15 Skin wound healing
Repair biology, wound, and scar treatment
Synopsis






Wound repair biology
Fast and uneventful wound closure restoring the integrity of the outer envelope is a criterion for survival for all living organisms. From prehistoric times up until today’s highly developed medicine, the aims of wound repair have not changed: restoring of the outer surface to protect the individual from infection and dehydration. Egyptian papyri document the treatment of wounds and recommend the use of different dressings depending on wound quality.1 The use of honey as an antiseptic has an astonishing history dating back more than 4000 years and has been rediscovered in recent years for the treatment of chronic wounds.2 Furthermore, different resins, myrrh, frankincense, and cinnamon were used for wound treatment. South American tribes applied antiseptic resins such as copaiba, Tolu or Peru balm on wounds.3 The oldest wound suture was detected on an Egyptian mummy dating from the 21st dynasty, around 1100 bc.1
Adult wound repair
The repair cascade consists of inflammatory, proliferative, and remodeling phases (Fig. 15.1). These overlapping phases act in highly coordinated relationships to heal skin defects. During the inflammatory phase, hemostasis occurs and an acute inflammatory infiltrate ensues. The proliferative phase is characterized by fibroplasia, granulation, contraction, and epithelialization. The final phase is remodeling, which is commonly described as scar maturation.
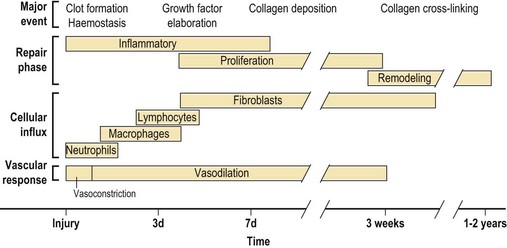
Fig. 15.1 The temporal patterns of repair phase, cellular influx, and vascular response during wound repair. The time points are approximate, and overlap occurs during these repair events.7
Inflammatory phase
The coagulation and complement cascades are initiated. The intrinsic and extrinsic coagulation pathways lead to activation of prothrombin to thrombin, which converts fibrinogen to fibrin, subsequently polymerized into a stable clot. As thrombus is formed, hemostasis in the wound is achieved (Fig. 15.2). The aggregated platelets degranulate, releasing potent chemoattractants for inflammatory cells, activation factors for local fibroblasts and endothelial cells, and vasoconstrictors. The adhesiveness of platelets is mediated by activated integrin receptors such as GpIIb-IIIa (αIIbβ3) on their surface.4,5 Interestingly, under thrombocytopenic conditions, macrophages and T cells at the wound site compensated for lack of platelet-derived growth factors (PDGFs) and initiation of the inflammatory phase.6
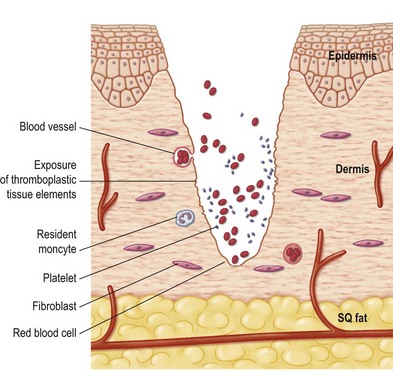
Fig. 15.2 Hemostasis is stimulated by platelet degranulation and exposure of tissue thromboplastic agents immediately after tissue injury.7 SQ, subcutaneous.
Immediately, the repair processes are initiated. After hemostasis, local vessels dilate secondary to the effects of the coagulation and complement cascades. Bradykinin is a potent vasodilator and vascular permeability factor that is generated by activation of Hageman factor in the coagulation cascade.8 The complement cascade generates the C3a and C5a anaphylatoxins, which directly increase blood vessel permeability and attract neutrophils and monocytes to the wound. These complement components also stimulate the release of histamine and leukotrienes C4 and D4 from mast cells. The local endothelial cells then break cell–cell contact and increase permeability, which enhances the margination of inflammatory cells into the wound site.8
The initial influx of white blood cells in the wound is composed of neutrophils (Fig. 15.3). This early neutrophil infiltrate scavenges cellular debris, foreign bodies, and bacteria. Activated complement fragments aid in bacterial killing through opsonization. The neutrophil infiltrate is decreased in clean surgical wounds compared with contaminated or infected wounds.
