Reactive Arthritis: Introduction
|
Historical Background
 Reactive arthritis (ReA) is an inflammatory syndrome that results after certain genitourinary or gastrointestinal symptoms. The most common inciting infections include Chlamydia trachomatis, Salmonella, Shigella, Campylobacter, and Yersinia. The term reactive arthritis was introduced in 1973 to describe this type of arthritis that occurs after a triggering infection.1 There is considerable ambiguity in the literature in terms of the nomenclature of this condition for many terms and eponyms have been utilized. In 1942 two Harvard researchers, Bauer and Engelmann, recognized a case of ReA and upon their review of the literature they discovered that Hans Reiter had described this same syndrome in 1916.2 Reiter had described a German officer who developed the clinical triad of arthritis, nongonococcal urethritis, and conjunctivitis after an episode of acute dysentery. It was at that point that Bauer and Engelmann coined the term Reiter syndrome and this eponym was often used to describe the condition.
Reactive arthritis (ReA) is an inflammatory syndrome that results after certain genitourinary or gastrointestinal symptoms. The most common inciting infections include Chlamydia trachomatis, Salmonella, Shigella, Campylobacter, and Yersinia. The term reactive arthritis was introduced in 1973 to describe this type of arthritis that occurs after a triggering infection.1 There is considerable ambiguity in the literature in terms of the nomenclature of this condition for many terms and eponyms have been utilized. In 1942 two Harvard researchers, Bauer and Engelmann, recognized a case of ReA and upon their review of the literature they discovered that Hans Reiter had described this same syndrome in 1916.2 Reiter had described a German officer who developed the clinical triad of arthritis, nongonococcal urethritis, and conjunctivitis after an episode of acute dysentery. It was at that point that Bauer and Engelmann coined the term Reiter syndrome and this eponym was often used to describe the condition.
 In recent years, the eponym Reiter syndrome became problematic for several reasons. First, most clinicians often demanded that a patient have the classic triad of symptoms involving the synovium, urethra, and conjunctiva before the diagnosis would be made. It is now known that most patients with ReA do not have the complete triad of symptoms.3 Further, many common features of ReA are not included in this “classic triad” including involvement of the skin and mucous membranes as well as other parts of the eye, namely the anterior uveal tract. More troubling is the acts of the man from which the eponym was coined. Although Hans Reiter was an erudite intellectual and a brilliant investigator who performed many useful studies early in his career including the discovery of the spirochete that causes leptospirosis and a nonpathogenic strain of Treponema, some of his later experimentation during World War II was deplorable. Reiter played an active role in the design of a study that inoculated concentration camp victims at Buchenwald with an experimental typhus vaccine, which directly resulted in hundreds of deaths.4 Reiter was arrested after World War II; he was tried and convicted at Nuremberg for these war crimes. Because of this, some have correctly argued that the term Reiter syndrome should be abandoned and should only be used in historical context.5
In recent years, the eponym Reiter syndrome became problematic for several reasons. First, most clinicians often demanded that a patient have the classic triad of symptoms involving the synovium, urethra, and conjunctiva before the diagnosis would be made. It is now known that most patients with ReA do not have the complete triad of symptoms.3 Further, many common features of ReA are not included in this “classic triad” including involvement of the skin and mucous membranes as well as other parts of the eye, namely the anterior uveal tract. More troubling is the acts of the man from which the eponym was coined. Although Hans Reiter was an erudite intellectual and a brilliant investigator who performed many useful studies early in his career including the discovery of the spirochete that causes leptospirosis and a nonpathogenic strain of Treponema, some of his later experimentation during World War II was deplorable. Reiter played an active role in the design of a study that inoculated concentration camp victims at Buchenwald with an experimental typhus vaccine, which directly resulted in hundreds of deaths.4 Reiter was arrested after World War II; he was tried and convicted at Nuremberg for these war crimes. Because of this, some have correctly argued that the term Reiter syndrome should be abandoned and should only be used in historical context.5
 It turns out that Reiter was not the first to describe ReA. In reality, it had been described by many physicians in different parts of the world many years before Reiter’s case in 1916. Several others have described similar cases in the literature including Pierre van Forest’s description of a case of “secondary arthritis and urethritis” in 1507,6 Thomas Sydenham’s association of arthritis with diarrhea in 1686,7 Stoll’s documentation of arthritis following dysentery in 1776,8 and Yvan’s description of a French captain who developed “ophthalmia” and inflammatory arthritis primarily of the lower extremities 15 days after a venereal infection.9 Sir Benjamin Brodie was an English physiologist and surgeon who pioneered research in joint disease. He described five patients with rather classic ReA in his treatise Pathological and Surgical Observations on the Diseases of the Joints.10 Importantly, he recognized the similar “train of symptoms” that all five patients experienced, and clearly noted the relapsing course in the few who developed chronic disease. Interestingly, in 1897 Launois clearly made the distinction of septic from aseptic arthritis and demonstrated that patients with the latter occasionally develop cutaneous lesions on the plantar surface of the feet (keratoderma blenorrhagicum).11 Finally, two French physicians, Fiessinger and Leroy, described this same syndrome in the exact same year as Reiter, i.e., 1916.12 Therefore, the term Fiessinger Leroy syndrome has occasionally been used in the past, especially in the French literature.
