Form of EB
Lin et al. [6]
Tong et al. [7]
Simplex EB
3 %
12 %
Junctional EB
39 %
40 %
Recessive dystrophic EB
51 %
51 %
Dominant dystrophic EB
17.6 %
4 %
Common ocular symptoms in EB patients are redness [9], tearing [9], photophobia [3, 10] and ocular pain [3, 10, 11]. Common ocular signs described include blepharoconjunctivitis [10, 12, 13], conjunctival injection [14], conjunctival oedema [9], conjunctival blister [9], corneal blister and corneal erosion [6, 7, 9, 10, 12, 15–17] and corneal scar [6, 7, 9, 12, 15]. Other ocular findings include blepharitis [12], ectropion [6, 7], eyelid blistering [12], lacrimal duct obstruction [6], symblepharon [13], exposure keratitis [7], corneal ulceration [3], corneal opacity [9, 13], limbal broadening [9, 13] and pannus formation [6, 7, 9].
73.2.1 Eyelid and Lacrimal Apparatus
Blepharitis, ectropion formation, blistering of the eyelid skin and lacrimal duct obstruction occur uncommonly in EB patients. Blepharitis is characterised by crusting at the eyelid margins and ulceration at the mucocutaneous junction [12]. This is associated with foreign body sensation, mucoid discharge and red eye. Chronic blepharitis is associated with the loss of eyelashes [12]. In the NEBR group, blepharitis was most commonly seen in JEB (6 and 7 % in the non-Herlitz and Herlitz subtypes), generalised severe RDEB (18 %) and RDEB-inversa (18 %) [8]. Ectropion is likely to be caused by blisters and scars around the eyelids [6]. It occurred most commonly in Herlitz JEB (14 %) and generalised severe RDEB (7 %) [8]. Cicatricial ectropion can subsequently lead to exposure keratitis and cornea perforation [18].
Lacrimal duct obstruction was reported in all subtypes of EB, with the greatest frequency of occurrence found in RDEB-I (12 %) [8].
73.2.2 Conjunctiva
Symblepharon is an uncommon complication in EB. In the NEBR group, symblepharon was observed only in JEB and RDEB patients, with the highest occurrence seen in RDEB-inversa (12 %) and generalised severe RDEB (10 %) (Fig. 73.1) [8]. Symblepharon in EB patients was reported by Lin et al. to gradually thicken overtime and to eventually immobilise the globe and eyelid [6]. Other conjunctival complications include conjunctival injection [14], conjunctival oedema [9] and blepharoconjunctivitis [10, 12, 13].
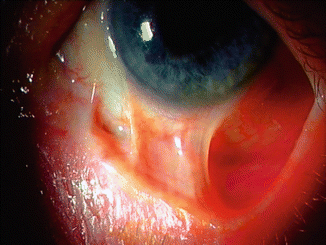
Fig. 73.1
Symblepharon in a young patient with RDEB
73.2.3 Cornea
Cornea erosion with or without scarring is a common and serious finding in EB (Fig. 73.2). The cornea of EB patients is vulnerable to friction-induced injuries. Cornea erosion occurs most commonly in the junctional and recessive dystrophic types of EB [8]. Fine et al. measured the cumulative risk for corneal blistering or erosion in the NEBR cohort and found that 71 % of all EB patients were predicted to have experienced this cornea complication by the age of 20. Cumulative risks were much higher in recessive dystrophic-GS and Herlitz junctional EB [8]. Cornea erosion typically presents with an acute onset of pain and photophobia in one or both eyes, associated with tearing and red eye [12]. These erosions heal spontaneously in 2–3 days. Other cornea lesions seen in EB include exposure keratitis [7], corneal ulceration [3], corneal opacity [9, 13], limbal broadening [9, 13] and pannus formation [6, 7, 9].
