Fig. 5.1
Malar rash of acute cutaneous lupus erythematosus. Photo courtesy of Oleg Akilov, M.D., Ph.D.
Subacute cutaneous lupus erythematosus (SCLE) (Fig. 5.2) is an eruption that lasts longer than ACLE, but similar to ACLE it does not result in scarring, with a relatively good prognosis [1, 3], affecting photosensitive areas on the sides of the face, upper trunk, and extremities. Since lesions can be annular, with raised red borders and central clearing, or scaly patches, they can be confused with figurate erythemas or dermatophyte infections, and dermatomyositis can often mimic SCLE. SCLE can also be induced by a wide variety of medications including numerous antihypertensives, terbinafine, and an assortment of other medications [2, 3].
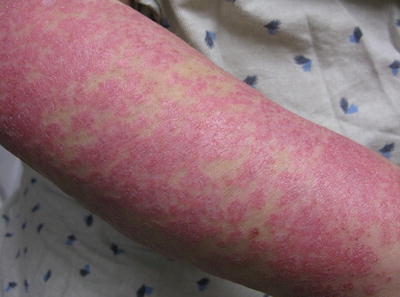
Fig. 5.2
Scaly erythematous plaques on the forearms consistent with SCLE. Photo courtesy of Joseph C. English, III, M.D.
Chronic cutaneous lupus erythematosus (CCLE) is subdivided into four subgroups: DLE, LET, LP, and chilblain lupus. DLE (Fig. 5.3) is characterized by chronic and inflammatory discoid lesions, which can lead to permanent disfiguring scars. It frequently involves the head (i.e., ears, scalp, face) and less frequently the mucosal surfaces (i.e., ocular, nasal, oral, and genital). Active lesions are more indurated, while healing lesions are more atrophic, which can heal with hypopigmentation or hyperpigmentation. One complication of DLE is development of squamous cell carcinoma within the lesions. DLE lesions are 1 of the 11 American Rheumatism Association (ARA) criteria for SLE and may be a presenting sign of SLE [2]. Approximately 5–10 % of patients will have a DLE-SLE overlap, with a relatively benign but chronic course [3].
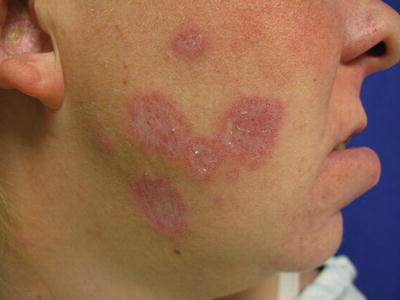
Fig. 5.3
Atrophic plaques with hypopigmentation and scarring characteristic of DLE. Photo courtesy of Joseph C. English, III, M.D.
A second subgroup of chronic lupus is LET (Fig. 5.4), which is seen less commonly. LET is characterized by indurated lesions, located on the sun exposed regions of the face and trunk [4]. Lesions can be reproducible with phototesting [5] and resolve without scarring or atrophy. LET is believed by some to be related to Jessner lymphocytic infiltrate [6], which is a benign lymphocytic infiltrate of the skin with similar clinical annular plaques and central clearing.
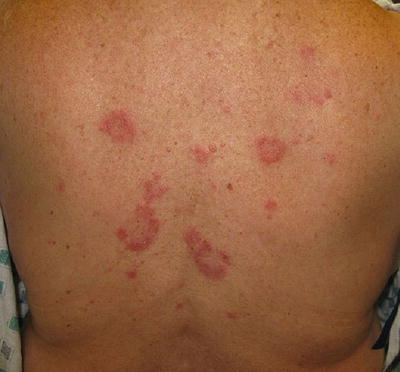
Fig. 5.4
Lupus erythematosus tumidus. Indurated erythematous plaques on the trunk. Photo courtesy of Joseph C. English, III, M.D.
The third subgroup of chronic lupus is lupus panniculitis (LP), an inflammation of the fat that results in erythematous indurated plaques characteristically located on the face, upper trunk, and arms, as well as thighs and buttocks [7]. Ulceration of the lesions has been reported in 6–28 % of patients in different studies, though to be caused by impaired circulation as a result of fibrin deposits or tissue hyaline necrosis of fat [7]. These lesions may result in lipoatrophy creating depressed skin regions (Fig. 5.5) [2, 7]. It is important to distinguish LP from the fatal subcutaneous panniculitis-like-T-cell lymphoma [8].
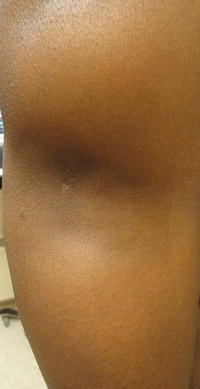
Fig. 5.5
Lupus panniculitis in the arm resulting in lipoatrophy. Photo courtesy of Oleg Akilov, M.D., Ph.D.