It turns out that Reiter was not the first to describe ReA. In reality, it had been described by many physicians in different parts of the world many years before Reiter’s case in 1916. Several others have described similar cases in the literature including Pierre van Forest’s description of a case of “secondary arthritis and urethritis” in 1507,6 Thomas Sydenham’s association of arthritis with diarrhea in 1686,7 Stoll’s documentation of arthritis following dysentery in 1776,8 and Yvan’s description of a French captain who developed “ophthalmia” and inflammatory arthritis primarily of the lower extremities 15 days after a venereal infection.9 Sir Benjamin Brodie was an English physiologist and surgeon who pioneered research in joint disease. He described five patients with rather classic ReA in his treatise Pathological and Surgical Observations on the Diseases of the Joints.10 Importantly, he recognized the similar “train of symptoms” that all five patients experienced, and clearly noted the relapsing course in the few who developed chronic disease. Interestingly, in 1897 Launois clearly made the distinction of septic from aseptic arthritis and demonstrated that patients with the latter occasionally develop cutaneous lesions on the plantar surface of the feet (keratoderma blenorrhagicum).11 Finally, two French physicians, Fiessinger and Leroy, described this same syndrome in the exact same year as Reiter, i.e., 1916.12 Therefore, the term Fiessinger Leroy syndrome has occasionally been used in the past, especially in the French literature.
 In light of the above issues, the more recent literature has made a concerted effort to standardize the disease terminology for this condition. Because Hans Reiter was not the first to describe the syndrome, and the fact that many clinicians are reluctant to diagnose this condition in those who do not display the complete triad of symptoms thereby missing the majority of cases, and ReA is a more descriptive term, the term ReA has become the appropriate terminology for this disease process regardless of whether their symptoms involve the three classic organ systems.
In light of the above issues, the more recent literature has made a concerted effort to standardize the disease terminology for this condition. Because Hans Reiter was not the first to describe the syndrome, and the fact that many clinicians are reluctant to diagnose this condition in those who do not display the complete triad of symptoms thereby missing the majority of cases, and ReA is a more descriptive term, the term ReA has become the appropriate terminology for this disease process regardless of whether their symptoms involve the three classic organ systems.
Epidemiology
 In a similar fashion to the ambiguity that previously involved the proper disease terminology, so too there has been no definitive set of diagnostic criteria. This can result in nonhomogenous patient populations in various studies. This coupled with the fact that the disease can often be mild, there has been a traditional overreliance on the complete clinical triad of symptoms, and the incidence of the trigger infections can vary over time, makes epidemiological studies problematic. There is a link between ReA and the human leukocyte antigen (HLA)-B27, but the role this antigen plays in ReA disease susceptibility might be overstated although this HLA antigen plays a role in those individuals who develop chronic disease.13 Nevertheless, the prevalence of HLA-B27 varies between populations and, as might be expected, the incidence and prevalence of ReA varies widely in studies. Because ReA has a definitive trigger as an etiologic agent, but not all patients exposed to these causative organisms develop ReA, the concept of attack rate is extremely important in ReA. The attack rate represents the percentage of patients who develop ReA after exposure to one of the causative organisms. Finally, many cases diagnosed as seronegative oligoarthritis or undifferentiated oligoarthritis may actually be ReA, thereby resulting in an underestimate of disease prevalence or incidence.