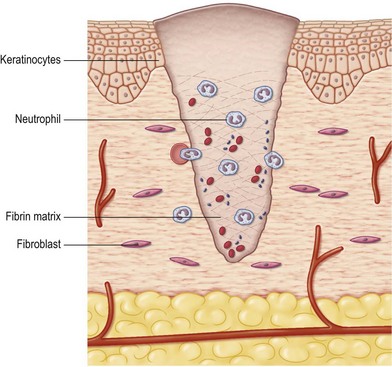
Fig. 15.3 A neutrophil influx into the wound occurs within 24 hours. The neutrophils scavenge debris and bacteria and secrete cytokines for monocyte and lymphocyte attraction and activation. Keratinocytes begin migration when a provisional matrix is present.7
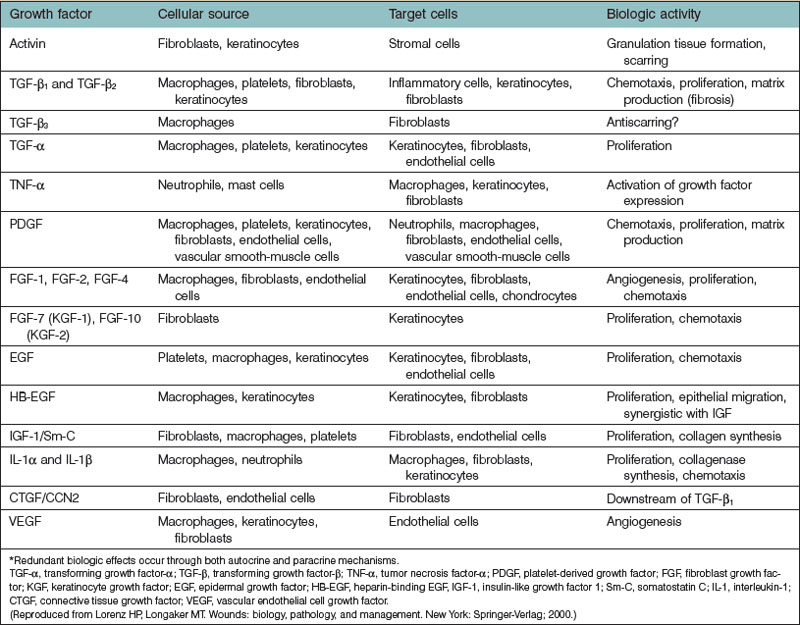
Within 2–3 days, the inflammatory cell population begins to shift to one of monocyte predominance. Circulating monocytes and mast cells are attracted and infiltrate the wound site.9,10 These monocytes differentiate into macrophages and, in conjunction with the resident tissue macrophages, orchestrate the repair process. Macrophages not only continue to phagocytose tissue and bacterial debris but also secrete multiple peptide growth factors. These growth factors activate and attract local endothelial cells, fibroblasts, and keratinocytes to begin their respective repair functions. More than 20 different cytokines and growth factors are known to be secreted by macrophages (Table 15.1).11
Depletion of monocytes and macrophages in combination with steroid therapy resulted in severe wound-healing deficiency with poor debridement, delayed fibroblast proliferation, and inadequate angiogenesis.12
In contradiction to the dogma that inflammation is essential for skin wound healing, mice deficient in the hematopoietic ETS family transcription factor PU.1 showed normal wound repair13 despite immunoincompetent leukocytes and nonfunctional macrophages.14 Although the growth factor and cytokine pattern was altered, incisional wounds healed uneventfully and scar-free with phagocytic fibroblasts clearing the wound debris. Obviously, attenuated inflammation is beneficial for wound healing.9
Proliferation phase
Extracellular matrix formation
During fibroplasia, fibroblasts synthesize and deposit replacement extracellular matrix (ECM) at the wound site. The proliferative phase begins with degradation of the initial fibrin–platelet provisional matrix. Three major classes of proteases are involved in wound repair. Proteases of the serine, cysteine, and matrix metalloproteinase (MMP) family are secreted to facilitate cellular migration through fibrin clot and provisional matrix. Both tissue and urokinase plasminogen activator are expressed to dissolve the blood clot.15 MMPs comprise endopeptidases such as collagenases, gelatinases, and stomelysins that can degrade virtually all matrix proteins, activate growth factors, and bind on cell surfaces.16,17 Cysteine proteases are present in epithelia during wound repair and are involved in signaling and basement membrane protein degradation.18
Macrophages, mast cells, and the adjacent ECM release growth factors that stimulate fibroblast activation.9 Local fibroblasts become activated and increase protein synthesis in preparation for cell division. As fibroblasts proliferate, they become the predominant cell type by 3–5 days in clean, noninfected wounds (Fig. 15.4). After cell division and proliferation, fibroblasts begin the synthesis and secretion of ECM products. The initial fibrin matrix is replaced by a provisional matrix of fibronectin and hyaluronan, which facilitates fibroblast migration. The control of ECM deposition by fibroblasts is complex and partially regulated by growth factors and interactions of fibroblast cell membrane receptors with the ECM. Integrins are regulators of cellular function during repair. They are transmembrane receptors with extracellular, membrane, and intracellular protein domains. Integrins are heterodimeric and composed of α and β subunits that interact to form the active protein receptor. Ligands to integrins include growth factors and ECM structural components such as collagen, elastin, and other cells.19,20 After ligands bind, phosphorylation occurs in the cytoplasmic domain of the integrin receptor, which starts a signal transduction cascade that ultimately changes gene expression, and new cellular function ensues.
