Pemphigus: Introduction
|
The term pemphigus refers to a group of autoimmune blistering diseases of skin and mucous membranes that are characterized histologically by intraepidermal blisters due to acantholysis (i.e., separation of epidermal cells from each other) and immunopathologically by in vivo bound and circulating immunoglobulin (Ig) directed against the cell surface of keratinocytes. The nosology of this group of diseases is outlined in Box 54-1. Essentially, pemphigus can be divided into four major types: (1) vulgaris, (2) foliaceus, (3) paraneoplastic (see Chapter 55), and (4) IgA pemphigus (see Chapter 54). In pemphigus vulgaris (PV), the blister occurs in the deeper part of the epidermis, just above the basal layer, and in pemphigus foliaceus (PF), also called superficial pemphigus, the blister is in the granular layer.
PEMPHIGUS SUBTYPES
|
INTRAEPIDERMAL BLISTERING DISEASES WITHOUT AUTOANTIBODIES
|
MOUTH ULCERS/EROSION WITHOUT AUTOANTIBODIES
|
SUBEPIDERMAL BLISTERING DISEASES WITH AUTOANTIBODIES
|
SUBEPIDERMAL BLISTERING DISEASES WITHOUT AUTOANTIBODIES
|
The history of the discovery of pemphigus, and its various forms, is covered in Walter Lever’s classic monograph Pemphigus and Pemphigoid.1 Both PV and PF display a spectrum of disease. Various points along these spectra have been given unique names, but because the presentation of these diseases is fluid, patients’ disease usually crosses these artificial designations over time. Thus, patients with PV may present with more localized disease, one form of which is called pemphigus vegetans of Hallopeau. This may become slightly more extensive and may merge into pemphigus vegetans of Neumann. Finally, with more severe disease, full-blown PV may appear. Similarly, patients with PF may present with more localized disease, represented by pemphigus erythematosus. However, these patients often go on to more widespread PF.
The discovery by Ernst Beutner and Robert Jordon in 1964 of circulating antibodies against the cell surface of keratinocytes in the sera of patients with PV pioneered our understanding that PV is a tissue-specific autoimmune disease of skin and mucosa.2 Ultimately, their work led the way to the discoveries of autoantibodies in other autoimmune bullous diseases of the skin.
Epidemiology
A few prospective and several retrospective surveys of patients with pemphigus clearly indicate that the epidemiology of pemphigus is dependent on both the area in the world that is studied as well as the ethnic population in that area.3–10 PV is more common in Jews and probably in people of Mediterranean descent and from the Middle East. This same ethnic predominance does not exist for PF. Therefore, in areas where the Jewish, Middle Eastern, and Mediterranean population predominates, the ratio of PV to PF cases tends to be higher. For example, in New York, Los Angeles, and Croatia, the ratio of PV to PF cases is approximately 5:1; in Iran the ratio is 12:1; whereas in Singapore it is 2:1; and in Finland, it is only 0.5:1. Similarly, the incidence of pemphigus varies by region. In Jerusalem, the incidence of PV has been estimated to be 1.6 per 100,000 people per year and in Iran approximately 10.0 per 100,000 people per year. Elsewhere in Europe, the incidences are lower, ranging from a high of 0.7 PV cases per 100,000 person years in the United Kingdom to tenfold less, 0.5–1.0 per million person years, in Finland, France, Germany, and Switzerland.
The prevalence and incidence of PF are also very dependent on its location, as best exemplified by the finding of endemic foci of PF in Brazil, Colombia, and Tunisia. The first recognition of endemic PF was in Brazil and is called fogo selvagem, which means “wild fire” in Portuguese. It is a disease that is clinically, histologically, and immunopathologically the same as sporadic PF in any individual patient, but its epidemiology is unique.11,12 Fogo selvagem is endemic in the rural areas of Brazil, especially along inland riverbeds. The geographic distribution of disease clustering is similar to that of a black fly, Simulium nigrimanum, thought by natives to be a vector of this disease. A study of potential environmental risk factors has also implicated the bite of this black fly, showing it to be significantly more frequent among those with the disease compared to an age-, sex-, and occupation-matched control population with unrelated dermatoses.13 The prevalence on some well-studied Indian reservations in rural Brazil can be as high as 3.4%, with the incidence up to 0.8–4.0 new cases per 1,000 people per year.12,14 On the reservation in Limao Verde, up to 55% of unaffected individuals have a low-level IgG1 antibody response against desmoglein 1, the PF autoantigen, which becomes an IgG4 response of higher titer against a more pathogenic epitope in disease.12 These results suggest that some environmental agent (e.g., insects or other infectious disease agent) may trigger a low-level autoantibody response that becomes pathogenic by intramolecular epitope spreading in genetically susceptible individuals. With this theory in mind, it is interesting that 40%–80% of patients from Brazil with the insect-borne diseases onchocerciasis, leishmania, and Chagas disease have low-level antidesmoglein 1 antibodies, but patients with other infectious diseases from Brazil rarely have such antibodies.15
Fogo selvagem occurs often in children and young adults, unlike sporadic PF, which is a disease of mostly middle-aged and older patients. Also unlike PF, fogo selvagem occurs not infrequently in genetically related family members, although it is not contagious. This fact probably implies a common exposure, as well as susceptibility. There is no known racial or ethnic predominance, and anyone moving into an endemic area may be susceptible to disease. Again supporting the presence of an environmental trigger, the development of the rural endemic areas of Brazil decreased the incidence of disease. Certainly, this fascinating disease holds clues to understanding how this autoimmune response is triggered.
