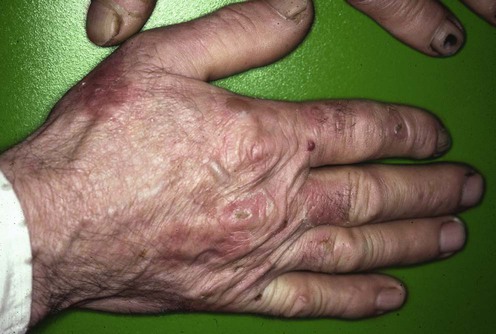
192 Porphyria cutanea tarda Maureen B. Poh-Fitzpatrick Evidence Levels: A Double-blind study B Clinical trial ≥ 20 subjects C Clinical trial < 20 subjects D Series ≥ 5 subjects E Anecdotal case reports The term porphyria cutanea tarda (PCT) encompasses several related inherited or acquired disorders in which insufficient hepatic uroporphyrinogen decarboxylase enzyme activity causes overproduction of polycarboxylated porphyrins. These porphyrins mediate cutaneous photosensitivity manifested as fragility, bullae, hypertrichosis, dyspigmentation, sclerodermoid features, and scarring. Multiple factors may contribute to disease expression: mutant uroporphyrinogen decarboxylase genes, hemochromatosis genes or other predisposing genetic determinants, ethanol abuse, tobacco smoking, medicinal estrogen, iron, hepatitis and/or human immunodeficiency viral infections, chronic dialysis treatment, toxic aromatic hydrocarbon exposure and, rarely, hepatic tumors. Increased tissue iron, commonly seen with alcoholism, hepatitis C infection, hemochromatosis, and end-stage renal disease, plays a central role in the pathogenesis of PCT. Iron-dependent partial oxidation of uroporphyrinogen to uroporphomethene, a competitive inhibitor of uroprophyrinogen decarboxylase, may be the mechanism by which its enzymatic activity is reduced in PCT (Phillips et al. Proc Natl Acad Sci USA 2007; 104: 5079–84). Iron-enhanced complete oxidation of uroporphyrinogen accumulated due to inhibited enzyme activity yields photoactive uroporphyrin. Hepatic siderosis or porphyrin crystallization in hepatocytes may lead to hepatocellular carcinoma, a known complication of chronic active PCT. Ferrodepletion by serial phlebotomy is preferred first-line therapy for PCT patients with demonstrable iron overload. Management strategy A precise diagnosis is essential because optimal management consists of induction of remission using strategies inappropriate for other porphyrias or pseudoporphyrias. Associated disorders that may influence management, such as viral infections, hemochromatosis or other causes of excess iron storage, lupus erythematosus, diabetes mellitus, and anemias, should be identified. Eliminating exacerbating factors and pursuing ferrodepletion by serial phlebotomy, iron chelation, or erythropoietin bone marrow stimulation can induce biochemical and clinical remissions. Porphyrin excretion can be increased using chloroquine or hydroxychloroquine, enteric sorbents, or metabolic alkalinization. Interferon-α or antiretroviral drugs may benefit PCT associated with hepatitis C or AIDS, respectively. Children can be treated by phlebotomy protocols adjusted for pediatric parameters. Rare reports of chloroquine or hydroxychloroquine treatment of children suggest that cautious low-dose schedules may be safe and effective. Vitamins E and C, plasmapheresis or plasma exchange, high-flux hemodialysis, and cimetidine have been reported as beneficial alternative or adjunctive therapies. Skin should be protected from sunlight exposure and mechanical trauma until full clinical remission is achieved. Hepatoerythropoietic porphyria, caused by co-inheritance of two uroporphyrinogen decarboxylase gene mutations, resists induction of remission, and so requires lifelong vigilant skin photoprotection. Photothermolysis using light with selected wavelengths may reduce persistent hypertrichosis. Specific investigations Porphyrin concentrations and types in erythrocytes, serum or urine, feces Hematological and iron profiles, serum ferritin, hemochromatosis gene analysis Liver function profile; serum α-fetoprotein level, liver imaging, and liver biopsy if clinically indicated Hepatitis A, B, and C viral serology HIV serology if risk factors are present Fasting blood glucose Serum antinuclear antibody Porphyria cutanea tarda: clinical features and laboratory findings in forty patients. Grossman ME, Bickers DR, Poh-Fitzpatrick MB, DeLeo VA, Harber LC. Am J Med 1979; 67: 277–86. Abnormalities of liver function, glucose tolerance, antinuclear antibody titers, other laboratory parameters, clinical manifestations, skin and liver histopathology, and experience with phlebotomy are surveyed in a large population. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Ryan Caballes F, Sendi H, Bonkovsky HL. Liver Int 2012; 32: 880–93. A current review, emphasizing pathophysiologic roles of hepatitis C virus, iron-facilitated oxidative stress, and hepcidin (a key regulator of iron absorption and metabolism that is downregulated in PCT). Treatment guidelines are offered. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America. Bonkovsky HL, Poh-Fitzpatrick MB, Pimstone N, Obando J, Di Bisceglie A, Tattrie C, et al. Hepatology 1998; 27: 1661–9. Of 70 American patients with PCT, 53% had evidence of hepatitis C infection, and 43% of 26 patients had HFE gene mutations associated with hereditary hemochromatosis. Hepatocellular carcinoma risk in patients with porphyria cutanea tarda. Gisbert JP, Garcia-Buey L, Alonso A, Rubio S, Hernandez A, Pajares JM, et al. Eur J Gastroenterol Hepatol 2004; 16: 689–92. These authors recommend that patients presenting with PCT should undergo serological viral hepatitis testing and liver biopsy, and those with concomitant hepatitis C infection or advanced fibrosis/cirrhosis should be monitored with semiannual ultrasonography and serum α-fetoprotein testing. The decision for liver biopsy should be weighed carefully. Patients without risk factors for hepatic siderosis or other liver pathology (i.e., women with estrogen use as the only identifiable PCT-inducing factor) may not need this invasive procedure. First-line therapies Serial phlebotomies B Chloroquine, hydroxychloroquine B Only gold members can continue reading. Log In or Register to continue Share this: Click to share on X (Opens in new window) X Click to share on Facebook (Opens in new window) Facebook Related Related posts: Cat scratch disease Hemangiomas Drug eruptions Erythropoietic protoporphyria Ichthyoses Nevoid basal cell carcinoma syndrome Stay updated, free articles. Join our Telegram channel Join Tags: Treatment of Skin Disease Comprehensive Therapeutic Strategies Aug 7, 2016 | Posted by admin in Dermatology | Comments Off on Porphyria cutanea tarda Full access? Get Clinical Tree
192 Porphyria cutanea tarda Maureen B. Poh-Fitzpatrick Evidence Levels: A Double-blind study B Clinical trial ≥ 20 subjects C Clinical trial < 20 subjects D Series ≥ 5 subjects E Anecdotal case reports The term porphyria cutanea tarda (PCT) encompasses several related inherited or acquired disorders in which insufficient hepatic uroporphyrinogen decarboxylase enzyme activity causes overproduction of polycarboxylated porphyrins. These porphyrins mediate cutaneous photosensitivity manifested as fragility, bullae, hypertrichosis, dyspigmentation, sclerodermoid features, and scarring. Multiple factors may contribute to disease expression: mutant uroporphyrinogen decarboxylase genes, hemochromatosis genes or other predisposing genetic determinants, ethanol abuse, tobacco smoking, medicinal estrogen, iron, hepatitis and/or human immunodeficiency viral infections, chronic dialysis treatment, toxic aromatic hydrocarbon exposure and, rarely, hepatic tumors. Increased tissue iron, commonly seen with alcoholism, hepatitis C infection, hemochromatosis, and end-stage renal disease, plays a central role in the pathogenesis of PCT. Iron-dependent partial oxidation of uroporphyrinogen to uroporphomethene, a competitive inhibitor of uroprophyrinogen decarboxylase, may be the mechanism by which its enzymatic activity is reduced in PCT (Phillips et al. Proc Natl Acad Sci USA 2007; 104: 5079–84). Iron-enhanced complete oxidation of uroporphyrinogen accumulated due to inhibited enzyme activity yields photoactive uroporphyrin. Hepatic siderosis or porphyrin crystallization in hepatocytes may lead to hepatocellular carcinoma, a known complication of chronic active PCT. Ferrodepletion by serial phlebotomy is preferred first-line therapy for PCT patients with demonstrable iron overload. Management strategy A precise diagnosis is essential because optimal management consists of induction of remission using strategies inappropriate for other porphyrias or pseudoporphyrias. Associated disorders that may influence management, such as viral infections, hemochromatosis or other causes of excess iron storage, lupus erythematosus, diabetes mellitus, and anemias, should