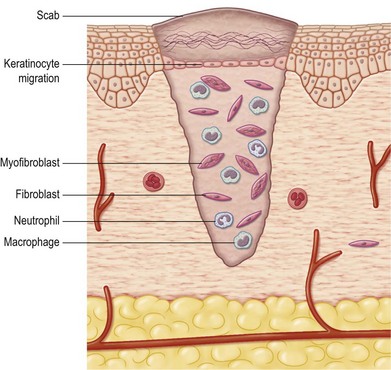
Fig. 15.4 Fibroblasts are activated and present at the wound by 3–5 days after injury. These cells secrete matrix components and growth factors that continue to stimulate healing. Keratinocytes migrate over the new matrix. Migration starts from the wound edges as well as from epidermal cell nests at sweat glands and hair follicles in the center of the wound.7
Fibronectin and the glycosaminoglycan hyaluronic acid compose the initial wound matrix.10 Hyaluronic acid provides a matrix that enhances cell migration because of its large water content. Adhesion glycoproteins, including fibronectin, laminin, and tenascin, are present throughout the early matrix and facilitate cell attachment and migration. Integrin receptors on cell surfaces bind to the matrix glycosaminoglycans and glycoproteins. As fibroblasts enter and populate the wound, they secrete hyaluronidase to digest the provisional hyaluronic acid-rich matrix, and larger, sulfated glycosaminoglycans are subsequently deposited. Concomitantly, new collagen is deposited by fibroblasts on to the fibronectin and glycosaminoglycan scaffold in a disorganized manner, resulting in scar formation. The major fibrillar collagens composing the ECM in skin and scar are collagen types I and III. The ratio of collagen type I to type III is 4 : 1 in both skin and wound scar. Although type III collagen is initially deposited in relatively greater amounts in wounds, its amount is always less than type I collagen in mature scar.21
At least 28 different types of collagen are currently known.22 Most collagen types of the ECM are synthesized by fibroblasts; however, some types are synthesized by keratinocytes.23 The collagens share common characteristics: the basic structural unit is a right-handed triple helix. Unique structural properties that distinguish the different collagen types include segments that interrupt the triple helix and fold it into other structures with unique properties.24
The major structural component of wound scar is collagen. Fibroblasts synthesize and secrete collagens through a complex intracellular and extracellular process. Coordinated transcription of genes on different chromosomes (2, 6, 7, 12, 13, 17, and 21) occurs. In addition, several intracellular and extracellular modifications are required to form the new collagen fiber. Collagens contain a high fraction of proline amino acid residues, and this is the basis of its triple helix structure. With regard to collagens I and III, tropocollagen is formed in the ECM.24 Tropocollagen molecules aggregate laterally and are covalently cross-linked by the enzyme lysyl oxidase to form collagen fibrils.25 The fibrils interact with other fibril types, which then aggregate into fibers. Fibers then aggregate into bundles and form a collagen-rich scar.
Granulation tissue formation and angiogenesis
Granulation tissue is a dense population of blood vessels, macrophages, and fibroblasts embedded within a loose provisional matrix of fibronectin, hyaluronic acid, and collagen. Granulation tissue is clinically characterized by its beefy-red appearance (i.e., “proud flesh”) and is present in open wounds. It is a consequence of the rich bed of new capillary networks (neoangiogenesis) that form by endothelial cell division and migration. The directed growth of vascular endothelial cells is stimulated by tissue hypoxia, and platelet and activated macrophage and fibroblast products.26 Examples are basic fibroblast and vascular endothelial growth factor (VEGF), which induce migration and proliferation of endothelial cells.27 A prerequisite for endothelial sprouting is the expression of the integrin avβ3 at the leading capillary tip28 and the secretion of proteases for matrix degradation. Granulation tissue is a clinical indicator that an open wound is amenable to skin graft treatment. Wounds that benefit from skin grafts are of sufficient size such that the healing time would be decreased after grafting. Because granulation tissue has a high level of vascularity due to the abundance of new capillary formation, it readily accepts and supports skin grafts.
Wound contraction
Contraction is the process in which the surrounding skin is pulled circumferentially toward an open wound (Fig. 15.5). This phenomenon does not occur with closed surgical incisions. Open wounds after trauma, burns, and previously closed wounds secondarily opened by infection are associated with contraction. Wound contraction decreases the size of the wound dramatically without new tissue formation. This repair process speeds wound closure compared with epithelialization and scar formation alone. In addition, the area of insensate scar is smaller. Animals have a much greater capacity for wound contraction than humans do. Most mammalian animals (e.g., rodents, cats, dogs, sheep, and rabbits) have a panniculus carnosus, which is a myofascial layer between the subcutaneous fat and musculoskeletal layers. This anatomy results in a plane of low resistance between two fascial layers, which allows enhanced skin mobility and therefore contraction. The amount of contraction is related to both the size of the wound and mobility of the skin. In humans, contraction is greatest in the trunk and perineum, least on the extremities, and intermediate on the head and neck. Eighty percent to 90% of wound closure can be due to contraction in the trunk and perineum.29 These regional differences in contraction are probably due to relative differences in skin laxity.
