If the child has symptoms and signs of elevated ICP caused by hydrocephalus, then a VP shunt is placed first. If the child is asymptomatic, it is best to wait until after the craniofacial reconstruction to reduce the risk of infection from a VP shunt in the field of surgery. Elevated ICP often causes severe erosion of the skull bone (Fig. 15.3). These erosions are seen as scalloping of the inner table of the calvarium on plain films or full-thickness defects on CT scanning (Fig. 15.4). The dura can erode through the bone, making it technically difficult to elevate the pericranium from the dura. Fig. 15.2 Widened scalp scar: (A) top view, (B) lateral view. Children with craniofacial dysostoses have skull base anomalies, the most common of which is the Chiari malformation, and associated distortions of the foramen magnum.12 Consequently, a thorough preoperative evaluation with magnetic resonance imaging (MRI) or CT is essential to evaluate the brain stem and cervicomedullary junction. Appropriate preoperative evaluation prevents postoperative quadriplegia from spinal cord or brain stem compression caused by inappropriate surgical positioning during craniofacial reconstruction (i.e., cervical hyperextension). Children with metopic synostosis commonly have a maldeveloped, foreshortened anterior fossa. A frontal lobe pseudoherniation often occurs, moving the lobes downward to a position between the orbits. In coronal synostosis, the temporal lobe tends to be located very anterior in the lateral orbital wall. These abnormalities increase the risk of dura and brain injury requiring careful brain retraction (Fig. 15.5). Preoperative computed tomography (CT) or magnetic resonance imaging (MRI) helps to discover such problems, allowing modification of the surgical approach as necessary to avoid these injuries. Fig. 15.3 Intraoperative view showing inner table calvarial scalloping. Most operative series for craniosynostosis have been associated with an infection rate of 3 to 5%.13,14 The duration of the operation is directly related to the relative risk of infection, with shorter operations (e.g., strip craniectomies) carrying a 3% risk of infection. Longer operations may lead to increases in rates of infection of up to 8%. A key factor in infection is the age of the child and the developmental status of the sinuses. If the frontal sinus communicates with the epidural space, the rate of infection increases greatly, especially when advancement procedures leave a dead space. In most cases, such operations are performed before pneumatization of the sinuses occurs at 3 years of age. Monobloc procedures have the highest risk, because they cause connection between the nasal and cranial cavities. Pericranial flaps and tissue sealants are used to reduce the risks. Osseo-distraction rather than acute advancement is also associated with lower risks. Surgical prophylaxis should target the typical offenders, mainly Streptococcus and Staphylococcus. Copious irrigation to remove bone debris and blood aids in the prevention of infections. Infection poses the risk of losing the osteotomized bone. Fig. 15.4 3D CT showing calvarial defects. Fig. 15.5 Brain protection during bandeau osteotomies. (Illustration by Robert Brown.) Dural tears with subsequent cerebrospinal fluid (CSF) leak occur most commonly at the bony sutures where the dura is tightly attached in children under the age of one year. Also, the skull erosion associated with elevated ICP leads to direct pericranium to dura contact that increases the risk of tears. These tears increase the incidence of postoperative CSF leakage and the risk of meningitis. Therefore, meticulous attention must be paid to closing such tears when leaks are present. If the dura is torn, a watertight repair must be performed. At the completion of the operation, it is advisable to check with a Valsalva maneuver for persistent CSF leakage. After surgery, large subgaleal collections can occasionally result from CSF pooling. Such collections rarely require treatment, because they usually resolve spontaneously. If a leak persists, a lumbar spinal drain can be placed for five to seven days; the pressure reduction of CSF is usually effective in correcting a CSF leak. Thereafter, the surgical site must be reexplored and the CSF leak site repaired. In the past, metallic wires and miniplates have led to extrusion through the skin or worse, migration into the brain. Most craniofacial surgeons now use resorbable miniplates or suture material (Fig. 15.6). Various commercial products are currently in use, primarily composed of polyglycolic acid (PGA), poly-L-lactic acid (PLLA), and PGA-PLLA copolymers. Miniplates provide additional structural support, particularly when advancement and transposition techniques are performed. The reported complications of resorbable plating systems include pronounced fibrous encapsulation and visibility,15 palpability,16,17 sterile sinus formation,18 and bone osteolysis.19 In 1,883 pediatric patients repaired with resorbable plates, Eppley et al. found that significant infectious complications occurred in 0.2%, device instability (primarily resulting from postoperative trauma) was noted in 0.3%, and self-limiting local foreign body reactions were seen in 0.7% of the treated patients.20 The overall reoperation rate attributable to identifiable device-related problems was 0.3%. Improved