and Iris Lim Trinh3
(1)
University of Florida, College of Medicine, Gainesville, FL, USA
(2)
Private Practice:, Orlando, FL, USA
(3)
Pediatrician, Private Practice, Orlando, FL, USA
2.1 Neonatal Dermatology
Transient Neonatal Pustular Melanosis (Figure 2.1A)
Onset at birth; common in darkly pigmented infants
Presents with small pustules or residual hyperpigmented macules with collarette of scale
Smear of sterile pustule shows numerous neutrophils
Histology: subcorneal pustules with neutrophils
Erythema Toxicum Neonatorum
Onset typically 24–48 hours after birth; occurs in half of all full term infants
Presents with blotchy erythematous macules, papules, pustules and wheals
Smear of sterile vesicle/pustule shows eosinophils
Histology: subcorneal pustules with eosinophils associated with pilosebaceous unit
Onset typically within first 30 days (vs. infantile acne between 1-12 months)
Presents with erythematous follicular comedones, papules and pustules on face; Malassezia spp. implicated
Histology: follicular pustules with neutrophils
Sclerema Neonatorum
Onset usually within first week of life; form of panniculitis in severely ill, premature infants; often fatal
Presents with diffuse woody hardening of skin; spares genitalia, palms and soles
Histology: needle-shaped clefts with necrotic adipocytes with little surrounding inflammation
Subcutaneous Fat Necrosis of the Newborn (Figure 2.1C)
Onset within first weeks of life; localized form of sclerema neonatorum in healthy infants
Presents with indurated subcutaneous nodules favoring cheeks, shoulders, back, buttocks and thighs
Associated with hypothermia, perinatal hypoxemia (from preeclampsia, meconium aspiration, etc), hypoglycemia
Calcification may occur; ± profound hypercalcemia with resolution so prudent to monitor calcium levels until 1 month after full resolution of lesions
Histology: panniculitis with prominent inflammatory infiltrate with needle-shaped clefts and fat necrosis
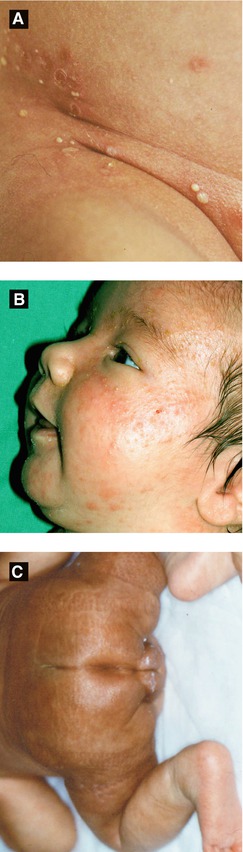
Figure 2.1:
A: Neonatal pustular melanosis*B: Neonatal cephalic pustulosis(Reprint from Boekhout T, Gueho-Kellerman E, Mayser P, Velegraki A. Malassezia and the skin. New York, NY: Springer; 2010.)C: Subcutaneous fat necrosis* *Reprint from Laxer RM, ed. The Hospital for Sick Children: Atlas of Pediatrics. Philadelphia, PA: Current Medicine, 2005.
Pedal Papules of Infancy
Soft, non-painful papules involving heels
Seborrheic Dermatitis
Onset typically 1 week after birth; lasts several months, mostly resolves by 1 year of age
Presents with ill-defined erythematous patches with waxy scale over scalp (‘cradle cap’), ± axillae and groin; lesions may appear psoriasiform
Miliaria Crystallina (MC) or Miliaria Rubra (MR)
Onset first few weeks of life; due to obstructed sweat glands and associated with ↑ temperature (ie. occlusion)
Presents with clear vesicles favoring head, neck and upper trunk (MC) or erythematous papules/vesicles grouped in intertriginous areas or occluded areas (MR)
Onset before birth; localized defect in epidermis, dermis and/or fat; variable appearance, typically along midline
Presents with erosion, ulceration, scar or membranous defect (ovoid lesion covered by an epithelial membrane)
Hair collar sign (Figure 2.2C): ring of dark long hair encircling lesion; ± marker of underlying neural tube defect
Typically isolated abnormality, but may be associated with developmental anomalies or following disorders:
Bart syndrome | ACC of lower extremities + epidermolysis bullosa (DDEB) |
Adams-Oliver syndrome | ACC on scalp (with skull ossification defect) + CMTC + limb defects (reductions, syndactyly) + cardiac abnormalities |
Seitles syndrome | Bilateral temporal ACC + abnormal eyelashes, ‘leonine’ facies, upward-slanting eyebrows |
Cutis Marmorata Telangiectatica Congenita (CMTC) (Figure 2.2D)
Onset at birth; typically improves with age
Presents with blanching reticulated vascular pattern on trunk/extremities with segmental distribution