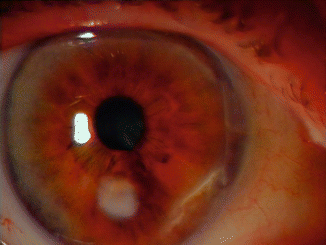
Fig. 73.2
Corneal scar in a patient with JEB
73.2.4 Visual Function
73.3 Hair
The anagen follicle expresses all basal membrane zone (BMZ) antigens found in the normal interfollicular epidermis. In normal conditions, the BMZ components show normal expression at the upper portions of the hair follicle and a gradual decrease in staining in the transient regions of the hair follicles, i.e. at the level of the hair bulb [19, 20]. The complete structure of the hemidesmosomes provides adhesion of the upper follicle to the surrounding connective tissues, while the incomplete hemidesmosome structure allows the movement of the transient region.
Blistering of the scalp involving the lamina lucida and below, as in junctional and dystrophic forms of epidermolysis bullosa (EB), usually leads to cicatricial alopecia.
73.3.1 EB Simplex
Most cases of autosomal dominant EBS are caused by dominant negative mutations in either KRT5 or KRT14. These keratins are different from those expressed in the hair shaft, and because blistering also occurs in the basal layer of the skin, there is generally no scarring unless there is a secondary infection. Even patients with recessive EBS caused by keratin 14 null mutations do not get a specific alopecia. If these patients however become anaemic because of blistering or severely ill from sepsis, they may develop a telogen effluvium, which is reversible if the cause is treated. Two patients with EBS Dowling-Meara type and loose anagen hair syndrome have recently been described. Both patients had the same keratin 14 N123S mutation and presented with sparse blond hair that did not grow long. Complete absence of hair, eyebrows, eyelashes and vellus hairs is a distinctive feature of lethal acantholytic EB, caused by truncation mutations in the desmoplakin gene that remove the C terminus of the protein [21]. In patients with suprabasal EB caused by plakophilin-1 deficiency (previously known as ectodermal dysplasia-skin fragility syndrome), the hair is sparse, short, dry and curly; eyebrows and eyelashes may be absent. Some of these patients have woolly hair, while others have hypotrichosis [22–24]. Lethal congenital epidermolysis bullosa (LCEB) is caused by a homozygous nonsense JUP mutation, leading to complete loss of plakoglobin. It is characterised by generalised epidermolysis, total alopecia and onycholysis [25]. EBS with muscular dystrophy (EBS-MD) is not usually responsible for alopecia.
73.3.2 Junctional EB
Scarring alopecia may be observed in Herlitz JEB [26], even if most patients do not survive beyond 1 year. Non-Herlitz JEB is genetically heterogeneous. When caused by defects in Lm332, the alopecia may be very severe because of exuberant granulation tissue formation and scarring. More subtle alopecia and loss of eyebrows can also be seen. In non-Herlitz JEB caused by Type XVII collagen deficiency, patchy or even diffuse scarring alopecia (with a typical male pattern baldness even in females) has been reported as a hallmark. Partial absence of eyelashes, eyebrows and pubic and axillary hair has also been reported [27–29]. Because of the thinning of the skin and loss of hair follicles, this type of alopecia has been referred to as atrophic [30]. Patients with reduced staining with 1A8C and 1D1 caused by COL17A1 mutations have normal primary hair and sparse secondary hair. Patients with lack of staining of the Type XVII collagen antibodies 1A8C and 1D1 caused by COL17A1 mutations have sparse primary and absent secondary hair and even nonscarring universal alopecia with almost complete absence of vellus hair, eyelashes, eyebrows and secondary hairs [31]. The COL17A1 knockout mice also develop sparse nonpigmented hair and subsequently alopecia [32]. JEB with pyloric atresia (PA) or due to reduced integrin beta 4 may result in scarring alopecia if the affected area of the scalp is eroded [33]. The only case of JEB, reported with alopecia and absence of axillary and pubic hair, was a long-term survivor without PA, who had homozygous missense mutations in ITGB4 [34].
73.3.3 Dystrophic EB
In patients with severe generalised recessive DEB, gradual onset of scarring alopecia in areas of trauma is very common [35]. Generally, the authors have observed that the hair loss in RDEB is worse in female patients who choose to plait their hair or tie it up in a ponytail, compared with males with short hair or females who do not regularly put their hair under tension (Fig. 73.3). Folliculitis-like lesions have also been reported in one patient with DEB pruriginosa. Scalp lesions consisted of multiple, nonpainful, red follicular papules and pustules [36].