Chilblain lupus erythematosus (CHLE) or SLE pernio represents the fourth subgroup of chronic cutaneous lupus, with characteristic dusky purple papules and plaques on the acral surfaces (i.e., fingers, toes, nose) (Fig. 5.6). CHLE is triggered by cold, and moist or damp conditions. Compromised perfusion can result in partially necrotic and malodorous superinfected ulcerations [9]. Some lesions with time may develop into a discoid lesion [2] and approximately 18 % may progress to SLE [9]. It is also possible that a patient may have concurrent chilblains (also known as perniosis), which is a cold-sensitive disorder resulting from abnormal inflammatory response to cold thought to be vascular in origin. CHLE can be sporadic or inherited via an autosomal dominant missense mutation. There are some reports describing tumor necrosis factor alpha (TNF-α)-antagonist-induced chilblain lupus [10].
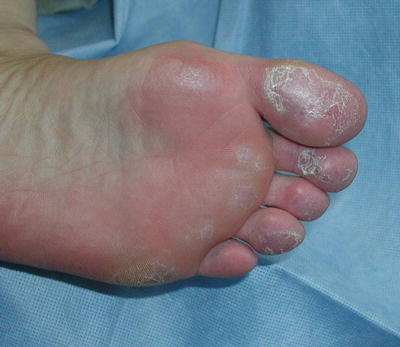
Fig. 5.6
Chilblain lupus. Without knowing the clinical history, chilblain lupus can be difficult to distinguish from simple cold-induced injury. Seen here are the dusky purple changes typical for this condition. Photo courtesy of Julia R. Nunley, M.D.
Additional, nonspecific lesions can be associated with, but not limited to, SLE. Blisters can be seen in lupus patients, which could represent bullous lupus erythematosus (BLE) lesions (found to be associated with active LE) (Fig. 5.7) or could represent a concurrent disease process (i.e., pseudoporphyria, immunobullous diseases, etc.) [3]. Patients with BLE have a good response to treatment with oral dapsone and often have antibodies to type VII collagen. Other nonspecific lesions seen in LE patients are vascular lesions [2], and low complement levels of C3 and C4 point to connective tissue vasculitis. Systemic involvement should be considered in patients with cutaneous lupus and other associated findings, such as livedo reticularis (“lacy” vascular pattern on extremities) (Fig. 5.8), Raynaud phenomenon (cyanotic finger tips or other acral sites, due an exaggerated vascular response to cold/emotion) (Fig. 5.9a, b), erythema of the palms and periungual telangiectasias. Other clues of systemic disease are purpuric lesions, ulcers and urticarial papules, and atrophie blanche (hypopigmented atrophic scars, usually on lower extremities) (Fig. 5.10) [2]. Of note, findings of livedo reticularis, thromboses, and ulcerations are also associated with antiphospholipid antibodies. Also, keep in mind syndromes that may be associated with some of these findings, such as Sneddon syndrome (associated with livedo reticularis and ischemic central nervous system (CNS) disease) as well as Hughes syndrome (associated with antiphospholipid syndrome).
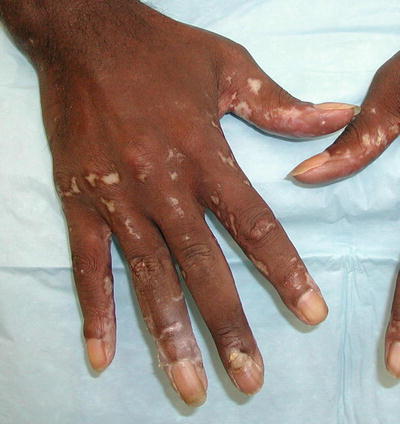
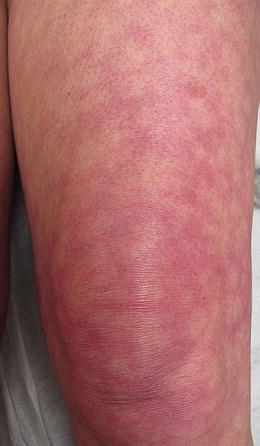
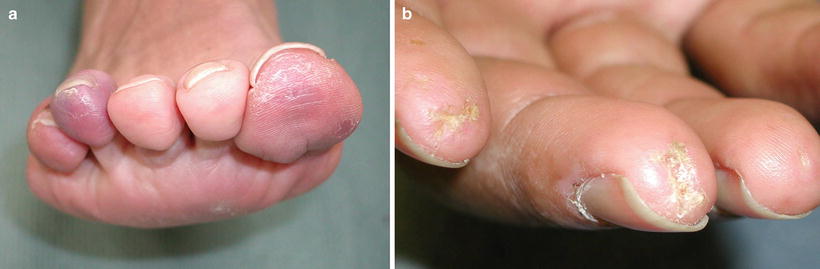
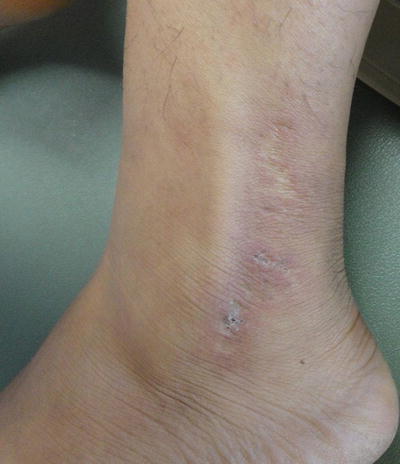
Fig. 5.7
Bullous lupus. Seen in this image are various stages of bullous lupus; some lesions are still vesicular, whereas others have resulted in a scar. Photo courtesy of Julia R. Nunley, M.D.