In a similar fashion to the ambiguity that previously involved the proper disease terminology, so too there has been no definitive set of diagnostic criteria. This can result in nonhomogenous patient populations in various studies. This coupled with the fact that the disease can often be mild, there has been a traditional overreliance on the complete clinical triad of symptoms, and the incidence of the trigger infections can vary over time, makes epidemiological studies problematic. There is a link between ReA and the human leukocyte antigen (HLA)-B27, but the role this antigen plays in ReA disease susceptibility might be overstated although this HLA antigen plays a role in those individuals who develop chronic disease.13 Nevertheless, the prevalence of HLA-B27 varies between populations and, as might be expected, the incidence and prevalence of ReA varies widely in studies. Because ReA has a definitive trigger as an etiologic agent, but not all patients exposed to these causative organisms develop ReA, the concept of attack rate is extremely important in ReA. The attack rate represents the percentage of patients who develop ReA after exposure to one of the causative organisms. Finally, many cases diagnosed as seronegative oligoarthritis or undifferentiated oligoarthritis may actually be ReA, thereby resulting in an underestimate of disease prevalence or incidence.
 The postdysentery form of ReA affects males and females with the same frequency, whereas the postvenereal form occurs at a male to female ratio of 9:1.14 Adults are more likely to develop ReA than children.15 In Finland, the annual incidence of ReA has been estimated to be 30/100,000 individuals,16 whereas in Norway the annual incidence is approximately 10/100,000.17 In this latter study, the incidences of postenteric and postchlamydial ReA were essentially equal at 5/100,000 and 4.6/100,000, respectively. In the United States, one study estimated the age-adjusted annual incidence of ReA in males younger than age 50 as 3.5/100,000.18 However, in this same study no female cases were identified, suggesting this represents an overall underestimate. In terms of overall prevalence of ReA, a German study suggested a prevalence of 10/100,000.19 Yet, in this study undifferentiated spondyloarthritis (uSpA) was the second most common type of spondyloarthritis diagnosed (after ankylosing spondylitis) and this same diagnosis was more than twice as common as psoriatic arthritis. Data described below suggests that many cases of uSpA are actually ReA. If true, the prevalence of ReA in this German study would be much greater.
The postdysentery form of ReA affects males and females with the same frequency, whereas the postvenereal form occurs at a male to female ratio of 9:1.14 Adults are more likely to develop ReA than children.15 In Finland, the annual incidence of ReA has been estimated to be 30/100,000 individuals,16 whereas in Norway the annual incidence is approximately 10/100,000.17 In this latter study, the incidences of postenteric and postchlamydial ReA were essentially equal at 5/100,000 and 4.6/100,000, respectively. In the United States, one study estimated the age-adjusted annual incidence of ReA in males younger than age 50 as 3.5/100,000.18 However, in this same study no female cases were identified, suggesting this represents an overall underestimate. In terms of overall prevalence of ReA, a German study suggested a prevalence of 10/100,000.19 Yet, in this study undifferentiated spondyloarthritis (uSpA) was the second most common type of spondyloarthritis diagnosed (after ankylosing spondylitis) and this same diagnosis was more than twice as common as psoriatic arthritis. Data described below suggests that many cases of uSpA are actually ReA. If true, the prevalence of ReA in this German study would be much greater.
 An analysis of the expected number of cases of ReA and those actually diagnosed rather strongly argues that ReA is underdiagnosed. Perhaps this is most true with postchlamydial ReA. Because many of the organisms responsible for causing ReA are reportable diseases, the annual incidence of these infections is well described. The expected number of cases of ReA that results from these infections should result in an annual incidence of ReA that greatly exceeds the annual incidence of rheumatoid arthritis.20 However, the number of patients diagnosed with rheumatoid arthritis greatly exceeds that diagnosed with ReA. One study demonstrated that 36% of ReA subjects went undiagnosed in the community clinic in spite of a clear antecedent gastrointestinal infection.21 These data suggest that awareness of this condition needs to improve and the burden of ReA on society might be significantly underrecognized.
