Single-Suture Craniosynostosis and Deformational Plagiocephaly
Gary F. Rogers
Stephen M. Warren
INTRODUCTION
Craniosynostosis is the premature fusion of one or more cranial sutures. This pathologic process occurs in 1 in 2,000 to 2,500 live births and can occur in association with more than 130 different syndromes (multi-suture craniosynostosis is discussed in Chapter 23).1 Any cranial suture can ossify prematurely, but fusion is most common in the sagittal suture (40% to 55%), followed by the coronal (20% to 25%), metopic (5% to 15%), and lambdoid (1% to 5%) sutures. Craniosynostosis results in characteristic changes to the cranial shape that indicates which suture(s) is involved (Virchow’s law). Patients with craniosynostosis, especially syndromic forms, may also have other physical findings such as midface hypoplasia, deafness, blindness, speech impairments, learning disabilities, nasopharyngeal airway obstruction, swallowing dysfunction, heart and lung abnormalities, and extremity anomalies.
The diagnosis, management, and treatment of craniosynostosis can be complex and requires coordinated care. This is best accomplished by an interdisciplinary team comprised of professionals from the following disciplines: anesthesiology, craniofacial surgery, genetics, hand surgery, intensive care, neurosurgery, nursing, ophthalmology, orthodontics, pediatrics, pediatric dentistry, prosthodontics, psychology, radiology, social work, and speech/language pathology.2 Implicit in the choice of a team is the understanding that the first procedure provides the best opportunity for an optimal surgical outcome. A good or excellent surgical outcome is more challenging to achieve if critical tissues are surgically damaged, malpositioned, or discarded. An experienced team is even more important when contemplating surgical revision.
PATHOGENESIS
Historically, three etiopathogenic theories have dominated the field of craniosynostosis. In his 1851 paper on cretinism and pathologic brain malformation, Rudolph Virchow suggested that cranial suture fate was independent of the neurocranial environment. He presumed that the osteogenic fronts of the calvarial suture possessed the autonomous capacity (i.e., independent of interactions with the dura mater or brain) to fuse or remain patent. Virchow based his deductions on the work of Sommering (1800), who first described cranial suture anatomy and proposed that premature suture fusion could alter the head shape. Virchow’s etiopathogenic theory also benefitted from the work of Otto, who used Sommering’s observations to develop the hypothesis that suture fusion in one region on the cranium leads to compensatory overgrowth in another. Virchow’s primary contribution was to expand and refine Otto’s proposal and provided more conclusive support for what is now known as Virchow’s law: premature suture fusion results in compensatory skull growth parallel to the fused suture and a decreased growth perpendicular to the suture (Figure 22.1).
Virchow’s hypothesis of skull maldevelopment remained unchanged for nearly 70 years until 1920, when Park and Powers postulated that craniosynostosis was caused by a primary defect in the cranial suture mesenchymal blastema. They alleged that an embryologic defect in the cranial suture mesenchyme leads to premature fusion. Their theory was pervasive until Van der Klaauw (1946) and Moss (1959) suggested that the dura mater acted as a conduit or “functional matrix” for cranial base biomechanical forces. Accordingly, transmitted tension from an abnormal cranial base would presumably alter normal cranial suture physiology. For example, in coronal synostosis, spatially malformed lesser sphenoidal wings were hypothesized to transmit aberrant tensile force upward through dural fiber tracts leading to premature fusion of the overlying cranial suture. In sagittal synostosis, abnormalities in the cribriform plate and crista galli could generate forces that, at the points of dural attachment, would promote premature suture fusion.
Contemporary research has fundamentally changed our understanding of cranial suture fate. Based on a wealth of evidence, it appears that conserved signaling pathways mediate cranial suture fate. Numerous in vitro and in vivo models have demonstrated that the subjacent dura mater shapes the cranial suture complex by temporally and spatially supplying growth factors (e.g., fibroblast growth factor-2) and cellular elements (e.g., osteoblastic cells) to the overlying osteogenic fronts and suture mesenchyme. Genetic findings in human syndromic and nonsyndromic craniosynostoses indirectly support this hypothesis. Using positional cloning, candidate gene approaches, and comparative genomic hybridization techniques, over 100 mutations have been identified in genes such as TWIST, NELL-1, MSX2, GLI3, AND FGFR1-3.1 Exactly how these mutations cause craniosynostosis is still being elucidated.3 The link between cranial suture fusion and facial hypoplasia appears to occur through a secondary cascade of growth impairment that extends from the cranial base through the facial skeleton. Findings by Mooney and others suggest that the calvarial dysmorphology can drive the basicranial and midface changes. Further supporting evidence for the primacy of cranial vault pathology comes from clinical observations by Marsh, Vannier, and others that early cranial vault remodeling can sometimes lessen the severity of cranial base and facial abnormalities.