be identified. Eliminating exacerbating factors and pursuing ferrodepletion by serial phlebotomy, iron chelation, or erythropoietin bone marrow stimulation can induce biochemical and clinical remissions. Porphyrin excretion can be increased using chloroquine or hydroxychloroquine, enteric sorbents, or metabolic alkalinization. Interferon-α or antiretroviral drugs may benefit PCT associated with hepatitis C or AIDS, respectively. Children can be treated by phlebotomy protocols adjusted for pediatric parameters. Rare reports of chloroquine or hydroxychloroquine treatment of children suggest that cautious low-dose schedules may be safe and effective. Vitamins E and C, plasmapheresis or plasma exchange, high-flux hemodialysis, and cimetidine have been reported as beneficial alternative or adjunctive therapies. Skin should be protected from sunlight exposure and mechanical trauma until full clinical remission is achieved. Hepatoerythropoietic porphyria, caused by co-inheritance of two uroporphyrinogen decarboxylase gene mutations, resists induction of remission, and so requires lifelong vigilant skin photoprotection. Photothermolysis using light with selected wavelengths may reduce persistent hypertrichosis. Specific investigations Porphyrin concentrations and types in erythrocytes, serum or urine, feces Hematological and iron profiles, serum ferritin, hemochromatosis gene analysis Liver function profile; serum α-fetoprotein level, liver imaging, and liver biopsy if clinically indicated Hepatitis A, B, and C viral serology HIV serology if risk factors are present Fasting blood glucose Serum antinuclear antibody Porphyria cutanea tarda: clinical features and laboratory findings in forty patients. Grossman ME, Bickers DR, Poh-Fitzpatrick MB, DeLeo VA, Harber LC. Am J Med 1979; 67: 277–86. Abnormalities of liver function, glucose tolerance, antinuclear antibody titers, other laboratory parameters, clinical manifestations, skin and liver histopathology, and experience with phlebotomy are surveyed in a large population. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Ryan Caballes F, Sendi H, Bonkovsky HL. Liver Int 2012; 32: 880–93. A current review, emphasizing pathophysiologic roles of hepatitis C virus, iron-facilitated oxidative stress, and hepcidin (a key regulator of iron absorption and metabolism that is downregulated in PCT). Treatment guidelines are offered. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America. Bonkovsky HL, Poh-Fitzpatrick MB, Pimstone N, Obando J, Di Bisceglie A, Tattrie C, et al. Hepatology 1998; 27: 1661–9. Of 70 American patients with PCT, 53% had evidence of hepatitis C infection, and 43% of 26 patients had HFE gene mutations associated with hereditary hemochromatosis. Hepatocellular carcinoma risk in patients with porphyria cutanea tarda. Gisbert JP, Garcia-Buey L, Alonso A, Rubio S, Hernandez A, Pajares JM, et al. Eur J Gastroenterol Hepatol 2004; 16: 689–92. These authors recommend that patients presenting with PCT should undergo serological viral hepatitis testing and liver biopsy, and those with concomitant hepatitis C infection or advanced fibrosis/cirrhosis should be monitored with semiannual ultrasonography and serum α-fetoprotein testing. The decision for liver biopsy should be weighed carefully. Patients without risk factors for hepatic siderosis or other liver pathology (i.e., women with estrogen use as the only identifiable PCT-inducing factor) may not need this invasive procedure. First-line therapies Serial phlebotomies B Chloroquine, hydroxychloroquine B Only gold members can continue reading. Log In or Register to continue Share this: Click to share on X (Opens in new window) X Click to share on Facebook (Opens in new window) Facebook Related Related posts: Cat scratch disease Hemangiomas Drug eruptions Erythropoietic protoporphyria Ichthyoses Nevoid basal cell carcinoma syndrome Stay updated, free articles. Join our Telegram channel Join Tags: Treatment of Skin Disease Comprehensive Therapeutic Strategies Aug 7, 2016 | Posted by admin in Dermatology | Comments Off on Porphyria cutanea tarda Full access? Get Clinical Tree







 Serial phlebotomies
Serial phlebotomies Chloroquine, hydroxychloroquine
Chloroquine, hydroxychloroquine