Fig. 5.8
Livedo reticularis. Lacy vascular pattern on extremities. Photo courtesy of Joseph C. English, III, M.D.
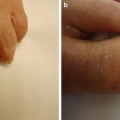
Fig. 5.9
Raynaud phenomenon is most common in patients with scleroderma. Seen here are two of the classic changes. Image a demonstrates the dusky blue that is transient; image b is a result of the chronic ischemia with skin necrosis. Photo courtesy of Julia R. Nunley, M.D.
Fig. 5.10
Hypopigmented stellate scarring on the lower extremities consistent with atrophie blanche. Photo courtesy of Joseph C. English, III, M.D.
Alopecia occurs in as high as 45 % of patients with lupus [1]. Scarring alopecia is more common with discoid LE lesions, whereas diffuse nonscarring alopecia is more associated with systemic disease (Fig. 5.11). Overall, lupus patients have increased risk of alopecia areata [11]. Moreover, systemic medical therapy for lupus may be responsible for some of the nonscarring alopecia [1]. Additionally, patients with lupus have periungual capillary abnormalities, but these are less prominent than seen in scleroderma or dermatomyositis [2].
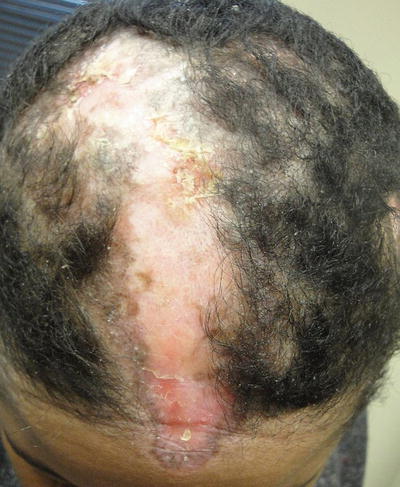
Fig. 5.11
Scarring alopecia in the setting of discoid lupus erythematosus. Photo courtesy of Joseph C. English, III, M.D.
Differential Diagnosis
The differential diagnoses for cutaneous lupus lesions depend on the subtype of lupus noted on exam [2]. Given that acute cutaneous lupus often presents with the malar rash, keep in mind the differential of other common entities, such as rosacea, atopic or seborrheic dermatitis, as well as phototoxic and photoallergic drug reactions, dermatomyositis, and pemphigus erythematosus. Annular scaly lesions of subacute cutaneous lupus can look like psoriasis, fungal infections, figurate erythemas (e.g., erythema annulare centrifugum), atopic or contact dermatitis, and dermatomyositis. Facial discoid lupus can look similar to tinea, lichen planus, and lichen planopilaris, Jessner lymphocytic infiltrate, polymorphous light eruption, sarcoidosis, lupus vulgaris, non-melanoma skin cancer and other lymphocytic infiltrations, granuloma faciale, and fungal infections. For the differential of LET, consider Jessner lymphocytic infiltrate, reticular erythematous mucinosis and polymorphous light eruption. When considering LP, one should consider other panniculitidies, such as erythema nodosum, subcutaneous panniculitis like T cell lymphoma. Finally, it is imperative to exclude drug-induced lupus, which is a systemic reaction that manifest with a rash, arthralgias and fevers, but which does not have CNS and kidney involvement seen with SLE, and which commonly resolves with discontinuation of the inciting drug.