stability was gained by using the longest plate geometries and configurations possible and by bone grafting any significant gaps across plated areas that were structurally important. Mackool et al.21 reported four cases in which a PLLA system—MacroPore (Cytori Therapeutics, San Diego, CA), a copolymer of 70:30 Poly (L-lactide-co-d,L-lactide)—resulted in long-term palpability and visibility, with underlying bone resorption and a significant foreign body giant cell reaction. Fig. 15.6 Resorbable plates used for cranial fixation. Calcium phosphate cements (CPC) are often used as bone graft substitutes for skull defects. Verret et al.22 reported their experience with CPC in craniofacial reconstruction in 102 patients (Fig. 15.7A,B). Twenty-four of the reconstructions were related to trauma and 78 arose secondary to neoplasm. Eleven implants required removal; six were related to placement within the frontal sinus or in contact with the frontal sinus. The remaining five were from other parts of the craniofacial skeleton. Dujovny et al.23 and Mathur et al.24 discussed the incomplete setting of CPC, partial resorption of paste, and subsequent volume loss in the presence of a water-based medium such as blood. Implant migration and micro-fragmentation related to dural pulsations have also been reported from postoperative CT scans. Avoidance of sinus contact and shielding from dural pulsations reduce these problems.25 Fig. 15.7 (A) Dural pulsation–shielding resorbable plates in preparation for cranioplasty. (B) Calcium phosphate cement cranioplasty. Persistent calvarial contour irregularities and defects are a potential problem in all craniofacial reconstructions. Temporal hollowing is a common problem in frontal advancement cases that can be avoided by advancing the body of the muscle forward and reattaching it to the lateral orbital rim. All cut bone edges have the potential to be palpable and even visible, particularly with the thinness of the young scalp. When extensive advancements are performed, sufficient bone for reconstruction of the calvarium may be lacking. In children under 1 year old, most bone defects less than 2.5 cm in size close with time. However, after age 1 the ability of the patient to close bone defects decreases significantly. If a persistent bone defect is encountered, it is reasonable to wait until the child is 3 years old before attempting to repair it. By the time the child is 3, the diploe has formed, and split-thickness bone grafts can be harvested and used to fill in the defects.26 Defects have also been repaired with distant bone grafts and a myriad of bone graft substitutes, such as the previously mentioned CPC (Fig. 15.7A,B). Visible contour irregularities may require burring to improve appearance. The frequency of postoperative head trauma tends to be age-related, being highest in children who have reached the crawling and toddler stages. Protective headgear is recommended at this stage if significant defects persist. In addition, helmets may be used postoperatively to re-shape the head further, or they may be used in lieu of surgery for nonsynostotic plagiocephaly cases. Potential health risks associated with the use of helmets include excessive skin breakdown and infection, inappropriate mechanical restriction leading to altered or impaired head growth and development, asphyxiation, and head and neck trauma as well as ocular trauma.27 The parents should be instructed on how to position their child in cases of plagiocephaly or advancement repair so that the child’s sleep position does not add to the forces for skeletal relapse. Techniques for positioning the child with rolled towels should be demonstrated. During the day, parents should be encouraged to keep the child in a walker-type stroller fitted with a circumferential rail that prevents the child from receiving an impact with hard objects. About 6–12 weeks of healing are required to achieve good structural support for the repositioned bone and brain protection. During this critical period, the child must be watched closely. Daycare may be a risk in this period. After 3–6 months, the bone is well fibrosed, and only a hard impact can disrupt the bone units. The visual system is often significantly involved in craniosynostosis.28 Impaired vision may be an inherent feature of the pathologic process or occur as a secondary complication. In particular, optic disc edema, optic atrophy, and progressive optic nerve dysfunction may accompany increased intracranial pressure without evidence of hydrocephalus and with open fontanels. Uncorrected refractive error (particularly anisometropia), strabismus, ptosis, and corneal exposure problems are an invitation to the development of amblyopia.29 If not reversed, this condition can lead to permanent visual disability. Proptosis and corneal exposure problems are a third potentially treatable cause of functional blindness. Prompt involvement of an ophthalmologist in the care of children with craniosynostosis is essential. Proptosis resulting in corneal exposure should be urgently managed with tarsorrhaphy. Tarsorrhaphy is also useful intraoperatively to protect the corneas. Papilledema and disc pallor both signal threatened vision and require urgent action. In a review of 141 patients with craniosynostotic syndromes of Apert, Crouzon, Pfeiffer and Saethre–Chotzen, Khan et al.30 demonstrated a poor visual outcome with an acuity of 6/12 Snellen or worse in the better eye in 39.8% of cases, while 64.6% of cases had this level of vision or worse in either eye. Tay et al.31 assessed the prevalence and causes of visual impairment in 71 patients with craniosynostotic syndromes of Apert, Crouzon, Pfeiffer, Saethre–Chotzen and CFND and confirmed Hertle’s findings.28 Causes of visual impairment were amblyopia (16.7%), ametropia (25%), optic atrophy (16.7%), exposure keratopathy (4.2%), and infantile nystagmus syndrome (4.2%). Overall, 6.3% of the patients had amblyopia that was attributable to a combination of factors: exotropia (two cases), astigmatism (two cases), and anisometropic hypermetropia (one case). 