It has become accepted that myofibroblasts play a key role in wound contraction. After wounding, dermal fibroblasts at wound margins are activated by growth factors released into the wound. Stimulated by mechanical tension and PDGF, they turn into stress fiber-expressing protomyofibroblasts. Protomyofibroblasts are found in early granulation tissue and in normal connective tissue with high mechanical load. Approximately 4 days after wounding, myofibroblasts appear in the wound.30 Mechanical tension, activated transforming growth factor-β (TGF-β1)16 and the splice variant ED-A fibronectin trigger protofibroblast differentiation into alpha-smooth-muscle actin-expressing myofibroblasts.31 Myofibroblasts exert their contractile forces by focal adhesion contacts that link the intracellular cytoskeleton to the ECM. Interestingly, intercellular myofibroblast contacts via connexin-containing gap junctions are important for contractility because connexin-43-depleted fibroblasts were incapable to contract collagen matrices.32 Furthermore, the role of keratinocytes in wound contraction should not be neglected. In vitro experiments showed higher contractile forces of keratinocytes compared with fibroblasts.33 Future research will shed further light on the biology of skin wound contraction.
Epithelial resurfacing
Morphologic changes in keratinocytes at the wound margin are evident within hours after injury. Marginal basal cells dissolve intercellular contacts, e.g., desmosomes and adherence junctions, and cell–matrix contacts, e.g., hemidesmosomes. Flattened migratory cells express cytoplasmatic actin filaments, promigratory integrins, e.g., α2β1, α5β1, αvβ5, αvβ6, and secrete proteases to enable movement.34 MMP-3 is localized to wound margin keratinocytes and cleaves the intercellular contact protein E-cadherin.35 MMP-9 cleaves basement proteins, e.g., collagen type IV and VII releasing keratinocytes from their substratum.36 MMP-1 degrades native collagen type I and III, smoothing the way for newly formed matrix. Furthermore, MMP-1 binds to α2β1 integrin on cell membranes, presumably acting as a migration promoter.37 Blocking MMP activity with synthetic broad-spectrum inhibitors abolished epithelial migration in vitro and in vivo.38,39
In contrast to keratinocytes are fibroblasts able to adopt an ameboid phenotype and squeeze through the matrix network in the presence of MMP inhibitors.40 Recently, cysteine proteases, e.g., cathepsin-B, C, K and L, entered the field of skin wound healing, acting as endopeptidases involved in the degradation of ECM proteins.18
After the re-establishment of the epithelial layer, keratinocytes and fibroblasts secrete laminin and type IV collagen to form the basement membrane.41 The keratinocytes become columnar and divide to restore the layering of the epidermis and reform a barrier to further contamination and moisture loss. Keratinocytes can respond to foreign-body stimulation with migration as well. Sutures in skin wounds provide tracks along which these cells can migrate. Fibrotic reactions, cysts, and sterile abscesses centered on the suture can occur. These are treated by removal of the inciting suture and epithelial cell sinus track or cyst.
Remodeling phase
The scaffold that supports cells in both the unwounded and wounded states is the ECM, which is the structural component of skin that must be repaired after injury. The ECM is dynamic and is constantly undergoing remodeling during repair, which can be conceptualized as the balance between synthesis, deposition, and degradation. Lysyl oxidase is the major intermolecular collagen cross-linking enzyme.25 Collagen cross-linking decreases its degradation and improves wound tensile strength. The balance of collagen deposition and degradation is in part determined by the regulation of MMP activity.42 MMPs are inactivated by tissue inhibitors of MMPs, α-macroglobulin, and degradation by other proteases.17,43 Except for reduced wound contraction in MMP-3-deficient mice,44 other MMP knockout mice have little or no wound-healing disturbances.36
Wound scar remodeling occurs during months to years to form a “mature” scar. The early scar appearance is red due to its dense capillary network induced at the injury site. When closure is complete, capillaries regress until relatively few remain. As the scar redness dissipates during a period of months, the true scar pigmentation becomes evident. Scars are usually hypopigmented after full maturation. However, scars can become hyperpigmented in darker-pigmented patients and in those lighter-pigmented patients whose scars receive excess sun exposure. For this reason, sun protection measures are recommended for patients with early scars on sun-exposed areas such as the scalp, face, and neck. During remodeling, wounds gradually become stronger with time. Wound tensile strength increases rapidly from 1 to 8 weeks after wounding and correlates with collagen cross-linking by lysyl oxidase. However, the tensile strength of wounded skin at best reaches only approximately 80% that of unwounded skin45 but can be increased by synthetic MMP inhibitors.46 In addition, scar is brittle and less elastic than normal skin. It is readily visible because of color, contour, and texture differences compared with unwounded skin. Although scars can be hidden well with proper surgical planning and uneventful healing, they may have aesthetically unacceptable appearances in nonelective wounds after trauma and burns and in wounds with healing problems.