Workup and Laboratory Abnormalities
Workup for cutaneous lupus includes performing a lesional punch biopsy for hematoxylin and eosin (H&E), and performing direct immunofluorescence (DIF), if histology is not definitive. If the skin biopsy is consistent with lupus, one needs to evaluate for systemic disease by questioning the patient about history of arthritis, noting any associated cutaneous findings on physical exam (discussed above) and checking for lymphadenopathy. Additional testing includes a urinalysis (UA), and blood tests such as complete blood count with diff, blood urea nitrogen (BUN), creatinine, antinuclear antibody (ANA) with profile anti-double stranded (ds) deoxyribonucleic acid (DNA) and anti-smooth muscle (Sm) antibodies, complement levels (C3, C4), and erythrocyte sedimentation rate (ESR). A diagnosis of SLE will be based on fulfilling the required 4 of 11 criteria [12, 13]. Of note, an inherited complement deficiency should be considered in a child with severe cutaneous disease and glomerulonephritis [14–16].
Histopathology
On histopathological evaluation there is presence of basal layer cell damage, lymphohistiocytic inflammation at the interface of dermal and epidermal junction, and periadnexal inflammation (Fig. 5.12). Overall, there is variable follicular plugging and scarring, but discoid lesions have more epidermal atrophy and follicular plugging [2, 17]. There is prominent dermal mucin deposition seen in LE tumidus, and LP has inflammation of the subcutis. Even though DIF evaluation of skin is not necessary for diagnosis, when it is performed one will see granular immunoglobulin G (IgG) and/or immunoglobulin M (IgM) antibody deposition at dermal–epidermal junction in lesional and nonlesional skin, and hair follicles.
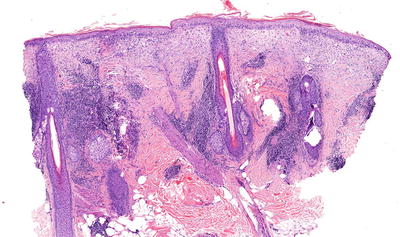
Fig. 5.12
Photomicrograph of a skin biopsy demonstrating basal layer cell damage with lymphohistiocytic inflammation and periadnexal inflammation. Photo courtesy of John Vu, M.D., Ph.D.
Laboratory Abnormalities
Abnormalities may be seen in the laboratory studies listed above in section labeled workup. In addition, appropriate laboratory studies should be ordered to evaluate for the possibility of antiphospholipid syndrome.
Treatment
When choosing the appropriate treatments for cutaneous lupus, which include both topical and systemic agents, one has to weigh the benefit and the side effect profile of the medications. Topical steroids are often safe to use, from mild potency steroids such as desonide 0.05 % ointment that can be used on the face, to moderate potency steroids such as triamcinolone 0.1 % ointment that can be used on the rest of body, both applied twice a day. Intralesional steroids (such as triamcinolone in a concentration of 10 mg/mL) are safe to use for active discoid and tumid lesions, sparing the patient side effects of systemic steroids. Of note, discoid lupus may be the rare instance when high potency steroids (such as clobetasol 0.05 % ointment) can be used on the face [2]. There is anecdotal evidence for use of topical immunomodulators such as calcineurin inhibitors or topical retinoids in cutaneous lupus [18–20]. Intravenous immunoglobulin (IVIG), systemic immunosuppressant therapy (see below), and biologic therapies such as belimumab (the first Food and Drug Administration (FDA)-approved targeted therapy for SLE) have been shown to have efficacy in mild to moderate disease [21].
Treatment of cutaneous lupus and associated systemic disease is accomplished with several agents [2]. The gold standard for systemic therapy are antimalarial agents such as hydroxychloroquine (200 mg PO daily to twice a day, max 6.5 mg/kg ideal body weight/day), chloroquine (125–250 PO daily; up to 3.5–4 mg/kg ideal body weight/day), or quinacrine (100 mg PO daily). For cutaneous disease that is resistant to antimalarials, one could use oral retinoids (e.g., acitretin, isotretinoin), thalidomide (initiate at 50–100 mg PO daily; after desired response drop to maintenance dose of 25–50 mg daily or if possible, every 3–4 days), or dapsone (for bullous SLE). Immunosuppressive agents such as systemic corticosteroids, mycophenolate mofetil, azathioprine, and sulfasalazine are used as well. Finally, immune response modifier agents used for treatment of cutaneous lupus resistant to other therapies are rituximab, abatacept, belimumab, and anti-interleukin (IL)-6 antibody and anti-IL-10 antibodies.
Patients should also be educated about sun protection and avoidance, which can act as triggering and exacerbating factors in the disease. Smoking has been shown to correlate with resistance to antimalarial therapy, and thus smoking cessation should be encouraged.