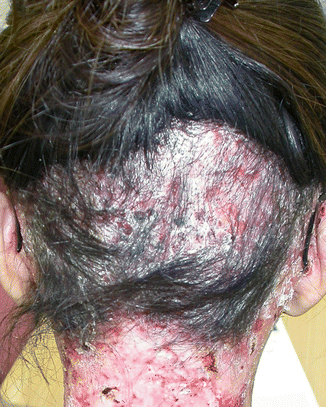
Fig. 73.3
Traction alopecia in an adult with RDEB intermediate
73.3.4 Kindler Syndrome
Kindler syndrome has not been associated with alopecia. This is perhaps because the defective protein, kindlin or FHH1 (fermitin family homolog 1), is in focal contacts rather than hemidesmosomes and are not expressed in hair follicles.
73.4 Nail
The antigenic expression of basement membrane zone (BMZ) components in the normal matrix, nail bed, proximal nail fold and hyponychium is similar to that of normal skin [37, 38]. This explains why nail changes are common albeit not specific to most epidermolysis bullosa (EB) subtypes (Table 73.1). Nail abnormalities may be the first or the only symptom of EB. They may precede the development of skin blistering as in the late-onset JEB and pretibial DEB or be an isolated finding as reported in some families with dominant dystrophic EB (DDEB) [39–43]. EB however is not necessarily associated with nail signs [1]. The presence of nail changes has recently been included among the criteria for scoring EB severity, as early nail dystrophy and loss are correlated with disease severity and progression, particularly in junctional epidermolysis bullosa (JEB) and recessive dystrophic epidermolysis bullosa (RDEB) [44].
73.4.1 EB Simplex
In severe EBS subtypes, blistering causes onycholysis and onychomadesis with regrowth of thickened dystrophic nails, with or without onychogryphosis (Fig. 73.4). Horn in 2000 [45] reported the clinical features of 130 EBS patients (7 patients with Dowling-Meara subtype, 69 with Koebner subtype and 54 with Weber-Cockayne subtype). In the Dowling-Meara EBS group, the infants had periodic shedding of the 20 nails and the adults had thickened great toenails. In the Koebner EBS group (nowadays classified by Fine and colleagues [1] as generalised EBS-other), 14 % of the patients had thickened great toenails. In the Weber-Cockayne EBS group (classified by Fine and colleagues [1] as localised EBS), thickened toenails were present in 12 % of individuals, including three children younger than 4 years. In EBS-Ogna, a dominant variant of EBS caused by plectin gene abnormalities, patients may have subungual blistering and bleeding, leading to nail dystrophy.
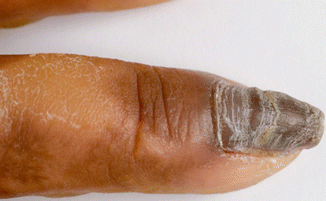
Fig. 73.4
Hyperkeratosis/thickening in EBS-DM
73.4.2 Junctional EB
Most JEB subtypes are associated with severe nail dystrophy. Subungual blistering causes onycholysis and thickened and abnormally shaped nails, which are sometimes onychogryphotic. In most patients with Herlitz EB, nail involvement occurs soon after birth, with typical paronychia-like lesions and nail loss with nail bed erosions due to extensive blistering. Development of subungual and periungual granulation tissue is typical and may cover the entire digit (Figs. 73.5 and 73.6). Patients with JEB caused by defects in lam332 may often have significant nail involvement leading to anonychia, while the patient’s systemic involvement can be mild. The new subtype of JEB, the laryngo-onycho-cutaneous syndrome (LOC), is characterised by early involvement of blistering under the nails. Eventually, the nails may be lost [46, 47]. In the late-onset JEB, symptoms usually appear between the ages of 5 and 8 years. Bullous lesions started from the hands and feet thus leading to nail dystrophy and loss of dermatoglyphic pattern [48].