Regulation
Growth factors in skin wound healing
Growth factors are the focal regulatory points of the repair process. They are polypeptides that are released by a variety of activated cells at the wound site (Table 15.1). They act in either a paracrine or autocrine fashion to stimulate or to inhibit gene expression by their target cells in the wound. In general, they stimulate cellular proliferation and chemoattract new cells to the wound. Myriad growth factors are present in wounds and many have overlapping biologic functions. Most growth factors exist in several isoforms with several receptor types present in wounds, which increases the complexity of growth factor function. In addition to growth factor ligands, their signaling receptors are another locus for regulation of repair. Growth factors do not have an effect on target cells without a functional signaling receptor present on the cell surface. The level of regulation is complex in that the growth factors have multiple different receptor types to which they can bind and induce cell signaling. In recent years, the development of transgenic and knockout mouse models including inducible and cre-lox technologies has generated new knowledge in the function of many growth factors in wound healing. Clinical trials applying various growth factors to chronic nonhealing wounds showed beneficial effects,47 although treatment efficacy should be confirmed by larger, randomized, double-blinded studies.48
Platelet-derived growth factor
PDGF is released from platelet alpha granules immediately after injury. PDGF attracts neutrophils, macrophages, and fibroblasts to the wound and serves as a powerful mitogen. Macrophages, endothelial cells, and fibroblasts also synthesize and secrete PDGF in the wound. PDGF stimulates fibroblasts to synthesize new ECM, predominantly the noncollagenous components such as glycosaminoglycans and adhesion proteins and to contract these matrices.49 PDGF also increases the amount of collagenase secreted by fibroblasts, indicating a role for this growth factor in tissue remodeling. PDGF strongly induces granulation tissue production and was the first growth factor to be approved for the treatment of diabetic nonhealing wounds.50
Transforming growth factor-β
The TGF-β family comprises TGF-β1–3, bone morphogenic proteins (BMP), and activins. TGF-β1 predominates in adult wound healing and is a promigratory and profibrotic growth factor that directly stimulates collagen synthesis and decreases ECM degradation by fibroblasts. It is released from all cells at the wound site, including platelets, macrophages, fibroblasts, and keratinocytes. TGF-β acts in an autocrine fashion to stimulate its own synthesis and secretion. TGF-β also chemoattracts fibroblasts and macrophages to the wound, promotes granulation tissue formation, myofibroblast differentiation, and wound contraction. TGF-β accelerates wound repair when it is applied experimentally to wounds that have no deficiency in repair. However, the increase in repair rate is at the expense of increased fibrosis, which could be a detriment during normal skin healing. In addition, increased TGF-β activity is associated with pathologic fibrosis in multiple different organ systems, including heart, lung, brain, liver, and kidney. TGF-β stimulates ECM synthesis by increasing collagen, elastin, and glycosaminoglycan synthesis. It increases integrin expression, which enhances cell–matrix interactions. TGF-β increases ECM accumulation by decreasing MMP and increasing tissue inhibitor of MMP expression. Through these mechanisms, exogenous TGF-β augments fibrosis at the wound site.51 Interestingly, studies in mice with a lack of endogenous TGF-β activity demonstrate accelerated healing with an impaired inflammatory response.52 In chronic wounds, however, significantly reduced TGF-β was found leading to clinical trials with TGF-β treatment that failed showing beneficial effects.53 Activins and BMP are promigratory proteins secreted by keratinocytes and fibroblasts during wound repair and promote keratinocyte differentiation.53 These findings underscore the complex effects of TGF-β and other growth factors during the repair process.
Fibroblast growth factors
The fibroblast growth factors (FGFs) are a group of heparin-binding growth factors that are secreted into the ECM, where they remain dormant until activated by tissue injury. They are bound by heparin and the glycosaminoglycan heparan sulfate. They have a broad range of biologic functions, specific to each isoform. FGF-1 (acidic FGF) and FGF-2 (basic FGF) stimulate angiogenesis.26 Endothelial cells, fibroblasts, and macrophages produce FGF-1 and FGF-2. Basement membrane serves as a storage depot for FGF-2, which is released on degradation of the heparin components of the basement membrane at sites of injury. FGF-1 and FGF-2 stimulate endothelial cells to divide and form new capillaries. They also chemoattract endothelial cells and fibroblasts. FGF-7 (keratinocyte growth factor 1 (KGF-1)), FGF-10 (KGF-2), and epidermal growth factor (EGF) stimulate epithelialization. KGF-1 and KGF-2 are expressed in wound fibroblasts and promote keratinocyte proliferation and migration in a paracrine fashion. Decreased expression of KGF-1 occurs in diabetic and reduced levels of FGF-2 in aged mouse wounds with decreased angiogenic response.54,55 In an ischemic rabbit wound model, exogenous KGF-2 treatment accelerates epithelialization without an increase in scar formation.32 Exogenous KGF-2 also increases wound tensile strength, collagen content, and epidermal thickness in animal models of normal healing.56 Hitherto, only one clinical trial has been conducted using recombinant human KGF-2 for treatment of chronic venous ulcers and showed accelerated wound healing.57
Vascular endothelial growth factor