The sex ratio of pemphigus cases is difficult to estimate accurately due to the overall low incidence. Larger epidemiologic studies (i.e., those identifying greater than 100 cases) have shown that the sex ratio of pemphigus in women versus men ranges from 1.33 or 2.25 to 1.7,9,16–20 Notable exceptions are the predominance of women (4:1) in an endemic focus of PF in Tunisia,6 and a predominance of men (19:1) in an endemic focus of PF in Colombia.21
The average age of disease onset also varies by region. In Turkey, Saudi Arabia, Tunisia, and Iran, the mean age of onset is approximately 40 years.6,16,18,22 Studies in the United States and elsewhere in Europe demonstrate an average age of onset between 50 and 70 years.5,6,9,10,17,19,23,24,25 Pemphigus rarely occurs in children,26 except in regions of endemic disease.
Etiology and Pathogenesis
The discovery of pemphigus as an organ-specific, autoantibody-mediated disease of desmosomes highlights the synergy between clinical care and basic science research. The development of light microscopy and electron microscopy allowed dermatologists to identify the morphology and immunopathology of disease. Patient serum IgG served as a key reagent to help identify both the PF and PV antigens.27–29 The cloning and characterization of the pemphigus antigens have subsequently led to the development of enzyme-linked immunosorbent assay (ELISA) tests to improve the sensitivity and specificity of disease diagnosis, and continued studies on pemphigus pathophysiology aim to develop safer and effective therapies for these potentially fatal diseases.
Pemphigus antigens are desmogleins, transmembrane glycoproteins of desmosomes (cell-to-cell adhesion structures, reviewed in Chapter 53).30,31 Desmogleins are part of the cadherin superfamily of calcium-dependent cell adhesion molecules. The original members of this family (e.g., E-cadherin) demonstrate homophilic adhesive interactions (binding between like molecules). Desmogleins similarly demonstrate homophilic binding but can also participate in heterophilic adhesion by binding desmocollins, the other major transmembrane glycoprotein of desmosomes.32,33
The PF antigen (as well as the fogo selvagem antigen) is desmoglein 1, a 160-kDa protein.27,28,34 The PV antigen is desmoglein 3, a 130-kDa protein that is 64% similar and 46% identical in amino acid sequence to desmoglein 1.29 All patients with PV have antidesmoglein 3 antibodies, and some of these patients also have antidesmoglein 1 antibodies.35,36 Patients with mucosal-dominant PV tend to have only antidesmoglein 3 antibodies, whereas those with mucocutaneous disease usually have both antidesmoglein 3 and antidesmoglein 1 antibodies.37–39 PF patients typically have antibodies against only desmoglein 1.