Prognosis
The prognosis of cutaneous lupus depends on the subtype, the associated progression of systemic involvement, and response to treatment. Patients with CCLE or SCLE primarily have skin involvement and generally lack organ involvement, which usually indicates a benign course [3]. Patients with ACLE have the highest risk of internal disease involvement; however, patients with any type of cutaneous LE can develop systemic involvement. Cutaneous diseases that are refractory to antimalarials are likely to be refractory other treatments as well [2]. Scarring is seen more commonly in SLE and discoid lupus, whereas is not typically seen in ACLE, SCLE, tumid lupus and LP. Late-onset SLE is associated with a worse prognosis [22].
Granulomatosis with Polyangiitis (Wegener Granulomatosis)
Introduction
Granulomatosis with polyangiitis (GPA), previously known as Wegener granulomatosis, is one of the antineutrophil cytoplasmic antibody (ANCA) associated vasculitides, which also includes microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA, previously known as Churg–Strauss syndrome). GPA is characterized by a triad of: (1) systemic vasculitis of skin and oral mucosa, (2) necrotizing granulomatous inflammation of the upper and lower respiratory tracts, and (3) pauci-immune glomerulonephritis [23–25]. GPA has a high mortality rate if untreated, and there is variation in severity, from focal involvement (i.e., only airway disease) to systemic or generalized disease (multiorgan) [25]. GPA has a predilection for white middle aged adults, affecting 5–12 cases per million and has a slight female predominance [2]. Primary systemic vasculitis is exceedingly rare in children; however, GPA is one of the most common vasculitides seen in this population [26].
Pathogenesis
The pathogenesis of GPA is thought to be multifactorial, and is influenced by the presence of ANCAs and environmental, as well as genetic factors. ANCA (particularly anti-PR3) have been shown to activate neutrophils presenting specific antigens (possibly infectious), activating alternative complement pathway and furthering an inflammatory vicious cycle that eventually leads to granuloma formation [27]. There are reports of Staphylococcus aureus nasal carriage correlating with disease relapse, which may play a role in nasal/pulmonary pathogenesis of GPA [28, 29]. ANCA antibodies are seen in 85–95 % of affected patients [30].
Clinical
Diagnosis of GPA can be made based on the 1990 ACR criteria, when two of the following four criteria are present: (1) nasal or oral inflammation, (2) abnormal chest radiograph, (3) abnormal urinary sediment (microhematuria >5 red blood cells per high-power field or red blood cell casts in urine sediment), and (4) granulomatous inflammation on biopsy. ACR criteria may not be appropriate for children with vasculitis; as such the European League Against Rheumatism/Pediatric Rheumatology European Society (EULAR/PRES) has proposed some additional criteria including not only nasal, but sinus inflammation, imaging studies to include chest computed tomography (CT), as well as other criteria. However, the new criteria have shown no improvement over the ACR criteria in disease classification [31].
Cutaneous involvement occurs in 46–66 % of GPA patients [32], but only 10 % of patients have it as a presenting feature [2]. The most common lesions are palpable purpura (Fig. 5.13a, b), followed by oral ulcers and red, friable gingival tissue and hyperplastic gums (“strawberry gums”). Oral involvement can be seen in 10–62 % of patients, but may be a presenting sign in 5–10 % of patients in GPA [32, 33]. Ten percent of patients will have painful subcutaneous nodules and ulcers found on the elbows, as well as the face and scalp, which resemble pyoderma gangrenosum. They may be mistaken for rheumatoid nodules, but GPA lesions can ulcerate unlike rheumatoid nodules (Fig. 5.14) [32].