Associated anomalies in ½ of patients (varicosities, nevus flammeus, macrocephaly, ulceration, hypoplasia and/or hypertrophy of soft tissue and bone)
Sucking Blister
Onset at birth or soon after; due to sucking
Presents with solitary blister (hand, wrist or lip)
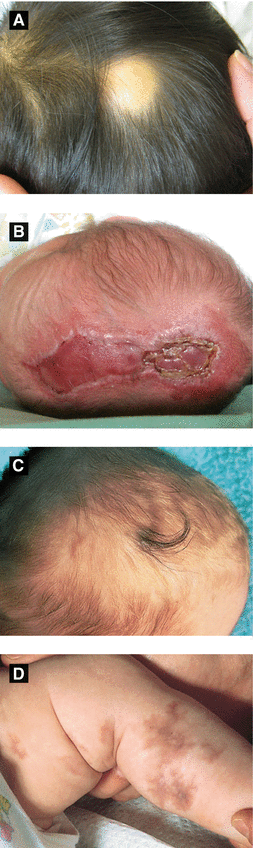
Figure 2.2:
A: ACC, cicatricial(Courtesy of Dr. Michelle B. Bain)B: ACC, ulcerated(Reprint from Lipsker D. Clinical Examination and Differential Diagnosis of Skin Lesions. Paris, France: Springer France;2013.)C: Hair collar sign in ACC*D: CMTC in Adams-Oliver syndrome* *Reprint from RM Trüeb. The Difficult Hair Loss Patient. Zug, Switzerland: Springer; 2015.
Table 2-1:
Congenital Infections of the Newborn
Infection | Clinical Findings | Extracutaneous Findings | Important Points |
|---|---|---|---|
Cytomegalovirus (CMV) | Petechiae, purpura, vesicles and ‘blueberry muffin’ lesions(Fig 2.3A) | Intrauterine growth retardation, chorioretinitis, intracranial calcification | ⇒ Leading infectious cause of deafness and mental retardation ⇒ Typical findings on histology: enlarged endothelial cells with intranuclear inclusions |
Blueberry muffin lesions: red-blue papules/nodules due to dermal erythropoiesis | |||
Herpes Simplex Virus (HSV) | Localized or disseminated skin lesions (vesicles, erosions, scarring) | Encephalitis (predilection for temporal lobes), multi-organ failure, ocular infection | ⇒ Majority HSV2, 85% acquired perinatally ⇒ 50-75% mortality if left untreated |
Rubella | ‘Blueberry muffin’ lesions | Cataracts, deafness, congenital heart disease, CNS findings (microcephaly, hydrocephaly), hepatosplenomegaly (HSM) | ⇒ 50% chance of deafness ⇒ Severe birth defects if within first 16 weeks of pregnancy ⇒ Non-immune pregnant woman transfer the virus to the fetus |
Toxoplasmosis | ‘Blueberry muffin’ lesions favoring the trunk | Ocular abnormalities (chorioretinitis, blindness), CNS (deafness, mental retardation, seizures), thrombocytopenia, intracranial calcification | |
Varicella | Cicatricial skin lesions | Ocular abnormalities (chorioretinitis, cataracts), cortical atrophy, psychomotor retardation, hypoplastic limbs | ⇒ Greatest risk in first 20 weeks ⇒ 2% risk of embryopathy in women with infection within first 2 trimesters |
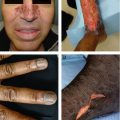
Syphilis, Early Congenital (Fig 2.3B-D) | Syphilitic pemphigus, rhagades (radial furrows/fissures in perioral area, turn into parrot lines), papulosquamous macules/papules (like secondary syphilis) | Snuffles (rhinitis, secondary to ulcerated mucosa), enlarged lymph nodes and spleen, neurosyphilis Be able to differentiate early and late congenital syphilis findings | ⇒ Early congenital syphilis occurs from birth to 2 years of age ⇒ Only congenital syphilis may show bullous lesions ⇒ Papulosquamous lesions common in the diaper area |
Syphilis, Late Congenital | Hutchinson’s teeth, Higoumenakis sign, mulberry molars, saddle nose, saber shins, parrot lines and furrows | Interstitial keratitis, gummas along long bones/skull, tabes dorsalis, generalized paresis | ⇒ Includes permanent sequelae of early congenital signs ⇒ Higoumenakis sign: congenital thickening of the medial aspect of the clavicle |
Table 2-2:
Differential Diagnosis for Diaper Dermatitis
Acrodermatitis Enteropathica(Fig 2.4A–B) | Erythematous crusted patches/plaques with flaccid bullae in perineal, periorificial, and distal extremities; due to ↓ zinc level (also ↓ alkaline phosphatase as zinc-dependent); may occur in following settings: 1. Premature infants (poor absorption and ↑ requirement of zinc) when weaned off breast milk (which has adequate zinc level) 2. Inherited form (AR) manifests when weaned off breast milk 3. Healthy infants if low zinc level in maternal milk 4. Acquired form if malabsorption or inadequate nutrition |
Allergic Contact Dermatitis | Rare in infants, ± related to topical preparations or foods |