Several lines of evidence indicate that antidesmoglein 1 and 3 antibodies in pemphigus patients directly cause blisters and hence are the etiologic agents of disease. Passive transfer of PV or PF IgG to neonatal mice or human skin causes blisters that clinically and histologically mimic the corresponding type of pemphigus in patients.40–42 The antidesmoglein antibodies are responsible for blister formation in the passive transfer model, since affinity purified antidesmoglein 1 and 3 autoantibodies cause PF and PV blisters, respectively, and adsorption of desmoglein-reactive autoantibodies from PF or PV IgG abrogates disease.43–46 Similar passive transfer “experiments” have been described in humans, where mothers with even mild PV can pass IgG autoantibodies to the fetus, causing blistering oral and skin disease that resolves by approximately 6 months, concurrent with the disappearance of maternal IgG from the circulation.47
Desmoglein 4, which is expressed in the developing hair cortex and the superficial epidermis, is a target of some pemphigus antibodies.48 However, the antidesmoglein 4 antibodies in mucocutaneous PV and in PF have been shown to be a result of cross-reactivity from desmoglein 1 autoantibodies, and the desmoglein 4 reactivity has not been shown to be necessary or sufficient for acantholysis.49 Other cell surface molecules such as acetylcholine receptors and E-cadherin have also been identified as immunologic targets of pemphigus autoantibodies, although their direct involvement in the pathophysiology of pemphigus is similarly unclear.50,51
 Early ultrastructural studies of the blisters in PV and PF focused on the appearance of desmosomes, because these are the most prominent cell-to-cell adhesion junctions in stratified squamous epithelia (see Fig. 52-2 and Chapter 52). Almost all studies confirm that at various time points during acantholysis, the desmosome is affected and ultimately destroyed, consistent with the cell biologic data discussed in Section “Pathophysiology of Acantholysis.” However, conclusions from electron microscopy studies as to the mechanism of desmosome destruction have varied. Several groups have proposed that the first pathologic event in pemphigus is intercellular widening of interdesmosomal cell membranes, with intact desmosomal junctions.52–55 Other studies have demonstrated half-split desmosomes without keratin tonofilament retraction, suggesting that pemphigus autoantibodies directly interfere with the trans-adhesive interface of desmosomes, and that keratin retraction is secondary to the loss of intercellular adhesion. Half-desmosomes without tonofilament collapse have also been observed in a mouse model of PV.56,57 Others have proposed that keratin retraction is a primary pathogenic event in pemphigus, triggered by cellular signaling after PV autoantibody binding.58 As a potential reconciliation of these findings, one study found that electron microscopic findings may differ depending on the site analyzed: in early blisters, half-desmosomes without keratin retraction are observed; in well-developed lesions, keratin retraction from half-desmosomes occurs; and in spongiotic nonblistered skin, intercellular widening with intact desmosomal junctions can be found, similar to electron microscopy findings in other spongiotic epidermal diseases. The conclusion from these studies is that desmosomal splitting is a primary ultrastructural change of acantholytic lesions in pemphigus.59 Currently, electron microscopy studies are not part of the clinical diagnostic workup for pemphigus.
Early ultrastructural studies of the blisters in PV and PF focused on the appearance of desmosomes, because these are the most prominent cell-to-cell adhesion junctions in stratified squamous epithelia (see Fig. 52-2 and Chapter 52). Almost all studies confirm that at various time points during acantholysis, the desmosome is affected and ultimately destroyed, consistent with the cell biologic data discussed in Section “Pathophysiology of Acantholysis.” However, conclusions from electron microscopy studies as to the mechanism of desmosome destruction have varied. Several groups have proposed that the first pathologic event in pemphigus is intercellular widening of interdesmosomal cell membranes, with intact desmosomal junctions.52–55 Other studies have demonstrated half-split desmosomes without keratin tonofilament retraction, suggesting that pemphigus autoantibodies directly interfere with the trans-adhesive interface of desmosomes, and that keratin retraction is secondary to the loss of intercellular adhesion. Half-desmosomes without tonofilament collapse have also been observed in a mouse model of PV.56,57 Others have proposed that keratin retraction is a primary pathogenic event in pemphigus, triggered by cellular signaling after PV autoantibody binding.58 As a potential reconciliation of these findings, one study found that electron microscopic findings may differ depending on the site analyzed: in early blisters, half-desmosomes without keratin retraction are observed; in well-developed lesions, keratin retraction from half-desmosomes occurs; and in spongiotic nonblistered skin, intercellular widening with intact desmosomal junctions can be found, similar to electron microscopy findings in other spongiotic epidermal diseases. The conclusion from these studies is that desmosomal splitting is a primary ultrastructural change of acantholytic lesions in pemphigus.59 Currently, electron microscopy studies are not part of the clinical diagnostic workup for pemphigus.
Unlike many other autoantibody-mediated diseases, such as pemphigoid and epidermolysis bullosa acquisita, in which the constant region of the antibody is required for blister formation to activate complement or bind antibody receptors on inflammatory cells, in pemphigus the variable region of the antibody is sufficient to cause blisters in neonatal mice or human skin.42,60–62 For this reason a significant amount of research on disease pathophysiology has focused on the epitopes bound by pathogenic autoantibodies, as these regions are likely critical for maintaining desmosomal cell adhesion.
Epitope mapping studies have shown that pathogenic PV and PF autoantibodies bind calcium-sensitive, conformational epitopes in the amino-terminal extracellular domains of desmogleins, whereas nonpathogenic antibodies tend to bind more membrane proximal extracellular domains.63–66 The amino-terminal domains bound by pathogenic autoantibodies are the same domains that are predicted to form the key molecular interactions for desmoglein intercellular adhesion, based on studies of cadherin ultrastructure.67,68 This evidence is the primary basis for the “steric hindrance” hypothesis, which proposes that pathogenic antibodies directly interfere with desmoglein adhesive interactions, causing acantholysis.