VEGF is also a potent angiogenic stimulus.58 It acts in a paracrine manner to stimulate vascular permeability and proliferation by endothelial cells after release from platelets, endothelial cells, neutrophils, macrophages, fibroblasts, and keratinocytes.59–61 Its expression is increased in hypoxic conditions, such as those found at the wound site.62 Intramuscular gene transfer of VEGF to patients with ischemic ulcers resulted in sprouting of collateral vessels and limb salvage.63 Despite these promising results, careful use of VEGF is recommended because exogenous VEGF administration yielded vascular leakage and malformation.64 Placental growth factor (PLGF) is also proangiogenic and acts synergistically to VEGF.65 PLGF is upregulated in migrating keratinocytes and promotes granulation tissue formation and migration. Unlike VEGF, PLGF does not interfere with lymphatic vessel formation66 and accelerates wound healing in diabetic mice.67
Other growth factors and cytokines
Multiple other growth factors affect wound repair. Epithelialization is also directly stimulated by members of the EGF family, e.g., EGF, heparin-binding EGF (HB-EGF), and TGF-α. These mitogens are released by keratinocytes to act in an autocrine fashion. Insulin-like growth factor 1 (IGF-1) stimulates cell proliferation and collagen synthesis by fibroblasts and interacts synergistically with HB-EGF, PDGF, and FGF-2 to facilitate fibroblast proliferation.51 Various cytokines comprising interleukins (IL), chemokines, and tumor necrosis factor-α (TNF-α) mediate inflammatory cell functions at the wound site and contribute to tissue remodeling and angiogenesis.51 IL-6 is crucial for initiation of the wound-healing response68 whereas overexpression leads to cutaneous scarring. TNF-α and interferon-γ have been shown to downregulate ECM protein synthesis. Granulocyte–macrophage colony-stimulating factor (GM-CSF) is a pleiotropic cytokine stimulating migration and proliferation during wound repair. Experimental treatment of diabetic and chronic wounds showed promising results with faster healing rates.69,70
Growth factor interactions with the extracellular matrix
In normal, unwounded conditions, the ECM is a depository of growth factors in latent forms. With injury and matrix destruction, growth factors are released from the ECM in active form and thereby assist in initiating and regulating the repair process. For example, TGF-β is bound in the ECM to the proteoglycan decorin and is inactive when bound. At sites of injury, TGF-β forms complexes with its binding protein, latency-associated protein (LAP), and is released. Under acidic conditions, such as at sites of hypoxia and tissue injury, LAP disassociates and active TGF-β is formed. LAP can also be proteolytically cleaved and released by MMPs and other proteases at the wound site. Active TGF-β immediately binds to its receptors (RI and RII), which are present on fibroblasts, macrophages, and endothelial cells.71 The TGF-β RI and RII receptors form heterodimeric complexes with each other, and TGF-β biologic activity is initiated in the target cell through the Smad pathway.72
Fetal wound repair biology
The therapeutic solution for the reduction and possible elimination of scar formation may be found in the mechanisms responsible for scarless wound healing in the fetus. The early-gestation fetus can heal skin wounds with regenerative-type repair and without scar formation.74,75 In scarless fetal wounds, the epidermis and dermis are restored to a normal architecture. The collagen dermal matrix pattern is reticular and unchanged from unwounded dermis. The wound hair follicle and sweat gland patterns are normal as well (Fig. 15.6) . The wound is not evident grossly unless a wound edge contour change is present, and this will cast a light shadow under appropriate circumstances.73 Scarless healing by the fetus does not depend on the fetal environment. Fetal wounds in marsupials76 or skin graft wounds heal without scar after transplantation to a postnatal environment. Thus, amniotic fluid, the intrauterine environment, and fetal serum factors are not required for scarless healing. Scarless repair appears to be inherent to the fetal tissue and probably depends on factors associated with skin development.75 Notably, fetal scarless repair in skin is organ-specific with scarring of intestinal wounds at the same gestational age.77 The fetal environment alone cannot induce scarless healing in adult skin. When adult skin is transplanted on to a fetus and later wounded at a point in gestation when fetal skin heals scarlessly, the adult skin grafts still heal with scar formation.78 The transformation of adult healing to scarless repair cannot be accomplished simply by perfusion of adult skin with fetal serum or by immersion in amniotic fluid. Thus, induction of scarless healing in adult skin will require more than recreation of the fetal environment. The adult repair processes will likely have to be modified to recapitulate skin development. An understanding of the biology of scarless fetal wound repair will help surgeons develop therapeutic strategies to minimize scar and fibrosis.
Transition from scar-free healing to scar formation
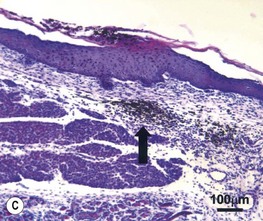
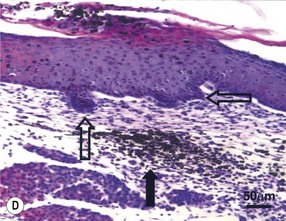
Both gestational age and wound size determine whether the fetus will heal a wound without scar.79 As gestation progresses, a transition from scarless healing to healing with scar formation occurs before birth. In large-animal models, such as the fetal lamb and monkey (Fig. 15.7), this transition occurs during the early part of the third trimester for incisional, closed wounds. In addition, wound size affects the temporal occurrence of this transition. Open, excisional wounds must be made earlier in gestation than closed, incisional wounds for scarless healing to occur. Also, larger open wounds must be made earlier in gestation than smaller open wounds for scarless healing.79 The exact reasons underlying these observations remain unknown but probably relate to whether the wound has healed before a certain threshold in development. The shift from scarless healing to scar formation is not abrupt but instead occurs gradually with an intermediate repair outcome that is neither regeneration nor scar: the transition wound. The transition wound has a normal reticular collagen and connective tissue matrix pattern but without restoration of epidermal appendages.73 Thus, it has features of both scar (no appendages) and scarless (normal ECM) healing.