The sequelae of craniosynostosis include both physical deformity and insufficient cranial volume to permit normal brain growth and development. While the effects of synostosis on brain development are unclear, some studies demonstrate neurocognitive deficiencies in children with synostosis; the etiopathogenesis of these neurocognitive impairments, however, remains unknown and may be the result of, or simply associated with, the fusion of a cranial suture.
HISTORY OF TREATMENT
The work of Otto and Virchow on the role of the cranial sutures in normal and abnormal calvarial growth provided the basis for early operative treatment of craniosynostosis. The first recorded operations removed the offending suture in an attempt to release the constricted brain. In 1890, Lannelogue described bilateral strip craniectomies for the correction of craniosynostosis. Lane subsequently described a similar and successful procedure in a 9-month-old infant with microcephaly. Two years later, Jacobi reported poor outcomes and high
morbidity and mortality in 33 patients with craniosynostosis treated with open strip craniectomy. He attributed these untoward consequences to major blood loss associated with the extensive surgical exposure. Interestingly, some authorities dispute whether many of these infants actually suffered from craniosynostosis and contend that some had microcephaly as a consequence of poor brain development. Either way, these “brain-releasing” procedures were abandoned in infants until 1927 when Faber and Town presented their successful experience using open craniectomy to treat severe forms of craniosynostosis in young infants. The success of these surgeons led to an acceptance of more extensive operative treatments that persist today.
morbidity and mortality in 33 patients with craniosynostosis treated with open strip craniectomy. He attributed these untoward consequences to major blood loss associated with the extensive surgical exposure. Interestingly, some authorities dispute whether many of these infants actually suffered from craniosynostosis and contend that some had microcephaly as a consequence of poor brain development. Either way, these “brain-releasing” procedures were abandoned in infants until 1927 when Faber and Town presented their successful experience using open craniectomy to treat severe forms of craniosynostosis in young infants. The success of these surgeons led to an acceptance of more extensive operative treatments that persist today.
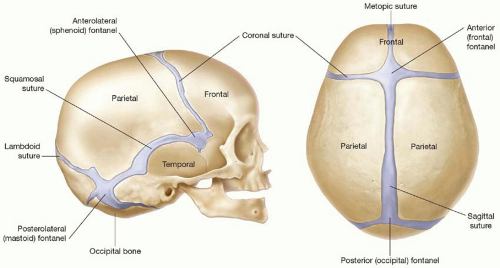 FIGURE 22.1. Schematic drawing of metopic, sagittal, coronal, and lambdoidal cranial sutures. The confluence points of the cranial sutures form the anterior, posterior, anterolateral (sphenoid), and posterolateral (mastoid) fontanelles. The fontanelles close sequentially and the sutures function as growth centers. The metopic suture fuses by 8 months of age in nearly all children. The remaining sutures fuse late in life. Virchow’s law states that premature suture fusion results in compensatory skull growth parallel to the fused suture and a decreased growth perpendicular to the suture. |
As anesthetic and blood management techniques improved, many surgeons became dissatisfied with the unpredictable results of simple suturectomy and began to use more extensive reshaping techniques. In 1967, Tessier presented his experience with cranial vault remodeling procedures that involved segmental bone removal, remodeling, and stabilization. The operations he described were much more extensive than the any previous methods; Tessier’s operations required more operative time, more blood loss, and observation in an intensive care unit. Nevertheless, because the bone segments were directly contoured and stabilized to achieve the desired shape, these operations generally had more predictable outcomes than simple release procedures. Furthermore, they could be done at any age since they did not rely on brain expansion to improve the cranial form or volume. These techniques remain the gold standard in most large craniofacial centers.