Studies on cultured keratinocytes have indicated that loss of intercellular adhesion by pathogenic autoantibodies leads to internalization and degradation of desmogleins,69–72 indicating that pemphigus antibody binding leads to loss of desmoglein function. If this is the case, then other model systems with loss of desmoglein function should mimic pemphigus. Indeed, mice genetically deficient for desmoglein 3 demonstrate suprabasal blisters in the oral mucosa histologically identical to PV patients.73 Additionally, cleavage of desmoglein 1 by staphylococcal exfoliative toxin (in bullous impetigo or staphylococcal scalded skin syndrome) causes blisters histologically identical to those seen in PF patients.74
If inactivation of desmoglein isoforms results in blistering, then why do blisters in PV and PF have specific tissue localizations that do not necessarily correlate with the sites at which the antibodies bind by immunofluorescence? In PF, for example, the antidesmoglein 1 antibodies bind throughout the epidermis and mucous membranes,75 yet blisters occur only in the superficial epidermis. This apparent paradox can be explained by desmoglein compensation, as outlined in Fig. 54-1. The concept of desmoglein compensation originates in the assumption that autoantibodies against one desmoglein isoform inactivate only that isoform and that another isoform coexpressed in the same area can compensate in adhesion.76–78 Desmoglein compensation explains why neonatal PF is so unusual, because even though the maternal antidesmoglein 1 antibodies cross the placenta, in neonatal skin, but not in adult skin, desmoglein 3 is coexpressed with desmoglein 1 in the superficial epidermis, thereby providing protection against the loss of desmoglein 1-based adhesion.77,79 Desmoglein compensation also offers an explanation for the differing sites of blister formation in PV and PF, both in regard to the histology (i.e., suprabasal or superficial), as well as the areas of involvement (mucosa and/or skin.)
Figure 54-1
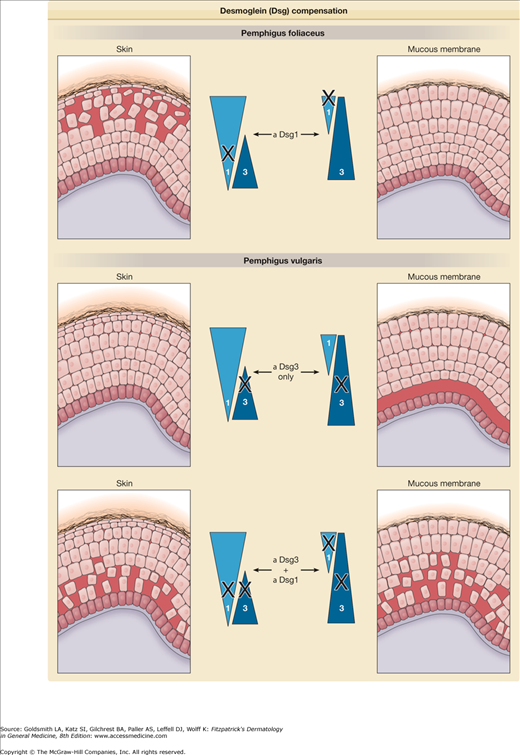
Desmoglein (Dsg) compensation. Triangles represent the distribution of Dsg1 and 3 in skin and mucous membranes. Anti-Dsg1 antibodies in pemphigus foliaceus cause acantholysis only in the superficial epidermis of skin. In the deep epidermis and in mucous membranes, Dsg3 compensates for antibody-induced loss of function of Dsg1. In early pemphigus vulgaris, antibodies are present only against Dsg3, which cause blisters only in the deep mucous membrane where Dsg3 is present without compensatory Dsg1. However, in mucocutaneous pemphigus, antibodies against both Dsg1 and Dsg3 are present, and blisters form in both mucous membrane and skin. The blister is deep probably because antibodies diffuse from the dermis and interfere first with the function of desmosomes at the base of the epidermis.
In potential challenge to the steric hindrance hypothesis, several studies have suggested that modulation of cell signaling pathways can prevent blister formation after passive transfer of pemphigus IgG in the neonatal mouse model, including p38 mitogen activated protein kinase (MAPK) and ρ GTPases, among others.80–82 Whether signaling is upstream or downstream of the loss of intercellular adhesion is controversial. Nevertheless, the current general consensus is that desmosomal adhesion is a dynamic process that is perturbed by pemphigus autoantibodies. Therefore, therapies that aim to strengthen keratinocyte adhesion by modulation of signaling pathways may have a beneficial effect on pemphigus, regardless of whether cell signaling is a primary pathologic cause of disease.