An analysis of the expected number of cases of ReA and those actually diagnosed rather strongly argues that ReA is underdiagnosed. Perhaps this is most true with postchlamydial ReA. Because many of the organisms responsible for causing ReA are reportable diseases, the annual incidence of these infections is well described. The expected number of cases of ReA that results from these infections should result in an annual incidence of ReA that greatly exceeds the annual incidence of rheumatoid arthritis.20 However, the number of patients diagnosed with rheumatoid arthritis greatly exceeds that diagnosed with ReA. One study demonstrated that 36% of ReA subjects went undiagnosed in the community clinic in spite of a clear antecedent gastrointestinal infection.21 These data suggest that awareness of this condition needs to improve and the burden of ReA on society might be significantly underrecognized.
Etiology and Pathogenesis
Bacteria that commonly cause ReA are Salmonella, Shigella, Campylobacter, Yersinia, and Chlamydia trachomatis. Indeed, these organisms represent the definitive triggers of ReA; however many other infectious agents have been implicated as potential causes (Box 20-1). Chlamydia trachomatis (Ct) is the most common etiologic agent causing ReA in the United States.14,22 Despite the obvious difference of initial route of infection (i.e., gastrointestinal vs. genitourinary), another distinction exists. The postdysentery form of ReA is always preceded by a symptomatic infection, and recent data suggest the more severe the initial gastrointestinal infection, the more likely ReA develops.23–25 However, an initial Ct infection is often asymptomatic.26–28 Recent data suggest that an initial asymptomatic Ct infection is a common cause of ReA.29 In this study, the majority of patients diagnosed with uSpA, because of no known preceding infection prior to the onset of their arthritis, were found to be polymerase chain reaction (PCR) positive for Chlamydiae on synovial tissue analysis. This is in keeping with previous data suggesting that 78% of subjects who develop ReA after a venereal infection had an asymptomatic infection.30 A number of published studies also indicate that Chlamydophila (Chlamydia) pneumonaie (Cpn), a related respiratory pathogen, is another causative agent in ReA, albeit at a lower frequency.31–33
DEFINITE CAUSES |
Postvenereal |
Chlamydia trachomatis |
Postenteric |
Salmonella (S. enteritidis, S. typhimurium, S. bovismorbificans, S. blockley) |
Shigella (S. flexneri, S. dysenteriae, S. sonnei, S. boydii) |
Campylobacter (C. jejuni, C. coli) |
Yersinia (Y. enterocolitica, Y. pseudotuberculosis) |
PROBABLE CAUSES |
Chlamydophila (Chlamydia) pneumoniae |
Ureaplasma urealyticum |
Bacille Calmette-Guérin (intravesicular) |
POSSIBLE CAUSES |
Bacillus cereus |
Brucella abortus |
Clostridium difficile |
Escherichia coli |
Helicobacter pylori |
Hafnia alvei |
Lactobacillus |
Neisseria meningitidis serogroup B |
Pseudomona |
Intestinal parasites (Strongyloides stercolis, Taenia saginata, Giardia lamblia, Ascaris lumbricoides, Filariasis, and Cryptosporidium) |
OTHER TYPES OF INFLAMMATORY ARTHRITIS IN WHICH BACTERIA MAY PLAY A CAUSATIVE ROLE |
Borrelia burgdorferi (Lyme disease) |
Propionbacterium acnes (SAPHO) |
Streptococcus sp. (poststreptococcal reactive arthritis) |
Trophyrema whippelii (Whipple’s disease) |
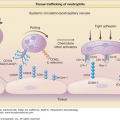