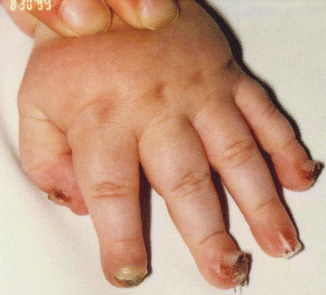
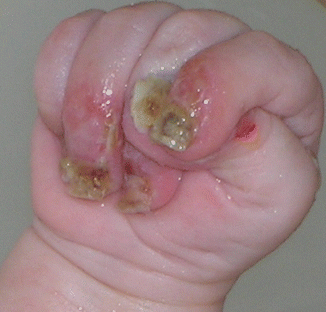
Fig. 73.5
Nail lifting with granulation tissue in JEB-LOC syndrome
Fig. 73.6
Periungual granulation tissue in Herlitz JEB
73.4.3 Dystrophic EB
Nail abnormalities in DEB range from a minor nail dystrophy to complete nail loss and mitten deformities. Repeated and extensive sub-basement membrane blistering followed by scarring leads to nail bed and matrix destruction with permanent anonychia. In localised DEB, only a few nails can be affected. Nails-only DDEB is a newly recognised subtype of DEB, in which nail dystrophy is the only clinical feature; the deformity, often limited to the toenails, can be mild and easily overlooked. This diagnosis should be considered in families with AD toenail dystrophy, even when there is no history of blistering. In the authors’ experience, the nail dystrophy is seen first in childhood where the great toenail is affected by onycholysis with subungual haemorrhages and mild nail bed hyperkeratosis. With time, the nail becomes thickened and dystrophic (pachyonychia) because of nail bed scarring and hyperkeratosis. In pretibial DEB, the nail dystrophy develops in childhood before the onset of skin lesions that typically occur after the age of 10 years. Nail dystrophy may occur in the absence of skin lesions in family members [40].
73.4.3.1 Clinical Features
Nail abnormalities observed in EB are not specific or pathognomonic, as they result from nail bed and matrix scarring. In the same way, there are nail changes characteristic of specific EB subtypes. The severity of the nail changes, however, usually is related to the severity of the skin lesions.
Pachyonychia (Nail Thickening)
Pachyonychia usually is reported in the literature as thickened dystrophic nails and is the most common nail finding in EB (EBS, DEB and JEB). The nail plate is shortened, thickened and yellow brown in colour. Onycholysis and nail bed haemorrhages are frequently associated. The nail abnormalities may affect several nails or be limited to the great toenail. In fingernails, pachyonychia is often associated with pincer nail deformity.
Onychogryphosis
The nail is thickened, opaque and yellow with an oyster-like appearance caused by excessive growth in the upward and lateral directions. The dystrophy is usually limited to the great toenails. It may be observed in EBS and JEB.
Nail Blistering
Periungual or subungual bullae produce haemorrhagic onycholysis and haemorrhagic paronychia with onychomadesis. Nail shedding may be followed by regrowth of normal or dystrophic nails (EBS, JEB) or produce loss of the nails (JEB, DEB) (Fig. 73.5).
Nail Erosions with Granular Tissue
The nail plate is absent, and the distal digit is covered by granulation tissue producing a drumstick appearance (JEB). This feature is characteristic of Herlitz EB, where patients usually have dystrophic or absent nails with periungual granulation tissue (Fig. 73.6).
Nail Atrophy
The nail plate is very thin, brittle and short. Nail changes result from nail matrix damage caused by repetitive blistering (JEB, DEB).
Anonychia
Nail loss caused by scarring is common in RDEB. Fusion of the digits by scar tissue leads to mitten-like appearance of hands and feet (pseudosyndactyly), which is typical of children with severe generalised RDEB, who usually develop the deformity by the age of 6–8 years. Anonychia is also a feature of Herlitz JEB and LOC syndrome [49], where it may be associated with thickening and occasionally periungual inflammation with granulation tissue of other nails [50].
Parrot Beak Nail Deformity
Recurrent blistering in the distal finger may result in soft tissue and bone reabsorption, with bending of the nail around the shortened fingertip. Satter [51] has reported this deformity in a 9-year-old boy with the AR Kindler syndrome.