Atopic Dermatitis | Increased incidence of diaper dermatitis in atopic patients |
Multiple Carboxylase DeficiencyBiotin Deficiency | Both resemble acrodermatitis enteropathica (periorificial dermatitis); treatment for both forms (listed below) is biotin 1. Neonatal form: AR, holocarboxylase synthetase deficiency, ± erythroderma with alopecia, fatal if not treated 2. Juvenile form: biotinidase deficiency, ± seizures, alopecia, hearing loss, developmental delay |
Bullous Impetigo | Honey-colored crusts and flaccid bullae |
Candidal Dermatitis (Fig 2.3E) | Bright red patches with pustules and satellite papules, ± intertriginous involvement (including scrotum), ± thrush |
Congenital Syphilis (Fig 2.3B-D) | Reddish-brown papulosquamous eruption, may be erosive or bullous |
Cystic Fibrosis | Resembles acrodermatitis enteropathica, also due to zinc deficiency ± pedal edema, failure to thrive, infections and malabsorption |
Granuloma Gluteale Infantum | Red to violaceous granulomatous nodules over the vulva, perianal area, buttocks, ± scrotum; due to irritation, occlusion, candidal infection |
Irritant Dermatitis | Poorly-demarcated erythematous plaques, spares inguinal folds |
Kawasaki Disease | Tender erythema in perineal area which later desquamates |
Langerhans Cell Histiocytosis (Fig 2.3F) | Yellow-brown crusted papules with purpura in seborrheic distribution; ± systemic involvement; Langerhans cells (CD1a +, S100+) |
Miliaria | Clear vesicles or erythematous papules/pustules due to blocked eccrine ducts from heat or humidity in diaper area |
Perianal Pseudo- verrucous Nodules | Erythematous nodules and papules in children with fecal incontinence |
Perianal Strep | Bright red, well-demarcated perianal erythema and involving creases |
Psoriasis | Sharply-demarcated bright pink to red plaques involving inguinal creases, minimal scale; most common psoriatic presentation in infants |
Scabies | Erythematous nodules involving diaper area, ± genitalia |
Seborrheic Dermatitis | Typical salmon-covered scaly patches and plaques involving the scalp, groin and other intertriginous areas |
2.2 Childhood Infectious Diseases
Table 2-3:
Childhood Infections
Disease | Exanthem | Etiology/Course |
|---|---|---|
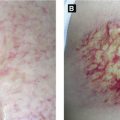
Acute Hemorrhagic Edema of Infancy(Finkelstein Disease) (Fig 2.5A) | Large circinate painful purpuric plaques involving face, ears, distal extremities → evolve into edematous targetoid lesions | Etiology: likely infectious(viral or bacterial) Age: 6 months – 3 years; self-limited Leukocytoclastic vasculitis seen on histology May be hypersensitivity reaction to infection (medication/vaccination less likely) |
Erythema Infectiosum (‘Slapped Cheek’ or Fifth Disease) | Bright red macular erythema over cheeks → lacy eruption mainly on the extremities | Etiology: parvovirus B19 (ssDNA) also causes hydrops fetalis; peaks in spring and winter Age: school-age children; self-limited Mild prodrome, 10% with arthralgias |
Abrupt onset of skin-colored to pink-red edematous papules to cheeks, buttocks, extremities | Etiology: likely infectious (HBV, EBV) Age: 6 months – 2 years; self-limited May have low grade fever and lymphadenopathy | |
Hand-Foot-Mouth Disease (Fig 2.5D) | Elliptical grayish vesicles, pustules, and erosions on hands, feet, and buttocks Oral: vesicles/erosions red base | Etiology: coxsackievirus A16 (enterovirus 71 less often) Age: children < 10 years (± adults); self-limited Fever, sore mouth, anorexia, abdominal pain; enteroviral infection may also cause myocarditis, pneumonia, meningoencephalitis |
Henoch-Schönlein Purpura (HSP) (Fig 2.4C) | Purpuric macules and papules favoring lower extremities and buttocks | Etiology: possibly infectious (viral, strep) Age: peaks at 4–7 years of age (± adults); self-limitedPresents 1–2 weeks after upper respiratory infection Arthralgias, GI bleeding, abdominal pain, nephritis with hematuria → IgA vasculitis |