Chlamydiae are Gram-negative, obligate intracellular organisms. The attack rate of ReA after a Ct infection is approximately 5%.30 Synovial tissue analyses from patients affected with postchlamydial ReA have shown that these organisms traffic from the initial site of infection to the synovium. These synovium-based Chlamydiae exist in a morphologically aberrant but metabolically active viable state termed chlamydial persistence.34,35 The pattern of gene expression is attenuated and significantly different than that seen during normal active infection. For example, during persistence of Ct expression of the major outer membrane protein (omp1) gene and several genes required for the cell division process are severely downregulated. This is coupled with differential regulations of the three paralog genes specifying Chlamydia trachomatis heat shock proteins (HSP)-60 [(1) Ct110, (2) Ct604, and (3) Ct755].36 The exact role that these synovium-based persistent Chlamydiae play in terms of disease pathogenesis or disease propagation is not completely understood. Of note, it has been demonstrated that these same persistent Chlamydiae traffic to other end organs, specifically the skin in patients with suspect keratoderma blenorrhagicum.37
Other recent data relating to Chlamydia-induced ReA force us to reconsider our traditional paradigms. Because these pathogens are responsible for genital infections, it was logical to assume that the genital strains of C. trachomatis were responsible for triggering ReA. However, there are several serovars of C. trachomatis, specifically serovars A through K. Serovars A, B, and C are ocular (trachoma) serovars and the remainders (serovars D through K) are genital. Remarkably, a recent study analyzing the chlamydial serovars of 36 subjects with known C. trachomatis-induced ReA demonstrated that all 36 synovial tissue samples were positive for the ocular serovars, not the genital serovars.38 It is known that genital infection with the ocular strains do occur, but are rare.39,40 The infrequent rate of genital infections with the ocular strain might explain the low attack rate of ReA in patients with acute chlamydial infections.
Reactive arthritis represents the classic interplay of host and environment. The environmental triggers outlined above play and undeniable role in disease genesis; the concept of bacterial persistence lends speculative support that these pathogens might also play a role in disease propagation. Certain bacterial serovars or species might be particularly arthritogenic or more prone to dissemination. However, genetic susceptibility also clearly plays a role. Because ReA is one of the spondyloarthritides, much of the focus on host genetics has centered on HLA-B27. There are also data indicating that patients with HIV are at increased risk for ReA and the symptoms can improve with antiretroviral HIV therapy.57
The prevalence of HLA-B27 and reactive arthritis varies around the world.58 In Caucasian populations, HLAB27 is present in 7%–9% of individuals. Older literature suggested that as many as 70%–80% of patients with ReA were HLA-B27 positive.59,60 However, several large epidemiological studies of ReA now dictate that, in reality, about 30%–50% of ReA patients are positive for this antigen.45,61–67 More recent data even suggest there might be no association with HLA-B27 and ReA.25,47,64 The vast majority of data regarding the prevalence of HLA-B27 in ReA comes from epidemiological studies of postenteric ReA after outbreaks with certain enteric pathogens. Therefore, the true prevalence of HLA-B27 in postchlamydial ReA is less well defined.
The possibility exists that HLA-B27 plays more of a role with phenotypic disease expression rather than a true genetic susceptibility locus. Several large ReA studies demonstrate that HLA-B27 positive patients have more severe symptoms, thereby making the condition more clinically apparent.23 This haplotype might also increase one’s risk for developing the complete triad of symptoms.68 It should also be noted that the variation in the prevalence of HLA-B27 in various studies described above could, at least in part, be explained by the potential role that HLA-B27 plays on phenotypic expression. The studies cited above suggesting that HLA-B27 has little to no role in disease susceptibility include patients with milder symptoms, whereas the previous studies demonstrating a higher HLA-B27 prevalence only include patients with more fulminate symptoms sometimes requiring the complete triad of symptoms.