Compared to a matched population, patients with PV have a markedly increased frequency of certain class II major histocompatibility complex (MHC) antigens. Among Ashkenazi Jews with PV, the serologically defined HLA-DR4 haplotype is predominant, whereas in other ethnic groups with PV, the DQ1 allele is more common.83 However, the association with disease susceptibility becomes even more striking in an analysis of these MHC alleles at a genetic level. Patients with the DR4 serotype almost all have the unusual allele DRB1*0402, and patients with the DQ1 serotype almost all have the rare allele DQB1*0503. Similar, but less restricted, HLA-DR alleles are associated with PF.84 The protein chains encoded by these PV MHC II alleles vary from those found in HLA-DR4 and DQ1 controls without disease by only a few amino acids.
MHC class II alleles encode cell surface molecules that are necessary for antigen presentation to the immune system; therefore, it is hypothesized that PV-associated MHC class II molecules allow presentation of desmoglein 3 peptides to T cells.85 Consistent with this hypothesis, certain peptides from desmoglein 3, predicted to fit into the DRB1*0402 peptide-binding pocket, were found to stimulate T cells from patients.86
Other studies have confirmed that the immune response in pemphigus is restricted to certain desmoglein peptides and MHC class II alleles.87–89 An unexpected observation was that T cells of normal people with the DRB1*0402 or DQB1*0503 respond just as well as those of pemphigus patients to the same desmoglein 3 peptides,85,90 indicating that T-cell reactivity to desmoglein 3 peptides is not sufficient for disease onset. The factor that may determine who gets pemphigus and who does not has been proposed to be the presence of regulatory T cells that can suppress the autoimmune response in those who do not.91
Cloning of antidesmoglein antibodies from PV and PF patients has indicated a marked restriction of antibody gene usage by the antidesmoglein antibodies, most notably for the heavy chain variable region.61,62,92 These studies also show that pathogenic antibodies from different patients bind at or near common epitopes on desmogleins and may share common idiotypes. In comparison to the restricted B-cell antibody variable region gene usage, there is more heterogeneity of the T-cell receptor variable gene usage in pemphigus patients.90,93 If specific antibody or T-cell receptor gene usage patterns are found to be shared among multiple pemphigus patients, these may serve as clinical markers for targeting disease-specific immune cell populations in pemphigus patients.
Clinical Findings
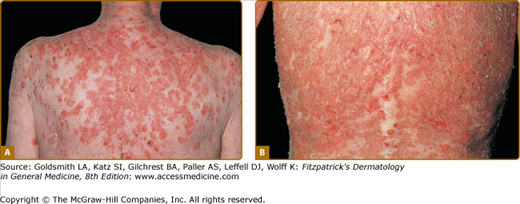
The skin lesions in PV can be pruritic or painful. Exposure to ultraviolet radiation may exacerbate disease activity.94,95 The primary lesion of PV is a flaccid blister, which may occur anywhere on the skin surface, but typically not the palms and soles (Fig. 54-2). Usually, the blister arises on normal-appearing skin, but it may develop on erythematous skin. Because PV blisters are fragile, the most common skin lesions observed in patients are erosions resulting from broken blisters. These erosions are often quite large, as they have a tendency to spread at their periphery (Fig. 54-3).
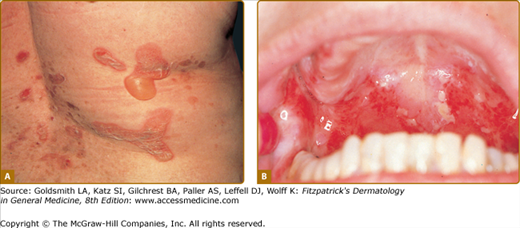
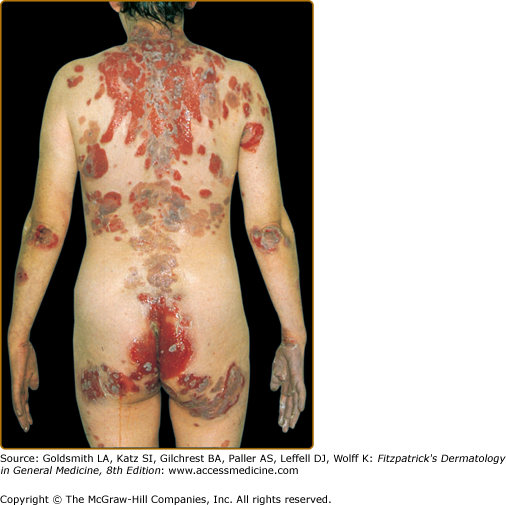
A characteristic finding in pemphigus patients is that erosions can be extended into visibly normal skin by pulling the remnant of the blister wall or rubbing at the periphery of active lesions; additionally, erosions can be induced in normal-appearing skin distant from active lesions by pressure or mechanical shear force. This phenomenon is known as the Nikolsky sign.96 This sign helps differentiate pemphigus from other blistering diseases of the skin such as pemphigoid (Box 54-1); however, similar findings can also be elicited in staphylococcal scalded skin syndrome, Stevens–Johnson syndrome, and toxic epidermal necrolysis.