6.3% patients were found to have optic atrophy. Tay et al. found complete resolution of papilledema in all six patients with it following decompressive surgery. Orbital repositioning may result in extraocular muscle dysfunction. In most instances, it resolves spontaneously within six months. Persistent problems may require strabismus surgery for correction. Pulsatile ophthalmopathy results from transmitted dural pulsations after fronto-orbital advancements without orbital roof reconstruction. In patients older than 9–12 months, the orbital roofs must have bone grafts. Diamond et al.32 reported the ocular and adnexal complications of unilateral orbital advancement for plagiocephaly. Ptosis was observed postoperatively in 29% of patients, and strabismus and amblyopia in 18% of the patients. For eye protection during craniofacial corrective surgery, tarsorrhaphy can be performed before surgical draping to protect the cornea. Otherwise, the eyes must be taped securely to avoid corneal injury. During surgery, retraction or pressure on the orbits can cause changes in the patient’s pulse rate and blood pressure. Accordingly, during orbital dissection the anesthesiologist should be alerted to monitor the vital signs with particular care, so that the surgical team can immediately be informed of oculocardiac stimulation. Fortunately, the reported number of complications involving the orbits that result in loss of vision is less than 1%. Distraction osteogenesis is becoming the treatment of choice for the surgical correction of many skeletal deficiencies because of its promise of new bone formation without donor sites, slow movements allowing for soft-tissue accommodation, and suitability for growing patients. Although initially there had been great emphasis on a several day postoperative latency period and a limited distraction rate of 1 mm per day, distraction is now often started on the first postoperative day after distracter insertion and at an increased rate of 2 mm per day without either fibrous union or premature consolidation. Mandibular distraction is less invasive than traditional methods of mandibular reconstruction with bone grafts and has a significantly reduced morbidity rate. It may also be done in infancy to avoid or shorten time with a tracheotomy in cases of micrognathia. Anterior cranial and maxillary distraction share the above benefits plus the lack of a large anterior cranial fossa defect with monobloc distraction compared with traditional advancement. In addition, larger movements are easier with distraction than with traditional techniques. Pitfalls include improper device and osteotomy placement, device failure, poor compliance with device activation, pin track scars, and difficulties with device removal after alteration of the anatomy. Complications can range from infection of the pin track to total failure of the procedure. Complications of osseodistraction can be divided into placement, osteotomy, and removal complications and distraction process complications. Surgical complications include those found with traditional osteotomies: infection, hematoma, brain or ocular injury, facial or mandibular nerve injury, damage to tooth buds, scars from visible incisions, and incorrect osteotomy or device placement (Fig. 15.8). Tooth bud injury can lead to eruption failure or odontogenic cyst formation.10,33–35 Careful planning and execution are the best ways to avoid these problems. Distraction process complications occur during activation of the device or in the latency phase. Device failure includes breakage of the distracter, screw or pin loosening, and device binding. Condylar displacement has also been reported.35,36 Distraction can cause pin migration through the soft tissues and pin tracks in the skin (Fig. 15.9). Compliance problems with BID or TID activation with a small tool that must be turned the correct way can lead to premature consolidation of the regenerate formation or lack of regenerate formation after what appears to be successful distraction. Lack of compliance with activity level restrictions can lead to device dislodgement. Other causes of failure to form regenerate are patient factors such as local vascular compromise and bone metabolism disorders. Fig. 15.8 Infected extruding fractured distracter. Fig. 15.9 External distracter pins extruded through the chin. The location of the cleft lip and palate anomaly leads to possible impairments of breathing, eating, hearing, speech, dental relationships, craniofacial growth, and facial appearance, along with possible psychosocial impairment. There is a lack of data on outcomes to direct cleft care. Several factors contribute to this