Whether HLA-B27 is more important in disease susceptibility or clinical expression, it clearly plays a role in ReA. Many theories exist regarding its pathophysiologic role, but none are proven. Since HLA-B27 is a Class I histocompatability antigen, it has been postulated that HLA-B27 presents arthritogenic microbial peptides to T cells stimulating an autoimmune response, so called molecular mimicry.69 Conversely, B27 itself may serve as the autoantigen that is targeted by the immune system.70 It is also possible that exposure to the triggering bacteria may subvert self-tolerance to the B27 antigen.71 Another theory suggests that the role of HLA-B27 may be to enhance invasion of the causative organisms into human intestinal epithelial cells in patients with postenteric ReA.72 Vast data demonstrate that misfolding of HLA-B27 can lead to the unfolded protein response enhancing the production of interleukin-23;73 however this unfolded protein response has not been definitively linked to ReA. HLA-B27 has multiple alleles that could influence host response and disease susceptibility. One study suggests that although HLA-B*2705 is the most common allele observed in B27-positive ReA patients, this allele is seen less frequently than in the other SpA’s and in B27 healthy controls.74
While HLA-B27 is important to pathogenesis in ReA, it is not the sole determinant. Clearly patients who are HLA-B27 negative can develop ReA. It has been suggested that HLA-B*5703 increases the risk for the classic triad of symptoms in patients who develop ReA.75 Gene expression analyses suggest that proangiogenic factors account for genetic susceptibility of ReA.76 Much remains to be learned about other HLA loci or even non-HLA genes that might be important in the pathophysiology of ReA.
Bacterial Triggers of ReA
 These recent data regarding the apparent causality of the ocular serovars raise intriguing questions about other features of ReA, namely the eye involvement with uveitis and/or conjunctivitis and the propensity of uveitis to recur in these subjects. As it turns out, we might have had clues to the apparent arthritogenicity of the ocular serovar years ago. In the 1960s and early 1970s, Bedsonia (Chlamydia) recovered from patients with ReA caused arthritis 100% of the time when injected into rabbits; in addition, intra-articular injection of these same organisms often caused ocular involvement.41,42 This suggests these particular organisms were not only arthritogenic, but had the ability to disseminate with particular affinity for the eye.
These recent data regarding the apparent causality of the ocular serovars raise intriguing questions about other features of ReA, namely the eye involvement with uveitis and/or conjunctivitis and the propensity of uveitis to recur in these subjects. As it turns out, we might have had clues to the apparent arthritogenicity of the ocular serovar years ago. In the 1960s and early 1970s, Bedsonia (Chlamydia) recovered from patients with ReA caused arthritis 100% of the time when injected into rabbits; in addition, intra-articular injection of these same organisms often caused ocular involvement.41,42 This suggests these particular organisms were not only arthritogenic, but had the ability to disseminate with particular affinity for the eye.
 The four enteric organisms known to cause ReA represent the largest bulk of the cases of ReA as a group. Salmonella is a Gram-negative, rod-shaped, motile bacterium widespread in animals and environmental sources. It is the most frequently studied enteric bacterium associated with ReA. The attack rate of Salmonella-induced ReA has ranged between 6% and 30% in different studies.43,44 All four of the species of Shigella (S. flexneri, S. dysenteriae, S. sonnei, and S. boydii) can cause ReA. The attack rate of Shigella-induced ReA was reported to be 7%–9% in one study.45 Campylobacter is another Gram-negative motile bacterium; it is a very common cause of enteric infections. C. jejuni is the main cause of bacterial food-borne disease in many developed countries.46 It spite of its frequency it appears to be somewhat less arthritogenic with an attack rate of 1%–5%.47 Although Yersinia infections are not as common as some of the other enteric pathogens, some data suggest that Yersinia is particularly arthritogenic. A study in Denmark suggests that it was the most likely organism to cause ReA with an attack rate of 23%.23 Other studies have suggested an attack rate of 12%.48,49 As with Chlamydiae, efforts have been made to detect these enteric pathogens in synovial tissue or fluid of patients with ReA. Bacterial DNA or bacterial degradation products have been detected, but, unlike Chlamydiae, no viable organisms have been demonstrated.50–52 However, one study did suggest viable Yersinia were present in synovial samples of these patients,53 yet other data contradict this finding.54 As is the case with Chlamydiae, the role these bacterial products play in the pathophysiology of ReA remains uncertain. However, the fact that these enteric bacteria do not appear to be viable, their role is likely different than that of the persistently viable Chlamydiae. It is also likely that different species from the same genus of the triggering enteric bacteria vary in their arthritogenic propensity.