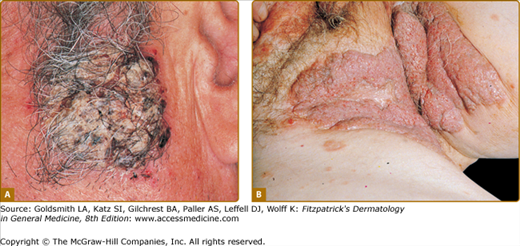
In certain patients, erosions have a tendency to develop excessive granulation tissue and crusting, referred to as vegetating lesions (Fig. 54-4). This type of lesion tends to occur more frequently in intertriginous areas, in the scalp, or on the face (see Fig. 54-4A). Historically, patients presenting with vegetating lesions have been split out into different disease designations: pemphigus vegetans of Hallopeau and pemphigus vegetans of Neumann. However, the subsequent analysis of vegetating skin lesions by histology and immunofluorescence suggests that these cases are simply clinical variants of PV.1,97 In the Hallopeau variant, vegetating and often pustular lesions are present from the outset of disease, are not preceded by bullae, and favor flexural regions (see Fig. 54-4B). Generally, the prognosis for these patients is thought to be better, with milder disease and a higher chance of remission compared to typical PV patients.98 In patients with the Neumann variant, ordinary PV erosions heal with papillomatous formations, with prognosis related to the extent of disease activity. The vegetating type of response may also appear in certain lesions that tend to be resistant to therapy and remain for long periods of time in one place. Thus, vegetating lesions seem to be one reactive pattern of the skin to the autoimmune insult of PV.
The mucous membranes most often affected by PV are those of the oropharyngeal cavity (see Fig. 54-2B). As with cutaneous lesions, intact blisters are rare. Oropharyngeal erosions can be so painful that the patient is unable to eat or drink. The inability to eat or drink adequately may require inpatient hospitalization for disease control and intravenous fluid and nutrient repletion.
In the majority of patients, painful mucous membrane erosions are the presenting sign of PV and may be the only sign for an average of 5 months before skin lesions develop.3 However, the presenting symptoms may vary; in a study from Croatia, painful oral lesions were the presenting symptom in 32% of patients.20 Most of these patients progressed to a more generalized eruption in 5 months to 1 year; however, some had oral lesions for more than 5 years before generalization. On the other hand, in Tehran, 62% of patients presented with oral lesions only.7 Skin involvement without mucous membrane involvement in PV is less common, accounting in one study for 11% of PV cases.99
Gastrointestinal tract involvement with PV has been described in the esophagus, stomach, duodenum, and anus, although only biopsies of the esophagus have been proven to be due to suprabasal acantholysis.7,100,101 Involvement of other mucous membranes can also occur, including the vulvovaginal, nasal, laryngeal, and conjunctival mucosa.102–106 In women, cervicovaginal lesions may be found in up to 51% of patients with active disease but these lesions may be asymptomatic. Even without obvious lesions, Pap smears may be positive in women with pemphigus and the acantholytic cells may be misinterpreted as indicative of cervical dysplasia.107,108 There are rare case reports on corneal erosions in PV patients, but no histologic confirmation of acantholysis.109
The characteristic clinical lesions of PF are scaly, crusted erosions, often on an erythematous base. In more localized and early disease, these lesions are usually well demarcated and scattered in a seborrheic distribution, including the face, scalp, and upper trunk (Fig. 54-5A). The primary lesions of small flaccid blisters are typically not found. Disease may stay localized for years, or it may rapidly progress to generalized involvement, resulting in an exfoliative erythroderma (Fig. 54-5B). Like PV, PF may be exacerbated by ultraviolet radiation.95,110,111 Patients with PF often complain of pain and burning in the skin lesions. In contrast to patients with PV, those with PF very rarely, if ever, have mucous membrane involvement, even with widespread disease.
The colloquial term for Brazilian endemic pemphigus, fogo selvagem (Portuguese for “wild fire”), takes into account many of the clinical aspects of this disease: the burning feeling of the skin, the exacerbation of disease by the sun, and the crusted lesions that make the patients appear as if they had been burned.