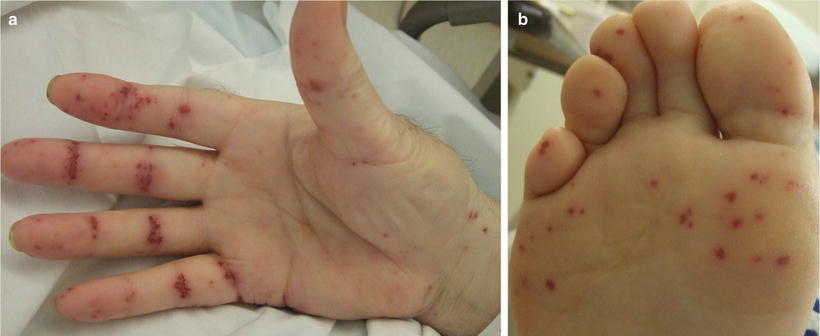
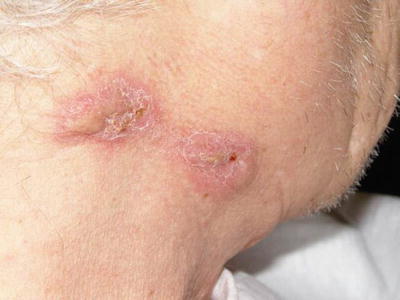
Fig. 5.13
(a, b) Palpable purpura on the extremities of a patient with granulomatosis with polyangiitis (Wegener granulomatosis)
Fig. 5.14
Subcutaneous nodules on the face of a patient with granulomatosis with polyangiitis (Wegener granulomatosis)
Airway involvement of the upper or lower respiratory tracts affects between 60 and 90 % of patients [2, 32], with more than 70 % of cases presenting with nasal, sinus, tracheal, and ear symptoms. Other signs and symptoms include recurrent epistaxis, mucosal ulcerations, nasal septal perforation, and saddle nose deformity. Furthermore, pulmonary involvement includes hemoptysis, dyspnea, cough, or pleuritis, with infiltrates and nodes on chest radiograph [2, 32]. Renal disease affects 18 % of patients at presentation and 77 % of patients eventually develop glomerulonephritis [23]. Necrotizing vasculitis of GPA also affects other organs, causing musculoskeletal (70 %), ocular (30–60 %), neurologic (20–50 %), and gastrointestinal (GI) (5–10 %) disease [32].
Differential Diagnosis
The differential diagnosis of GPA includes other ANCA-associated vasculitides, especially EGPA, which is another necrotizing granulomatous vasculitis. EGPA can also present with the papulonecrotic lesions seen in GPA, but on biopsy the former has eosinophils and pink instead of blue necrobiosis as well as evident peripheral eosinophilia (>109/L) [32].
Workup and Laboratory Abnormalities
The workup for GPA includes inflammatory labs, such as erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP), as well as CBC to check for anemia and elevated white blood cell count [2]. Fifty percent of patients have a positive rheumatoid factor. Cytoplasmic ANCA (c-ANCA) with anti-proteinase 3(anti-PR-3) specificity and antimyeloperoxidase (anti-MPO or p-ANCA) antibodies should be ordered. C-ANCA with anti-PR3 is positive in 75–80 % of patients with classic or severe GPA, whereas ANCA negative patients usually have focal disease and better prognosis [32].
Histopathology
Treatment
The goal of treatment of GPA is prompt recognition and early institution of treatment to prevent severe complications. Standard treatment of active severe GPA includes systemic glucocorticoids (1 mg/kg/day of prednisone) plus oral daily cyclophosphamide for 3–6 months. This treatment results in a remission in up to 75 % of patients, improvement in 91 % and survival in 80 %, but relapses occur 50 % of the time [2, 23]. Treatment with cyclophosphamide is associated with several serious side effects, including but not limited to transitional carcinoma of the bladder in 6–15 % of patients, myelodysplasia, and skin cancer [34]. There is evidence for intravenous cyclophosphamide pulses inducing remissions, with multiple low-dose treatments may be required to prevent relapses [35].
Alternatives to cyclophosphamide for induction or maintenance therapy for classic GPA include azathioprine, methotrexate (MTX), and mycophenolate mofetil [36, 37]. Methotrexate has been shown to have similar efficacy in inducing remission as compared to cyclophosphamide in active but not severe disease [38], and has the same rate of relapse with lower toxicity as maintenance therapy [38, 39]. Besides MTX and cyclophosphamide, maintenance therapy can also be accomplished with azathioprine or mycophenolate mofetil for 2 years or more. However, azathioprine maintenance therapy has been linked to higher relapse rate in anti-PR-3 positive patients. There are also reports of use of IVIG or plasmapheresis in severe, refractory vasculitis or when contraindications to cyclophosphamide exist [34]. Trimethoprim–sulfamethoxazole is recommended during the immunosuppressive regimen, for Pneumocystis jiroveci pneumonia prophylaxis, and some believe to decrease relapses associated with S. aureus respiratory infections and nasal carriage.
There is also promising evidence for use of biologic agents in GPA. The combination of rituximab in addition to corticosteroids may be as effective as cyclophosphamide plus corticosteroids in inducing remissions, but there are unclear effects on relapses and granulomatous manifestation [34, 40, 41]. Infliximab has been used in addition to standard therapy with remission in 88 % and flares in 20 %, but was associated with more infectious complications in 21 % of patients [42]. The role of adalimumab with prednisolone and cyclophosphamide in a phase II study revealed encouraging results with similar efficacy and adverse events, and reduced prednisolone exposure in ANCA-associated systemic vasculitis with renal involvement [43]. Etanercept was not shown to be effective for the maintenance of remission and even associated with increased risk of malignancy [2, 37]. Mechanisms of action of above therapies involve immunosuppression via depletion of B cells or suppression of other immune cells.