Recently, several less invasive methods to treat craniosynostosis have been introduced. In 1999, Jimenez and Barone presented their experience with endoscopic strip craniectomy and postoperative orthotic helmet therapy. The suturectomy was performed with the assistance of an endoscope through small incisions. The patients were fitted with a postoperative cranial orthosis to guide changes in cranial shape. The authors reported significant reductions in blood loss and transfusion, hospital stay, and cost compared with larger cranial vault remodeling procedures. Other authors have noted similar results.4 The effectiveness of this procedure, however, is limited to infants in the first several months of life and the outcomes of certain types of craniosynostosis, such as metopic, can be variable.2 In 2003, Lauritzen introduced the use of internal spring distractors to improve cranial shape.5 When compared with a modified pi procedure for sagittal synostosis, spring-mediated skull reshaping had comparable clinical outcomes with appreciably less morbidity. The application has been expanded to most forms of craniosynostosis with good reported outcomes.6 Since this procedure does not rely on the brain to expand the bone segments, it can be done successfully in older infants who would not be candidates for simple suturectomy.
The addition of three-dimensional computed tomography (CT), computer-guided modeling, improved pediatric anesthesia and blood conservation/salvage techniques, critical care, and intraoperative monitoring have improved the safety and effectiveness of craniofacial surgery and decreased the morbidity and mortality. Fixation using resorbable plates and screws has also greatly improved the stability and longevity of the correction. The adaptation of distraction osteogenesis to the craniofacial skeleton plays a small role in skull remodeling, but it has radically altered and expanded our surgical armamentarium for the treatment of midface hypoplasia in syndromic patients.
CRANIAL ANATOMY AND THE DEVELOPMENT OF ANOMALIES
The morphogenetic path between craniofacial embryogenesis and pathogenesis is extremely narrow. Cephalic development involves exceedingly complex mechanisms built on conserved elements that have undergone enormous evolutionary change. Cranial plates of the membranous neurocranium develop
through the coalescence of ossification centers that arise from the primitive mesenchyme overlying the brain (membranous ossification). The majority of the cranial base, or chondrocranium, begins as a cartilaginous anlage that becomes ossified gradually during embryologic development (endochondral ossification). Cranial sutures and fontanelles are the mesenchyme that persists between the calvarial plates. The major cranial sutures are the metopic, sagittal, coronal, and lambdoid (Figure 22.1). A list of minor sutures includes the temporosquamosal, frontonasal, sphenoethmoidal, and frontosphenoidal.
through the coalescence of ossification centers that arise from the primitive mesenchyme overlying the brain (membranous ossification). The majority of the cranial base, or chondrocranium, begins as a cartilaginous anlage that becomes ossified gradually during embryologic development (endochondral ossification). Cranial sutures and fontanelles are the mesenchyme that persists between the calvarial plates. The major cranial sutures are the metopic, sagittal, coronal, and lambdoid (Figure 22.1). A list of minor sutures includes the temporosquamosal, frontonasal, sphenoethmoidal, and frontosphenoidal.
The cranial sutures are important for two reasons. First, they allow the head to deform during parturition so that the infant can pass through the pelvis. Second, the sutures couple rapid brain expansion early in life to the growth of the cranium. Coordinated allometric growth of the cranium is achieved through a series of tissue interactions between the brain, dura mater, suture mesenchyme, and calvarial bones. Growth of the cranium is passive and occurs in response to outward expansion of the brain. This creates tension across the sutures and stimulates formation of new bone along the edge of the adjacent bony plates (osteogenic fronts of the cranial sutures). Thus, the cranial sutures permit the cranium to grow and expand as rapidly as the underlying brain.