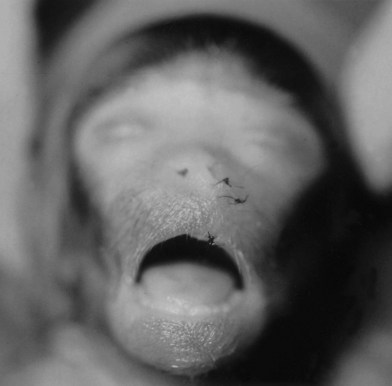
Fig. 15.7 Fetal monkey full-thickness wedge excision lip wound made at the beginning of the third trimester and harvested 2 weeks later. The cutaneous wound is identified by the sutures. No scar was present histologically, and the orbicularis oris muscle reformed across the defect.73 Interestingly, the naris was deformed compared with the contralateral side.
Differences between fetal and adult repair
Cellular differences
Because fetal fibroblasts deposit matrix in a scar-free pattern, they are crucial for scarless repair. A number of studies have defined the functional differences between fetal and postnatal dermal fibroblasts. First, fetal fibroblasts synthesize more collagen type III and IV with simultaneous proliferation, in contrast to their postnatal counterparts in vitro.80 Fetal fibroblasts failed to respond to TGF-β stimulation with collagen synthesis compared to postnatal fibroblasts in vitro.81 Furthermore, higher gene expression of MMP-1, -2, -3, -9, and -14 is found during fetal wound healing with virtually absent myofibroblasts during early gestation.82,83 Fetal fibroblasts also migrate at a faster rate than postnatal fibroblasts.84 Increased migration velocity during repair is likely to affect collagen deposition and crosslinking.
In postnatal wounds, epidermal continuity is restored by proliferating stem cells derived from the epidermal basement membrane and the hair follicle bulge.85 Because immature fetal skin does not have hair bulges, the stem cell origin is likely to be different to adult wound repair. Indeed, a new stem cell was recently discovered and named dot cell because of its extremely small size.86 Dot cells are found in basement membranes and in the blood during fetal development and have a strong affinity to wounded tissue. After transplantation of dot cells into adult murine wounds, scarless healing was noted. Despite their small size, these cells seem to have a great future in wound repair.
Differences in wound repair
Several parameters of repair are different in fetal wounds compared with postnatal wounds. The rate of repair for wounds of equal size is faster in the fetus. The collagen synthesis rate is greater in fetal wounds. In addition, the rate of epithelialization is faster in the fetal wound.80 Fetal wound repair may be more rapid because there is little or no period of activation for the fetal fibroblast to synthesize ECM at the wound site.
A key difference between fetal and postnatal tissue repair is a reduced inflammatory cellular infiltrate in fetal wound repair.87 Inflammation plays a prominent role in postnatal repair, but it is not present in significant amounts during scarless fetal healing. This may be due to the immature fetal immune system with decreased platelet degranulation, PDGF and TGF-β production.87 However a marked inflammatory cellular infiltrate occurs later in gestation after the transition when fetal wounds form scar. The amount of inflammation correlates strongly with the amount of scar formation.
Growth factor expression in fetal wound repair
Few growth factors associated with adult repair have increased expression in scarless fetal wounds. This is likely due to inherent major differences in the regulation between the two repair systems. Growth factors that are upregulated in scarless wounds are increased more rapidly than in postnatal wounds. One example is VEGF, which is expressed rapidly and threefold higher after injury in scarless wounds but is delayed in postnatal wounds.88 The expression of TGF-β isoforms, receptors, and bioactivity modulators has been analyzed in fetal wounds. Compared with scarring wounds, scarless fetal wounds have more TGF-β3, receptor type II, and fibromodulin expression.89 In contrast, increased TGF-β1, receptor type I, and decorin expression is associated with the transition to scar formation. Thus, the expression profile of isoforms, receptors, and modulators for a growth factor can be different in scarless and scarring wounds. By understanding the scarless repair expression profile for a growth factor, information is obtained that can permit recapitulation of this profile during postnatal repair to reduce scar formation. Exogenous addition of several cytokines to fetal wounds has also been performed and in every case has resulted in accelerated scarring. The cytokines tested include TGF-β1, TGF-β2, PDGF, and BMP-2, each of which has increased expression during scar formation in postnatal wounds.90–92 Thus, growth factors with increased expression in scarring wounds probably modulate scarless wounds in a similar fashion, and this results in scar formation.
Differences in gene expression
Because fetal skin is growing and differentiating, genes associated with development are likely to have an important role during scarless healing. Their expression may not occur during adult healing because of inactivation at the end of development. Homeobox genes are transcription factors that govern gene expression during embryogenesis. PRX2 is a homeobox gene that has increased expression during scarless fetal wound healing and only weak expression during adult skin healing.93 New data are available for Wnt proteins involved in skin maturation, fetal and adult wound repair.94,95 Wnt signaling is increased in postnatal but not in fetal murine wound healing with a TGF-β1-like profibrotic influence on postnatal but not on fetal fibroblasts.96 Differential signaling in fetal and adult cells in response to wounding seems to direct the way to either tissue regeneration or scarring.