problem. The time lapse between treatment intervention and final growth result is significant. There are no standardized national treatment protocols. Most surgeons follow a protocol that places primary cleft lip repair and tip rhinoplasty between three and six months of age. Millard and Tennison techniques tend to be the ones most commonly used. That is followed by palatoplasty at six to twelve months, usually with some variation of a Langenbeck, Veaux-Wardill-Kilmer, or Furlow technique. Secondary speech surgery, if needed, is performed between the ages of three and seven years. At the age of seven to eleven years, alveolar bone grafting is frequently necessary. Some patients require orthognathic surgery, and in selected cases distraction osteogenesis is performed. Between the ages of 12 and 18 years, secondary rhinoplasties are indicated in most patients. Initial problems with bleeding, infection, or dehiscence are rare. Occasionally, significant respiratory distress may be noted from nasal obstruction, particularly in the repair of bilateral clefts. The problem responds acutely to insertion of nasal stents. Twenty-three hour postoperative observation is recommended for patients younger than four months of age. Avoidance of lip trauma in the first seven to ten days postoperatively with gentle feeding techniques and handling helps ensure good healing with minimal scars. Cleft lip repair generally involves recruiting tissue flanking the cleft to fill the cleft-related defects. Complications generally result in the need for a revision. Revisions can be divided into small touch-up procedures for minor deficiencies and total lip repair revisions for major problems. Careful analysis of the residual deformity leads to the choice of the appropriate approach. Residual deficiencies can be thought of as tissue deficiency problems, tissue distortion problems, or some combination of both. As is typical, the best management is proper initial technique to avoid residual problems. However, even the best hands still have revisions. Notching of the lip margin (whistle deformity) is a frequently encountered secondary deformity in cleft lip patients (Fig. 15.10). It represents some degree of vertical deficiency in the lip skin, vermilion, muscle, or mucosa. Central mucosal deficiency may be minimized at the time of the initial unilateral lip repair by back-cutting the mucosa in the gingivobuccal sulcus along the medial lip element, where the deficiency is most common, and advancing from lateral to medial and inferiorly. The resulting mucosal defect in the sulcus quickly heals secondarily. There is a natural vermilion deficiency in clefts (worse in bilateral clefts) as the wet line fuses with the white roll at the cleft margin. This is best managed during primary repair with vermilion flaps from the lateral segments being brought into the philtral tubercle. Vermilion deficiency can occur under the lip scar(s) or centrally as a philtral tubercle deficiency. Minor deficiencies may be addressed with dermal fat grafts or V–Y mucosal advancement, but mucosa outside of the wet line can lead to chapping and color mismatch. More significant deficiency is due to vertical skin or muscle deficiency, and depending on degree, it requires a scar revision or total lip revision. Severe bilateral cleft lip vermilion deficiencies can be corrected with a lower lip vermilion flap. Fig. 15.10 Whistle deformity. Cleft-side high peaking of Cupid’s bow or vertical skin deficiency without lip notching is commonly seen early after the Millard rotation advancement repair due to scar contracture.37 This usually resolves with gentle massage and scar maturation. If the problem persists beyond one year postoperatively, then it should be surgically corrected. In cases in which the lip is short by up to 2 mm, a diamond-shaped excision of the scar and reclosure can add adequate length. When the whole lip is short due to scar contracture, total excision of the scar eliminates contracture and allows for normal alignment. If additional lengthening is needed, Z-plasties, rotation-advancement, or triangular flap techniques can be used just in the skin. Greater vertical deficiency or a shortage of subcutaneous tissue and muscle necessitates complete revision. If the orbicularis muscle is dehiscent, it should be released from its abnormal insertions and approximated. The most severe defects may require an Abbe flap. Less commonly, excess in the vertical dimension results in a long upper lip. Reduction is achieved with an excision of the skin and muscle just below the nasal ala and sill.
Surgical Correction of Congenital Anomalies and Associated Complications
 Complications of the Surgical Treatment of Craniosynostosis
Complications of the Surgical Treatment of Craniosynostosis
Preventing Airway Complications
Complications Related to Timing of Surgery
Blood Loss, Air Embolism, and Pneumocephalus
Hydrocephalus and Intracranial Pressure
Complications Due to Altered Anatomy
Infection
Cerebrospinal Fluid Leak
Problems with Foreign Materials
Skull Irregularities and Defects
Postoperative Head Injury or Deformity
Ocular Complications
 Distraction Osteogenesis
Distraction Osteogenesis
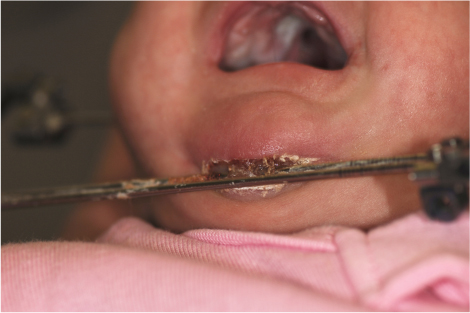
 Cleft Lip and Palate
Cleft Lip and Palate
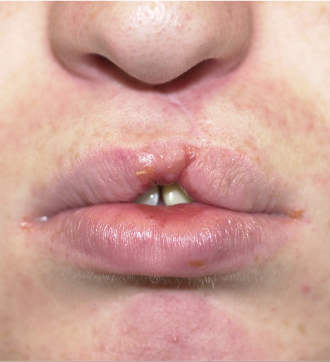
Cleft Lip Repair
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