73.5 Oral
The craniofacial and oral manifestations of the different EB types vary markedly in both character and severity depending largely on the EB type [52, 53] and can lead to significant morbidity. The tissues affected and the phenotypes displayed are closely related to the specific abnormal or absent proteins resulting from causative genetic mutations. For example, Type VII collagen is critical for maintaining the integrity of the oral mucosa in the same manner as in the skin but is not essential for normal development in the forming tooth bud. Consequently, individuals with Type VII collagen mutations typically have a developmentally normal dentition but can have severely affected oral soft tissues.
73.5.1 EB Simplex
The EB simplex subtypes are caused by mutations in the PKP1, DSP, KRT5, KRT14, PLEC1, ITGA6 and genes [1]. These genes all cause intra-epidermal cleavage in the skin and are all expressed by the oral mucosa which, like skin, also is comprised of a stratified epithelium [54–56]. Individuals with EB simplex therefore exhibit an increased fragility of the oral mucosa with blistering and ulceration [52]. In most cases, these will be localised and occur most often secondary to trauma or tissue manipulation; however, some individuals can experience significant oral blistering and severe mucosal involvement (Fig. 73.7). Typically oral soft tissue lesions heal without scarring although some severely affected EB simplex subtypes can display some oral scarring (e.g. Dowling-Meara).
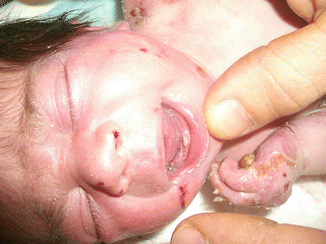
Fig. 73.7
Natal tooth in a baby with recessive EBS
73.5.2 Junctional EB
The junctional forms of EB are caused by mutations in LAMA3, LAMB3, LAMC3, COL17A1, ITG6A and ITGB4 that are important in basement membrane-mediated cell adhesion [1, 59, 60]. The proteins transcribed from these genes mediate epithelial cell adhesion in both the oral mucosa and the developing tooth bud [61, 62]. Almost all individuals with the junctional EB have an increased fragility of the oral mucosa that is accompanied by blister formation and ulceration [52, 63] which can be severe in some individuals. Most affected individuals however do not have significant oral scarring, and the soft tissue mobility and architecture remain relatively normal. One notable exception to this is the Herlitz EB subtype that is characterised by exuberant perioral granulation tissue. This frequently results in a reduction in the oral opening (microstomia) and some loss of tissue mobility in the lips and perioral tissues [52].
The genes that are causative of the junctional EB subtypes are all critical for normal tooth formation [61, 62]. Specifically, these genes produce proteins that are involved in cell adhesion of the odontogenic epithelium that gives rise to the cells that produce the dental enamel, ameloblasts. The enamel lesions can vary from generalised pitting to a generalised hypoplasia leaving only a very thin layer of enamel on the tooth surface. Some cases of Herlitz EB subtype also exhibit abnormal tooth eruption [64]. This is often most notable in the molar regions but can affect anterior teeth as well. Heterozygous carriers of COL17A1 mutations have been shown to have enamel defects that range from horizontal hypoplastic bands to white mottled enamel [65].
Individuals with junctional EB are at increased risk for developing dental caries [66], thought to be due to their marked enamel defects. The presence of extensive pitting over the tooth surface creates non-cleansable areas that are ideal for microbial growth and substrate retention that are known to cause dental caries. Generalised thin enamel that is frequently rough in nature also reduces the tooth’s primary resistance to the development and progression of caries.
73.5.3 Dystrophic EB
The dystrophic EB types are caused by mutations in COL7A1 gene that codes for anchoring fibril proteins that are located below the basal lamina at the dermal-epidermal basement membrane zone [67]. Type VII collagen is present in the basement membrane zone of the oral mucosa and is present during the early stages of tooth formation [68]. The COL7A1 gene is not expressed by the ameloblasts.