Craniosynostosis can impair brain growth and development. Normally, the brain attains about 83% of its final volume by 2 years of age and the remaining 17% is acquired between 2 and 8 years of age.7 With the exception of the metopic suture, which normally closes by 8 months of age, the other cranial sutures are patent during this period.3 If a single suture fuses prematurely, compensatory growth in the remaining patent sutures will lead to alterations in cranial shape (as predicted by Virchow’s law) but rarely leads to significant neurologic impairment. Single-suture fusions are occasionally associated with elevated intracranial pressures (ICPs) (-7.7%) and subtle learning disabilities that go largely unrecognized.8 The most common single suture to fuse prematurely is the sagittal suture, followed by the coronal, metopic, and lambdoid. Although most patients with single-suture synostosis have no associated syndrome or identifiable genetic cause, approximately 25% of patients with unilateral coronal synostosis will have a causative mutation (FGFR3 Pro250Arg, FGFR2, TWIST, EFNB1, and NELL-1) and nearly 30% of patients with metopic synostosis have an associated syndrome or a chromosome abnormality.1 Furthermore, familiar patterns of inheritance have been observed in sagittal synostosis. Thus, it is inaccurate to use the terms nonsyndromic and syndromic craniosynostosis synonymously with single and multiple-suture craniosynostosis.
Multiple-suture fusions significantly raise the possibility of cerebral compression and developmental effects (see section on Increased Intracranial Pressure). The most common multiple-suture fusion is bilateral coronal, but other unusual patterns have been described. Multiple-suture fusion that occurs early in utero can result in a cloverleaf, or Kleeblattschädel, deformity. In rare instances, all of the cranial sutures can be patent at birth and fuse later in infancy, a process termed progressive postnatal pansynostosis. These infants have small, but normally shaped heads and the only clinic sign may be a relentless decline in head circumference percentile. Most patients with multiple-suture fusions have an associated syndrome and a molecular basis for their craniosynostosis. The most common mutation associated with bilateral (and unilateral) coronal synostosis is the FGFR3 Pro250Arg mutation (Muenke syndrome), followed by mutations in TWIST (Saethre-Chotzen syndrome), FGFR2 (Apert, Crouzon, and Pfeiffer syndromes), and EFNB1 (craniofrontonasal malformation). The incidence of developmental and neurocognitive problems in this group is much higher than is seen in patients with single-suture fusion. It is unclear if this observation is a result of the craniosynostosis, or the effect of the genetic aberration on brain development.
FUNCTIONAL ASPECTS
Increased Intracranial Pressure
It has long been observed that changes in calvarial shape can induce compensatory changes in the shape of the underlying brain. With premature fusion of the cranial sutures and continued brain growth, surgeons have long speculated that such a mismatch in shape and volume between the cranium and the brain could lead to elevated ICP and neuropsychosocial retardation. Lannelongue (1890) suggested that craniosynostosis resulted in microcephaly with secondary mental retardation. He thought that excision of the fused sutures could reverse or prevent intellectual impairment. Shillito and Matson also advocated craniectomy in infancy to prevent elevated ICP and subsequent brain damage. Marchac and Renier measured the ICP in 121 craniosynostosis patients with an epidural sensor.8 They detected elevated ICP in 42% of patients with multiple-suture involvement and in 7% to 13% of patients with single-suture involvement.8 They noted a decrease in ICP in patients who underwent cranial surgery.8 Gault et al. also demonstrated that raised ICP was most frequent in those children with more than one suture fused prematurely (complex, oxycephaly, Crouzon, brachycephaly, and Apert syndromes).
Craniocerebral disproportion, however, is not the only cause of elevated ICP in patients with craniosynostosis. Sleep apnea resulting from midfacial retrusion can induce episodic nocturnal elevations in ICP secondary to the dilating effects of hypercapnia on the cerebral vasculature. Another potential cause is venous hypertension resulting from stenosis or complete closure of the sigmoid/jugular sinus complex.
The gold standard for detecting elevated ICP is direct monitoring. Intraparenchymal and intraventricular monitoring is more accurate than epidural measurements. The reliability of lumbar puncture is questionable. One difficulty in interpreting these numbers is that they fluctuate significantly with patient position, activity, blood pressure, and sleep. The most meaningful results are obtained when patients are monitored for a period of time, usually overnight. Significant elevations (>20 mm Hg) have been considered an absolute indication for intracranial expansion. Nevertheless, interpreting the significance of borderline pressure elevations (15 to 20 mm Hg) has been more problematic, and there is little consensus even among neurosurgeons.