Prognosis
Before the 1950s, no treatment for GPA was available and patients would die from pulmonary or renal complications after a median of 5 months [44], which was increased to 12 months after introduction of corticosteroids [45]. Today, the prognosis of GPA is considered good, given the recent advances in treatment, but depends on severity of the disease, organ involvement, and early diagnosis. Even though, remission is seen in 75 % of patient with standard therapy, at least half of patients still relapse, and many are afflicted with the associated malignancies and other adverse effects of their therapies. Further investigation needs to be done into the promising new therapies such as the biologics, with hopes to decrease toxicity and adverse events, as well as prolong remission rates.
Polyarteritis Nodosa (PAN)
Introduction
Polyarteritis nodosa (PAN) is a multisystem, medium sized vessel segmental necrotizing vasculitis, with either only cutaneous (also referred to as organ limited, benign, or cutaneous PAN) or with associated extracutaneous manifestations (also referred to as classic or systemic PAN).
Pathogenesis
The pathophysiology of PAN is unknown, but immune complex deposition has been proposed as one possible mechanism of pathogenesis [46]. PAN has many associations, including infections, like the hepatitis B virus (HBV) infection (seen in 7 % of PAN, and may be associated with increased gastrointestinal symptoms and worse prognosis [47, 48]), hepatitis C virus infection [49], streptococcal (in children), parvovirus B19 and human immunodeficiency virus (HIV) infection [2]. Malignancy, such as hairy cell leukemia, is another reported association [50]. Inflammatory conditions like inflammatory bowel disease (IBD), SLE, and familial Mediterranean fever have been observed with PAN, as well as certain medications, such as minocycline [2].
Clinical
PAN is a difficult diagnosis to make, given the nonspecific clinical features, controversial diagnostic inclusion criteria and low prevalence of the disease [51]. Several authors and reports categorized PAN into subtypes [52], including classic PAN (associated with hepatitis B and with more widespread disease [53]), ANCA negative PAN [46], benign cutaneous PAN (associated with fever, neutropenia and cutaneous ulcers, with possible association with IBD), Kawasaki disease associated [54], and MPA PAN (ANCA positive PAN in 10 % cases). Some authors argue that, even though PAN is a medium vessel vasculitis, there must still be some involvement of arterioles, capillaries or venules, which makes it all that more difficult to separate from entities such as MPA which includes small and medium sized vessels [51]. Several attempts have been made to reclassify vasculitides, but due to difficulty in diagnostic testing and other factors, no ideal criteria exist [52].
Symptoms of PAN result from ischemic damage to various vital organs, such as the skin, nervous system, muscle, heart, and kidney. Approximately 10 % of all PAN cases are cutaneous PAN or organ-limited PAN [2], which is distinguished from systemic PAN by lack of visceral involvement by angiography [51]. Cutaneous manifestations include palpable purpura (pustular or ulcerative), livedo racemosa, retiform purpura, “punched out” ulcers, subcutaneous nodules, and peripheral gangrene (Fig. 5.15) [52]. Cutaneous PAN is most commonly seen in children, which rarely progresses to systemic PAN [52], and is associated with streptococcal infections [55]. Skin involvement in PAN can be associated with symptoms of joint pain and ocular disease, as seen in MPA and GPA [2].
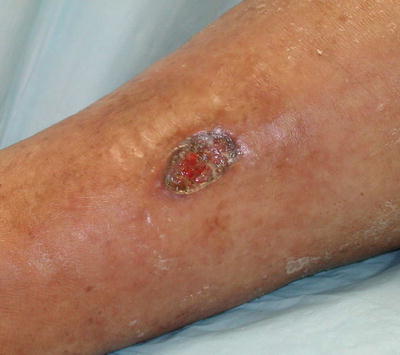
Fig. 5.15
Cutaneous polyarteritis nodosa. Histologic evaluation of this leg ulcer demonstrated changes diagnostic for polyarteritis nodosa; the systemic evaluation was negative. This lesion healed with low-dose corticosteroid therapy. Photo courtesy of Julia R. Nunley, M.D.