The four enteric organisms known to cause ReA represent the largest bulk of the cases of ReA as a group. Salmonella is a Gram-negative, rod-shaped, motile bacterium widespread in animals and environmental sources. It is the most frequently studied enteric bacterium associated with ReA. The attack rate of Salmonella-induced ReA has ranged between 6% and 30% in different studies.43,44 All four of the species of Shigella (S. flexneri, S. dysenteriae, S. sonnei, and S. boydii) can cause ReA. The attack rate of Shigella-induced ReA was reported to be 7%–9% in one study.45 Campylobacter is another Gram-negative motile bacterium; it is a very common cause of enteric infections. C. jejuni is the main cause of bacterial food-borne disease in many developed countries.46 It spite of its frequency it appears to be somewhat less arthritogenic with an attack rate of 1%–5%.47 Although Yersinia infections are not as common as some of the other enteric pathogens, some data suggest that Yersinia is particularly arthritogenic. A study in Denmark suggests that it was the most likely organism to cause ReA with an attack rate of 23%.23 Other studies have suggested an attack rate of 12%.48,49 As with Chlamydiae, efforts have been made to detect these enteric pathogens in synovial tissue or fluid of patients with ReA. Bacterial DNA or bacterial degradation products have been detected, but, unlike Chlamydiae, no viable organisms have been demonstrated.50–52 However, one study did suggest viable Yersinia were present in synovial samples of these patients,53 yet other data contradict this finding.54 As is the case with Chlamydiae, the role these bacterial products play in the pathophysiology of ReA remains uncertain. However, the fact that these enteric bacteria do not appear to be viable, their role is likely different than that of the persistently viable Chlamydiae. It is also likely that different species from the same genus of the triggering enteric bacteria vary in their arthritogenic propensity.
 Many other organisms have been implicated as potential causes of ReA. These include Ureaplasma urealyticum, H. pylori, and various intestinal parasites. The majority of these reports exist in the form of single cases or small series. Reports of ReA secondary to E. coli,23–25 C. difficile,55 and intravesicular Bacillus Calmette-Guérin (BCG)56 have garnered recent recognition. A recent study in Denmark suggested that a gastrointestinal infection with E. coli was just as likely to cause ReA as Shigella.23
Many other organisms have been implicated as potential causes of ReA. These include Ureaplasma urealyticum, H. pylori, and various intestinal parasites. The majority of these reports exist in the form of single cases or small series. Reports of ReA secondary to E. coli,23–25 C. difficile,55 and intravesicular Bacillus Calmette-Guérin (BCG)56 have garnered recent recognition. A recent study in Denmark suggested that a gastrointestinal infection with E. coli was just as likely to cause ReA as Shigella.23
Pathophysiology
It is apparent that the causative bacteria of ReA are incorporated intracellularly, either in-part or in-whole, then taken from the site of initial infection and trafficked to the synovium. However, what governs this process is not yet evident. It is also not clear if their presence in the affected organs represents a trigger for an autoimmune response, or if these organisms are the source for the inflammatory process. It appears that this phenomenon of cellular uptake, trafficking, and host tolerance is multifactorial in nature.
Although the causative organisms are intracellular pathogens, the process of cellular uptake is not entirely apparent. Indeed the specific mechanisms for cellular uptake of these microbes are probably different for each organism. In the case of Chlamydiae, the quest to find a specific extracellular ligand has been unsuccessful. Intriguing recent data suggest that there might not be a specific chlamydial ligand, rather these pathogens might utilize a different ligand entirely. Apolipoprotein E (ApoE4) that is adherent to the surface of chlamydial elementary bodies (EB) attaches to the host cell low-density lipoprotein (LDL) receptor family carrying the EB with it.77 However, this only appears to be true of C. pneumoniae, not C. trachomatis. In the case of Salmonella, type 1 fimbriae mediate the attachment of this organism to the cell leading to internalization.78
Toll-like receptors (TLR) are a key component of the innate immune system. Because they are among the first line of defense against microbes and they recognize extracellular pathogens, they could play a role in ReA. All of the definitive triggering organisms are Gram-negative with a lipopolysaccharide (LPS) component to their cell wall, and TLR-4 recognizes LPS. TLR-4 deficient mice exposed to Salmonella demonstrate dramatically increased bacterial growth and demise.79 Other animal data have shown that effective host clearance of Ct depends on appropriate TLR-4 expression by neutrophils.80 However, recent human data suggest that TLR-2, not TLR-4, is important in determining ReA disease susceptibility after a Salmonella infection.81
In terms of cytokine response in ReA, it appears that both Th1 and Th2 are important, but the Th2 pathway predominates. Although ReA patients have higher serum TNF-α (Th1) than normal controls,82 lower levels of TNF-α have been demonstrated in ReA compared to other types of inflammatory arthritis.83–85 Synovial fluid analyses from patients with ReA demonstrate higher levels of IL-10 and lower levels of TNF-α and IFN-γ (favoring a Th2 profile).84,85 Interestingly, in terms of postchlamydial ReA, lower levels of these same two cytokines (TNF-α and IFN-γ) have been associated with increased chlamydial replication and overall bacterial burden in vitro.86–88 The suppression of Th1 cytokines is likely mediated through suppression of IL-12 synthesis.