Differences in matrix composition
Fetal skin and wound matrix are composed of more hyaluronic acid than are postnatal skin and scar. Hyaluronic acid stimulates fibroblast migration and probably affects the ECM deposition pattern.84 Elastin, present in adult skin, is absent in fetal dermis, although no differences were found in basement membrane proteins or vessel formation.97 Fetal skin and wound ECM have a relatively greater amount of collagen type III, but how this affects scar formation is not known. The collagen fiber thickness is increased in scarring fetal wounds compared with unwounded fetal skin as measured by confocal microscopy. This distance between collagen fibers is also greater in scarring fetal wounds, but the mechanisms causing these earliest changes in healing associated with scar formation remain to be elucidated.74 Scarless fetal skin repair is the blueprint for ideal repair. With regard to TGF-β3, an antiscarring product was developed98 but failed to prove efficacy in clinical phase III trials. However, the mechanisms responsible for scarless repair still remain to be fully determined and when delineated will likely lead to innovative antiscarring treatment strategies.
Adult wound pathology
Nonhealing wounds
Chronic or nonhealing wounds are open wounds that fail to epithelialize and close in a reasonable amount of time, usually defined as 30 days. These wounds typically are clinically stagnant and unable to form robust granulation tissue. Many factors contribute to inhibit healing in these patients, but no unifying theory can explain the etiopathogenesis of each individual nonhealing wound (Table 15.2). Medical conditions such as diabetes, arterial insufficiency, venous disease, lymphedema, steroid use, connective tissue disease, and radiation injury inhibit wound healing. Nonhealing wounds can also be due to pressure necrosis, infection (especially osteomyelitis), skin cancer, malnutrition, chronic dermatologic disease, and other metabolic conditions. In each case, treatment begins with debridement of any necrotic tissue present.99 However, despite optimal treatment for each clinical problem, these wounds frequently still do not heal and surgical intervention is required.
Table 15.2 Factors that may impair normal healing and lead to chronic nonhealing wounds
| Etiopathology | Examples | |
|---|---|---|
| Vascular | Arterial | |
| Lymphatic | ||
| Venous | ||
| Mixed arteriovenous | ||
| Vasculitis | Wegener granulomatosis, Churg–Strauss vasculitis, Henoch–Schönlein purpura, Sneddon syndrome, systemic lupus erythematosus, rheumatoid arthritis, Felty syndrome, Takayasu arteriitis, polyarteritis nodosa, Kawasaki syndrome, pyoderma gangrenosum, necrobiosis lipoidica diabeticorum, thrombangiitis obliterans (Buerger syndrome), allergic reactions | |
| Vasculopathic syndromes | Raynaud syndrome, systemic scleroderma, CREST, Klippel–Trenaunay syndrome, proteus syndrome145,146, CLOVE syndrome,147 Kasabach–Merritt syndrome | |
| Physical, chemical, and biological causes | Pressure | |
| Trauma | Lacerations, any type of soft-tissue and bone injury, vascular rupture | |
| Thermal | Burns / frostbite, electrical injury (electrical current/high voltage/lightning) | |
| Radiation | Radiation therapy | |
| Chemical-toxic | Extravasation, chemical burns (acids/bases), sclerotherapy | |
| Infections | ||
| Neuropathic | Posttraumatic | |
| Congenital | Spina bifida, syringomyelia, multiple sclerosis, neurological syndromes | |
| Systemic neuropathic diseases | ||
| Hemopathological | Systemic diseases | |
| Disturbed hemostasiology | ||
| Neoplastic diseases | Cutaneous tumors | Basal and squamous cell carcinoma, melanoma, Bowen syndrome, Marjolin ulcer (scar carcinoma),148 tumors with cutaneous metastasis or penetration (e.g., Paget syndrome) |
| Therapeutic modalities | ||
| Systemic diseases | ||
| Other causes |
CREST, calcinosis, Raynaud’s syndrome; esophageal dysmotility, sclerodactyly, and telangiectasia; CLOVE, congenital lipomatous overgrowth, vascular malformations, and epidermal nevi; BCG, bacillus Calmette-Guérin; NSAIDs, nonsteroidal anti-inflammatory drugs.
Pressure sores
Wounds that develop over a bone prominence, usually in the immobile patient, are termed pressure sores. They are also called decubitus ulcers or bed sores. The sacrum, ischium, and greater trochanter are the most common locations affected.100 However, the metatarsal heads, ankles, heels, knees, and occiput are susceptible under certain conditions. Another problem is malleolar skin pressure necrosis due to constricting cast placement. The amount of tissue pressure necrosis is determined by both the degree and duration of the pressure. When the tissue pressure is greater than 25–30 mmHg and capillary perfusion pressure is blocked, microcirculation is compromised. Necrosis can occur with as little as 2 hours of sustained pressure at this level.101
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