Twenty-five percent of patients with classic, or systemic, PAN can have the above mentioned cutaneous findings. Extracutaneous manifestations of systemic PAN have been described to include fever, arthralgias, myalgias (myopathy with increased CPK levels), paresthesias (“stocking and glove distribution,” mononeuritis multiplex foot drop or wrist drop), abdominal pain (sometimes from mesenteric ischemia), orchitis (associated with HBV infection), renovascular hypertension (HTN)/renal failure (not glomerulonephritis), congestive heart failure causing shortness of breath, and cerebral infarcts, but often spares the lungs [2]. A recent large single center retrospective study of pediatric PAN cases revealed most patient presented with fevers, fatigue, weight loss, and myalgias, with skin involvement in 88 %, musculoskeletal in 75 %, renal in 19 %, as well as gastrointestinal (GI) in 10 % and neurological manifestations in 10 % of patients [56]. This study also revealed possible novel link between severe GI disease and systemic PAN relapse, with relapse being lower in children than adults, pointing to the possibility that childhood PAN has a higher chance of permanent remission.
Differential Diagnosis
There is a broad differential for PAN, including cryoglobulinemic vasculitis and autoimmune connective tissue disease [2]. PAN should be a diagnosis of exclusion, after GPA, MPA and EGPA have all been excluded. Given that PAN can cause aneurysms, one needs to consider diseases with aneurysmal dilatation, such as atherosclerosis, fibromuscular dysplasia, lupus, emboli and left atrial myxoma, neurofibromatosis, and Ehlers–Danlos syndrome [2]. Also on the differential, are etiologies that cause cutaneous necrosis, such as emboli and disorders of vasculopathy. Finally, one needs to rule out nodular tuberculosis before starting immunosuppressive treatment.
Histopathology
The histology of PAN is characteristic for necrotizing vasculitis in affected skin located in the deeper dermis and subcutaneous fat, as well as leukocytoclastic vasculitis of small to medium sized vessels of visceral sites [17]. There is infiltration of neutrophils within and around arterial walls, with rare eosinophils. Vessel walls eventually become necrotic, and the development of aneurysmal dilatation is common due to vessel wall weakening. Perivascular deposits of C3, IgM, and fibrin can be seen on DIF [2].
Workup and Laboratory Findings
There is no test to definitively make the diagnosis of PAN, but rather tests are performed to rule out other conditions such as cryoglobulinemias, autoimmune connective tissue disease, MPA, GPA, and EGPA and to determine the extent of organ involvement. For example, high ANA titers are more suggestive of autoimmune connective tissue disease, and ANCA positivity is more suggestive of another condition such as GPA. Blood cultures are usually performed to exclude endovascular infections. Tissue biopsies of involved organs, including skin, muscle, nerve, kidney, and testes can be performed to support the diagnosis. Nonspecific leukocytosis, elevated ESR, and thrombocytosis can be seen on serum testing.
Imaging Studies
In addition to tissue biopsies, imaging is becoming increasingly used to aid with diagnosis. Angiography allows detection of microaneurysms in the renal, celiac, and mesenteric arteries [57], and CT or magnetic resonance imaging (MRI) can be used to visualize luminal narrowing or wall thickening, and even follow-up response to therapy [58].
Treatment
First line therapy for PAN involves systemic corticosteroids with prednisone, at a dose of 1 mg/kg/day, tapered slowly over 6–12 months [59]. Approximately half of patients are expected to have a complete response to systemic corticosteroids, while others may require additional immunosuppressants or immune-modulators [60]. Cyclophosphamide, at doses of 2 mg/kg/day PO or pulses IV of 0.5–1.0 g/m2/month, have been used successfully in patients with internal organ involvement or patients with steroid refractory disease [60, 61]. In patients with cutaneous limited disease, MTX has also been shown to be an option for treatment [62]. While not a first line treatment, there is evidence that tumor necrosis factor (TNF) inhibitors can also provide relief in refractory disease [63, 64]. Treatment of diseases associated with PAN, such as HBV infections, is also important in the management of these patients.
Prognosis
Prognosis in PAN depends on systemic involvement of the disease, timing of initiation of treatment, and ability to control the progression of systemic involvement. Without treatment there is a less than 20 % 5-year survival, and with treatment this can be improved to 80 % 5-year survival [65].
Henoch–Schönlein Purpura (HSP)
Introduction
Henoch–Schönlein Purpura (HSP) is a small vessel vasculitis, most often presenting with a tetrad of symptoms including purpura, arthritis, abdominal pain, and hematuria. It is thought to occur secondary to immunoglobulin A (IgA) deposition in vessel walls, often following respiratory tract infections in children. In children it is the most common vasculitis, and 90 % of HSP occurs in children less than 10 years of age.
Pathogenesis
Disease pathogenesis is secondary to IgA deposition in the venules of the skin and mesangium of the kidney. HSP often presents 1–2 weeks following an upper respiratory infection in children. Approximately 50 % of children will have positive anti-streptolysin O (ASO) titers, suggestive of prior streptococcal infection. Other infections such as methicillin-resistant Staphylococcus aureus (MRSA) have also been associated with presentation of HSP [66




Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