In 1926, Francis Senear and Barney Usher described eleven patients with features of a pemphigus–lupus erythematosus overlap (Senear–Usher syndrome).112 Over the next several decades, debate over whether these patients had lupus erythematosus, pemphigus, seborrheic dermatitis, or features of all three disorders continued, with Senear concluding that the disease is best considered a variant of pemphigus, termed pemphigus erythematosus.113 As these observations were made prior to the development of immunofluorescence testing for both pemphigus and lupus, the diagnosis was primarily based on the clinical presentation: crusted erosions in a seborrheic distribution, at times concurrent with more lupus-like discoid lesions with “carpet-tack” scale. Walter Lever noted that many patients initially categorized as pemphigus erythematosus went on to develop systemic lupus, or more widespread PF, or even PV, in some cases due to incorrect initial diagnosis. Therefore, rather than perpetuate the use of one term for different diseases, he proposed that pemphigus erythematosus be used to describe a localized form of PF with better prognosis.1 After the development of immunofluorescence and antinuclear antibody testing for pemphigus and lupus, it was discovered that pemphigus erythematosus patients demonstrate immunologic overlap features; by definition all demonstrate the cell surface staining pattern classic for pemphigus, approximately 30% have positive antinuclear antibody titers, and 80% have positive lupus band tests, although the latter test is only positive in 20%–40% of biopsies on nonsun-exposed skin.114 As most patients with pemphigus erythematosus do not develop systemic signs or symptoms of lupus, and some may progress from localized disease to generalized PF,115 the diagnosis of pemphigus erythematosus is largely one of historic, rather than clinical, significance.
Infants born to mothers with PV may display clinical, histologic, and immunopathologic signs of PV.47,116 The degree of involvement varies from none to severe enough to result in a stillbirth. If the infant survives, disease tends to remit as maternal antibody is catabolized. Mothers with PF may also transmit their autoantibodies to the fetus, but, as discussed in Section “Pathophysiology of Acantholysis,” neonatal PF occurs only rarely.117–119 Neonatal pemphigus should be distinguished from PV and PF that occur in childhood, which are similar to the autoimmune diseases seen in adults.120
Although there are sporadic case reports of pemphigus associated with the use of several different drugs, the association with penicillamine, and perhaps captopril, is the most significant.121 The prevalence of pemphigus in penicillamine users is estimated to be approximately 7%. PF (including pemphigus erythematosus) is more common than PV in these penicillamine-treated patients, although either may occur. The findings of direct and indirect immunofluorescence are positive in most of these patients. Three patients with drug-induced PF and one with drug-induced PV have been shown to have autoantibodies to the same molecules involved in sporadic pemphigus, namely, desmoglein 1 and desmoglein 3, respectively.122 Therefore, by immunofluorescence and immunochemical determinations, these patients with drug-induced pemphigus resemble those with sporadic disease.
Both penicillamine and captopril contain sulfhydryl groups that are postulated to interact with the sulfhydryl groups in desmoglein 1, 3, or both, thereby causing pemphigus either by directly interfering with these adhesion molecules or, more likely, by modifying them so that they become more antigenic. The use of these drugs may also lead to a more generalized dysregulation of the immune response, allowing production of other autoantibodies such as those resulting in myasthenia gravis. Most, but not all, patients with drug-induced pemphigus go into remission after they stop taking the offending drug.
Additionally, rare anecdotal reports have suggested the association of dietary intake and pemphigus, proposing the hypothesis that thiol-containing foods such as garlic, leeks, and onions may precipitate disease.123,124 Some patients may note that certain foods aggravate oral lesions, but it is unlikely that dietary intervention alone will remit disease in most patients.
Interestingly, anecdotal case reports have reported improvement of PV with cigarette smoking,125 as well as with the cholinergic agonists pyridostigmine, carbachol, and pilocarpine.126,127 Studies suggest that activation of cholinergic receptors may regulate signaling pathways modulated by PV IgG, thereby affecting cell adhesion.128 These results are intriguing given the clinical benefit of nicotine noted in other inflammatory diseases, such as ulcerative colitis.129
Myasthenia gravis, thymoma, or both have been associated with PV and PF.130 Approximately one-half of associated pemphigus cases are vulgaris; one-half, foliaceus or erythematosus. Most of these data, however, were reported before the recognition of paraneoplastic pemphigus as a distinct entity. Therefore, although thymoma may clearly be associated with PV and PF, it may also be associated with paraneoplastic pemphigus (see Chapter 55). Myasthenia gravis is a tissue-specific autoantibody-mediated disease leading to skeletal muscle weakness. Early disease usually affects facial muscles, leading to symptoms of dysarthria, dysphagia, ptosis, or diplopia. Disease may then progress to affect the larger muscles of the trunk and extremities, with potential fatal complications from respiratory muscle involvement. Thymoma, in contrast, is typically asymptomatic in adults. In children, thymomas are more likely to be symptomatic with cough, chest pain, superior vena cava syndrome, dysphagia, and/or hoarseness from localized tumor encroachment.