Direct ICP monitoring is invasive and rarely used for routine screening. Moreover, the measurement is only a snapshot in time: a normal pressure measurement early in life does not imply that it will remain so as the brain continues to grow. Consequently, many surgeons resort to less invasive, but less reliable, indicators of ICP. Conventional clinical symptoms of acute ICP elevation, such as headache, somnolence, and dizziness, are often lacking even in affected children. Papilledema and subsequent optic atrophy is strongly suggestive of elevated ICP, but has limited sensitivity in children under 8 years of age. Findings can include blurring of the disk margins and obliteration of the optic cup, elevation of the nerve head (“champagne cork” appearance), capillary congestion, hyperemia, venous engorgement, loss of venous pulse, peripapillary exudates, retinal wrinkling, and punctate nerve fiber layer hemorrhages. As optic atrophy progresses, the disk becomes pale, the capillaries and hyperemia disappear, and significant secondary arteriolar narrowing occurs. Reducing ICP can reverse early changes, but more advanced degeneration may be permanent.
Radiographic evidence suggestive of elevated ICP includes loss of subdural space, often with effacement of the basal cisterns and vertex sulci, ventricular compression, and scalloping of the cranial endocortex. This latter finding has been termed the “copper-beaten” skull and can be visualized on both conventional radiography and CT. It is a late finding caused by pressure remodeling of the inner table of the skull
by the gyral convolutions. The predictability of this finding for ICP elevation has been questioned. While CT is the standard imaging technique, new imaging modalities are on the horizon. For example, one day it may be possible to diagnose and monitor increased ICP using transcranial ultrasound and resistive index calculations to assess the peak systolic and diastolic velocities of the major cerebral vasculature (these velocities increase as ICP rises). Similarly, magnetic resonance elastography may also be used in the future to measure ICP.
by the gyral convolutions. The predictability of this finding for ICP elevation has been questioned. While CT is the standard imaging technique, new imaging modalities are on the horizon. For example, one day it may be possible to diagnose and monitor increased ICP using transcranial ultrasound and resistive index calculations to assess the peak systolic and diastolic velocities of the major cerebral vasculature (these velocities increase as ICP rises). Similarly, magnetic resonance elastography may also be used in the future to measure ICP.
Hydrocephalus
Hydrocephalus is an infrequent finding in craniosynostosis. It is more common in patients with Crouzon syndrome. CT scans provide an accurate and noninvasive method of assessing ventricular size; however, assessment of ventricular size alone may not provide a true picture of hydrocephalus. For instance, ventriculomegaly is a common finding in patients with Apert syndrome, but is usually unrelated to increased ICP. More consistent findings include elevation of ICP on direct monitoring, the presence of enlarged or enlarging ventricles by serial CT scans, and periventricular lucency resulting from transependymal flow of cerebrospinal fluid (CSF).
Mental Impairment
Children with craniosynostosis can have cognitive delay and learning disability. However, intellectual development and learning are affected by many variables, including the presence of an associated syndrome, concurrent ICP elevation or hydrocephalus, prematurity, or family history. Patients with single-suture fusion and without an associated syndrome generally have near normal intelligence, but, as noted above, they may exhibit subtle learning disabilities. Patients with an associated syndrome have a significantly higher incidence of cognitive delay than the general population. This is loosely correlated with the type of syndromic diagnosis, but there is typically wide variability within any given patient population.
It is still unclear if the neurocognitive findings in patients with craniosynostosis are the result of the deleterious effects of early growth restriction from the suture fusions, or if the molecular process that lead to the suture fusion negatively impacted central nervous system development. In support of the former contention, Marchac and Renier found that overall intelligence was better in patients who underwent an earlier cranial release compared with those who had a later procedure.8 The findings are somewhat limited by the fact that the study was not controlled or randomized. Conversely, Starr and coworkers demonstrated that surgery did not favorably affect neurocognitve development in patients with single-suture synostosis.9 The parameter studied, however, was developmental quotient, not a sensitive indicator of intellectual performance and of questionable validity in younger age groups. Similarly, Camfield and Camfield concluded that mental impairment (IQ < 70) in children with single-suture craniosynostosis was usually the consequence of a primary brain malformation rather than brain distortion from the craniosynostosis. A major limitation of most prior neurocognitive studies in this patient population is that the instruments most commonly used (e.g., Bayley Scales and IQ testing) lack sufficient sensitivity and specificity to detect subtle cognitive differences, such as perceptual abnormalities. More refined testing is needed to provide a more global and comprehensive understanding of cognitive function in these patients.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