Data also exist suggesting that these cytokine levels can change over time. ReA patients with a disease duration of greater than 6 months secreted significantly less TNF-α.83 Temporal relationships of these different Th1 and Th2 cytokines might also be important in disease manifestations. Slight changes in the Th1/Th2 balance may explain the relapsing course that is frequently seen chronic ReA. Animal data also suggest that blunting of the initial cytokine response of TNF-α, IFN-γ, and Interleukin-4 (IL-4) to an acute chlamydial infection leads to decreased bacterial clearance.89 Therefore, lower initial responses of these Th1 cytokines may increase the likelihood of developing ReA. These animal data provide an interesting potential correlate to the fact that asymptomatic chlamydial infections in humans can often lead to ReA.
Clinical Manifestations
In spite of the fact that ReA has several distinct etiologic agents and the apparent differences in the pathophysiology depending on the triggering microbe, the clinical features of ReA are congruent regardless of the initiating infection. The clinical syndrome that encompasses ReA can involve many different organ systems, although it has a predilection for the synovium, urethra, eye, and the skin (Box 20-2). The symptoms start within 1–4 weeks of the initial infection. However, as stated with Chlamydiae, the inciting infection could be asymptomatic obfuscating the diagnosis. It has traditionally been felt that the vast majority of cases of ReA resolve spontaneously within weeks to months, but more recent data suggest that 30%–50% of cases can become chronic.45,61–67 One series suggested that 63% of patients develop chronic symptoms.18 In general, if a patient experiences symptoms of longer than 6 months duration, then they are felt to have chronic disease (Box 20-3). Typically, patients have more significant symptoms during the acute phase; these can include constitutional symptoms (malaise, fatigue) and fever. If the symptoms persist, then the long-term manifestations tend to be milder. In these chronic cases, it is not unusual for the symptoms to wax and wane.
ACUTE SYMPTOMS | |
Articular | |
Most commonly present with oligoarthritis, but can also present with polyarthritis or monoarthritis | |
Axial | Frequently involved Sacroiliac joints Lumbar spine Occasionally involved Thoracic spine (usually seen in chronic ReA) Cervical spine (usually seen in chronic ReA) Cartilaginous joints (symphysis pubis; sternoclavicular and costosternal joints) |
Peripheral | Frequently involved Large joints of the lower extremities (especially knees) |
Dactylitis (sausage digit) | Very specific for a spondyloarthropathy |
Enthesitis | |
Hallmark feature: inflammation at the transitional zone where collagenous structures such as tendons and ligaments insert into bone. Common sites: plantar fasciitis, Achilles tendonitis; but any enthesis can be involved. | |
Mucosal | |
Oral ulcers (generally painless) Sterile dysuria (occurs with both postvenereal and postdysentery forms) | |
Cutaneous | |
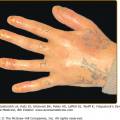
Keratoderma blenorrhagicum | Pustular or plaque-like rash on the soles and/or palms Grossly and histologically indistinguishable from pustular psoriasis Can also involve nails (onycholysis, subungal keratosis, nail pits), scalp, extremities |
Circinate balanitis | Erythema or plaque-like lesions on the shaft and/or glans of penis |
Ocular | |