Myasthenia gravis would be best evaluated by a neurologist, who can complete a full neurologic examination and may test for the presence of serum acetylcholine receptor autoantibodies. The course of myasthenia gravis and pemphigus appear to be independent of each other. Likewise, thymic abnormalities may either precede or follow the onset of pemphigus. Thymic abnormalities include benign or malignant thymoma and thymic hyperplasia. Posteroanterior and lateral chest radiographs with or without computerized tomography follow-up can detect most thymomas. Irradiation of the thymus or thymectomy, although clearly beneficial for myasthenia gravis, may not improve the pemphigus disease activity. Although this association is reported in at least 30 cases, the finding of thymoma or myasthenia gravis in a patient with PV or PF is still unusual.
Laboratory Tests
Diagnosis of pemphigus relies on skin biopsy of a fresh lesion for histology to determine the site of blister formation, as well as a confirmatory immunochemical study to document the presence of skin autoantibodies, either by direct immunofluorescence of perilesional skin, or indirect immunofluorescence or ELISA of patient serum.
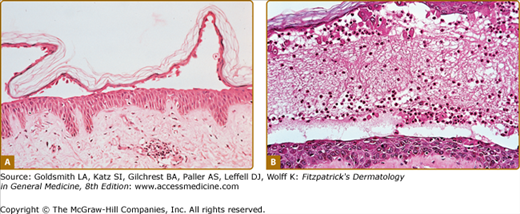
The characteristic histopathologic finding in PV is a suprabasal blister with acantholysis (Fig. 54-6). Just above the basal cell layer, epidermal cells lose their normal cell-to-cell contacts and form a blister. Often, a few rounded up (acantholytic) keratinocytes are in the blister cavity. The basal cells stay attached to the basement membrane, but may lose the contact with their neighbors; as a result, they may appear to be a “row of tombstones,” symbolic of the potentially fatal prognosis of this disease. Usually, the upper epidermis (from one or two cell layers above the basal cells) remains intact, as these cells maintain their cell adhesion. Pemphigus vegetans shows not only suprabasilar acantholysis, but also papillomatosis of the dermal papillae and downward growth of epidermal stands into the dermis, with hyperkeratosis and scale-crust formation. In addition, pemphigus vegetans lesions may show intraepidermal abscesses composed of eosinophils and/or neutrophils.131 Early PV lesions may show eosinophilic spongiosis.132
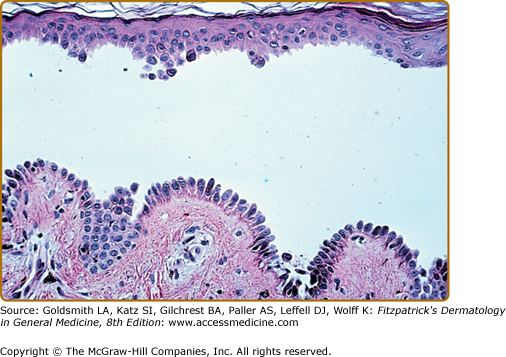
The histopathology of early blisters in PF patients demonstrates acantholysis (loss of cell-to-cell contact) just below the stratum corneum and in the granular layer (Fig. 54-7A). The stratum corneum is often lost from the surface of these lesions. The deeper epidermis, below the granular layer, remains intact. Another frequent finding is subcorneal pustules, with neutrophils and acantholytic epidermal cells in the blister cavity (Fig. 54-7B). Histologic findings in PF are often indistinguishable from those seen in bullous impetigo/staphylococcal scalded skin syndrome, because blisters in these latter diseases also result from dysfunction of desmoglein 1, in these cases due to proteolytic cleavage by staphylococcal exfoliative toxins.31 Therefore, immunochemical studies are essential to confirm a diagnosis of PF, as these would be negative in staphylococcal-mediated skin blisters. The site of blister formation in pemphigus erythematosus is identical to PF. As in PV lesions, very early PF lesions may show eosinophilic spongiosis.132
The hallmark of pemphigus is the finding of IgG autoantibodies against the cell surface of keratinocytes. These autoantibodies were first discovered in patients’ sera by indirect immunofluorescence techniques and soon thereafter were discovered by direct immunofluorescence of patients’ skin.133
Essentially all patients with active PV or PF have a positive finding on a direct immunofluorescence study, which tests for IgG bound to the cell surface of keratinocytes in perilesional skin (Fig. 54-8A).134 This is a nonquantitative test (either negative or positive). The diagnosis of pemphigus should be seriously questioned if the test result of direct immunofluorescence is negative. It is important that the biopsy for direct immunofluorescence be performed on normal-appearing perilesional skin, as the immune reactants can be difficult to detect in blistered inflamed epidermis (leading to a false negative result). In some cases of pemphigus erythematosus, IgG and C3 are deposited at the basement membrane zone of erythematous facial skin, in addition to the epidermal cell surface IgG, representing a positive lupus band test in addition to the typical pemphigus intercellular pattern.135
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