The soft tissue manifestations of dystrophic EB range from relatively mild to extremely severe [52, 69–72]. In dominant dystrophic EB, the oral manifestations can involve increased tissue fragility but infrequent blistering. Blistering can often be induced relatively easily, and dental treatments in even mildly affected dominant dystrophic EB should be approached with extra diligence to reduce soft tissue trauma. Individuals with severe generalised recessive dystrophic EB typically have extreme fragility of their oral and perioral mucosa. This is usually evident shortly after birth and can interfere with the neonate’s ability to suckle. The oral ulcerations can affect all areas of the oral mucosa including the tongue. The lesions heal with scarring. The continual process of blister formation and healing with scarring results in marked changes in the oral architecture. The tongue loses the lingual papillae and becomes bound down to the floor of the mouth (ankyloglossia). Anatomical structures such as the palatal rugae are ablated. The oral vestibules that normally form corridors for food clearance between the teeth and lips and cheeks become obliterated with the soft tissue attachment advancing till it is just below the crowns of the teeth. The soft tissues defining the oral opening fail to grow normally due to scarring resulting in a typically markedly restricted oral aperture. The presence of severe microstomia can impede the degree to which affected individuals can open their mouth dramatically limiting the distance between the teeth even when they are fully open. Milia are frequently observed on the skin of individuals with EB, and these can occur intra-orally as well, with the highest prevalence seen in dystrophic EB [52].
Individuals with certain forms of EB are known to have an increased risk for developing squamous cell carcinomas on the skin, but these can occur intra-orally as well [52, 63, 73, 74]. In particular, individuals with severe generalised recessive dystrophic EB are at increased risk of oral squamous cell carcinoma formation [74, 75] and should therefore be closely monitored for changes in oral ulcerations such as the development of raised, indurated borders.
Type VII collagen is not expressed by ameloblasts, and the enamel appears to generally form normally in individuals with the dermolytic forms of EB [57, 76]. Despite having relatively normal tooth formation, the prevalence of dental caries and resulting dental morbidity in severe generalised recessive dystrophic EB can be severe [66, 77, 78]. The tremendous oral soft tissue involvement results in the need to consume relatively soft diets that are frequently high in calories to meet the nutritional needs of the individual. In the presence of marked oral blistering, affected individuals frequently eat slowly and with increased frequency. The loss of normal tongue mobility and obliteration of the oral vestibule decreases the normal food clearance causing additional prolongation of the dental surfaces to potentially cariogenic substrates. Despite normal salivary secretion in most people with dermolytic EB subtypes, the oral cavity tends to be inoculated with high numbers of bacteria, and there tends to be excessive tooth plaque formation that further promotes the formation of dental caries [78]. Taken together, these factors produce an extremely high risk for dental caries in individuals with severe generalised recessive dystrophic EB that can be challenging to prevent and difficult to treat. Many severely affected individuals have tremendous difficulty performing normal oral hygiene due to their extreme soft tissue fragility, and even the use of anticariogenic mouth rinses can be unpleasant due to the presence of alcohol or strong flavouring agents [79].
73.5.4 Kindler Syndrome
Kindler syndrome is an autosomal recessive genodermatosis caused by mutations in the KIND1 gene that encodes for Kindlin-1 that is a component of focal contacts in basal keratinocytes [80].. Kindlin-1 is known to be expressed by the oral epithelium including the surface of the tongue [53, 81]. Oral blistering in the neonate and infant can be severe in Kindler syndrome [1], although the severity of tissue fragility diminishes with age. The Kindlin-1 protein is expressed by the epithelium that attaches the oral mucosa to the tooth [82]. The abnormal functioning of Kindlin-1 in forming normal focal adhesion in the basal keratinocytes appears to cause abnormal attachment and a predisposition to early-onset periodontal disease. Individuals affected with Kindler syndrome are therefore at risk for developing marked periodontal disease that can have its onset during the teenage years. Inflammation and gingival hyperplasia have been noted even in young children with Kindler syndrome suggesting that periodontal health may be compromised well before clinical signs of periodontal disease and alveolar bone loss [83]. It is not known if Kindlin-1 is expressed by the odontogenic epithelium; however, the dentition of affected individuals appears to be normal. Similarly, the salivary function and risk of dental caries is unknown in this rare syndrome.
Related posts:
 Kindlin-1 and Its Role in Kindler Syndrome
Kindlin-1 and Its Role in Kindler Syndrome
 Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
 Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
 Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
 Dermatitis Herpetiformis
Dermatitis Herpetiformis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


