Subcorneal Pustular Dermatosis (Sneddon–Wilkinson Disease): Introduction
|
Subcorneal pustular dermatosis (SPD) is a rare, chronic, recurrent, pustular eruption characterized histopathologically by subcorneal pustules that contain abundant neutrophils. The condition was originally described in 1956 by Sneddon and Wilkinson,1 who separated SPD from other previously unclassified pustular eruptions. Until 1966, when the first comprehensive review appeared, more than 130 cases had been reported, but not all fulfilled the clinical and histopathologic criteria required for this diagnosis.2 A considerable number of additional cases have since appeared in the literature, and a subtype with intraepidermal deposits of immunoglobulin (Ig) A directed against desmocollin 1 has been recognized.3 Today, these cases are usually classified as SPD-type IgA pemphigus and it is a matter of debate whether the finding of epidermal IgA deposits define a subset of SPD or a new pemphigus variant that is otherwise indistinguishable from “classic” SPD.
Epidemiology
There is no racial predilection. Most of the reported cases have been in whites, but the disease has also been observed in Africans, Japanese, and Chinese. The condition is more common in women and in persons older than 40 years of age, but SPD may occur at any age.2 A pustular eruption that is clinically and histologically similar to the human disease, which also responds to dapsone treatment, has been observed in dogs.4
Etiology and Pathogenesis
The cause of SPD is unknown. Cultures of the pustules consistently do not reveal bacterial growth. The role of trigger mechanisms such as preceding or concomitant infections, though repeatedly discussed, has remained speculative. Immunologic mechanisms have been implicated in the pathogenesis and in a subset of patients, whose disease clinically resembled SPD, intraepidermal IgA deposits have been detected. Some of these patients also had circulating IgA antibodies against the same sites within the epidermis. Desmocollin 1 and in a single case also desmocollins 2 and 3 have been described as autoantigens in these cases and the disease has been classified as a rare pemphigus variant (SPD-type IgA pemphigus).3,5–7 The pathogenetic role of these antibodies is still to be demonstrated.8
The occasional association of SPD with certain other diseases may represent more than a mere coincidence. Increased serum IgA has been detected in a number of patients, and the disease has been reported to occur in cases of IgA-paraproteinemia and IgA multiple myeloma.9–12 In addition, SPD is associated with pyoderma gangrenosum,13,14 ulcerative colitis,15 and Crohn disease.16 On the other hand, pyoderma gangrenosum is not uncommon in patients with inflammatory bowel disease, paraproteinemia, and myeloma (see Chapter 33). Whether or not the coexistence of these conditions reflects common pathogenetic mechanisms remains to be clarified, but an additional common denominator linking these disorders is their response to sulfone and sulfonamide therapy.
Clinical Findings
The primary lesions are small, discrete, flaccid pustules, or vesicles that rapidly turn pustular and usually arise in crops within a few hours on clinically normal or slightly erythematous skin (Fig. 35-1). In dependent regions, pus characteristically accumulates in the lower half of the pustule (see Fig. 35-1B); as the pustules usually have the tendency to coalesce, they often, but not always, form annular, circinate, or bizarre serpiginous patterns. After a few days, the pustules rupture and dry up to form thin, superficial scales and crusts, closely resembling impetigo. Peripheral spreading and central healing leave polycyclic, erythematous areas in which new pustules arise as others disappear (see Fig. 35-1A). There is no atrophy or scarring, but an occasional brownish hyperpigmentation may mark previously affected sites. Variable intervals of quiescence, lasting from a few days to several weeks, may be followed by the sudden development of new lesions. The eruptions tend to occur symmetrically, affecting mainly the axillae, groin, abdomen, submammary areas, and the flexor aspects of the limbs. In rare cases, the face,28 palms, and soles29 may be involved. Scalp and mucous membranes invariably remain free of lesions. Episodic itching and burning represent subjective symptoms in a small number of patients, but there are no systemic symptoms or abnormalities in routine laboratory parameters.
Figure 35-1
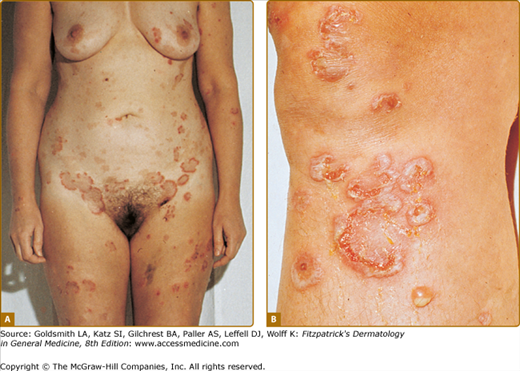
Subcorneal pustular dermatosis. A. Typical distribution. Note accentuated involvement of groin and abdomen. Hyperpigmented macules mark previously affected areas. B. Close-up showing coalescence of pustules, which form annular and circinate patterns. Lesions of different developmental stages are seen side by side. At the lower right, newly formed pustule with characteristic hypopyon formation.
Histopathology
The hallmark of the disease is a strictly subcorneal pustule filled with polymorphonuclear leukocytes,1 with only an occasional eosinophils.2 Acantholysis is not involved in pustule formation, but a few acantholytic cells may be found in older lesions (secondary acantholysis). Surprisingly, the epidermal layers underlying the pustule exhibit little pathology, and, apart from a variable number of migrating leukocytes, there is little evidence of spongiosis or cytolytic damage to the epidermal cells. The dermis contains a perivascular infiltrate composed of neutrophils and rarely mononuclear cells and eosinophils (Fig. 35-2).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