Herpangina | Exanthem: often absent Oral: painful gray vesicles on tonsillar, palate, buccal mucosa | Etiology: various enteroviruses (often coxsackie group A/B and echovirus) Age: 3–10 years old; self-limited |
Kawasaki Disease (Fig 2.5B) (Mucocutaneous Lymph Node Syndrome) | Polymorphous eruption (morbilliform, erythema multiforme-like or bullous); ± edema and erythema of distal extremities; can be generalized or localized (groin, LE) Oral: red swollen or dry fissured lips; strawberry tongue; pharyngeal erythema | Etiology: unknown but likely infectious Age: children < 5 years of age Arthritis, abdominal pain, GI symptoms Complications: cardiac aneurysm (in ¼ of untreated patients), myocarditis, pericarditis Diagnosis: fever > 5 days + 4 out of 5 of following (CRASH): conjunctivitis • rash • adenopathy (mainly cervical) • swollen lips/strawberry tongue • hand/foot change (edema/desquamation) |
Measles(Rubeola or First Disease) | Erythematous macules/papules over forehead, hairline and behind the ears → spreads downward Oral: Koplik spots (gray papules on buccal mucosa) | Etiology: measles virus (paramyxovirus) Age: unvaccinated children Prodrome: fever, cough, nasal congestion, rhinorrhea, conjunctivitis; rash appears after Koplik spots Complications: encephalitis, otitis media, pneumonia, myocarditis, ± subacute sclerosing panencephalitis |
Infectious Mononucleosis | Polymorphous: morbilliform (common), urticarial, petechial or erythema multiforme-like lesions Of note, morbilliform eruption may occur after treatment with ampicillin | Etiology: infectious (EBV) Age: children, young adults (15–25 years); self-limited Fever, pharyngitis, fatigue, myalgias, headaches, hepatosplenomegaly, lymphadenopathy Complications: splenic rupture, airway obstruction, hepatitis |
Papular Purpuric Gloves and Socks Syndrome (Fig 2.5F) | Erythema, edema, petechiae, and purpura on palms/soles (± extension to dorsal aspect), + burning and pruritus | Etiology: parvovirus B19 Age: children and young adults; self-limited Mild prodromal symptoms, occurs mainly in young adults; peaks in spring |
Roseola (Exanthem Subitum) (Sixth disease) | Circular to elliptical ‘rose red’ macules or papules involving trunk, occasionally surrounded by white halo | Etiology: human herpesvirus 6 (HHV6) Age: 6 months – 3 years Sudden-onset high fever; rash begins as fever subsides Complications in healthy patient: mainly seizures |
Rubella (German Measles) (Third Disease) | Erythematous macules and papules on face → spreads acrally, accompanied by tender lymphadenopathy (occipital, postauricular, cervical) | Etiology: togavirus (ssRNA) Age: unvaccinated children/adults; self-limited Usually mild prodrome Complications: arthralgia/arthritis, hepatitis, myocarditis, pneumonia |
Scarlet Fever (Second Disease) | Erythema of axilla, neck, chest → evolve to pink papules with erythematous background (sandpaper-like) → hand and foot desquamation (7–10 days later); Pastia’s lines (linear petechial streaks in body folds) Oral: ‘red strawberry’ tongue | Etiology: group A β-hemolytic streptococci (erythrogenic toxin A, B, C) Age: children (1–10 years old) Extracutaneous: sore throat, headaches, chills, fever, nausea, abdominal pain, anorexia Treatment: PCN 10–14 days (erythromycin in PCN- allergic pts) |
Unilateral Laterothoracic Exanthem | Morbilliform or eczematous eruption in axilla and lateral trunk with unilateral dominance (± bilateral involvement) | Etiology: likely viral Age: children (6 months – 10 years of age); self-limited |
Varicella(Chickenpox) (Fig 2.5E) | Pruritic, erythematous macules/papules of scalp, face → spreads to trunk and extremities, evolves into vesicles with narrow red halo (‘dew drops on rose petal’), central crust or necrosis seen within lesions | Etiology: varicella zoster virus (VZV) Age: children and adults; self-limited in healthy children Complications in children: secondary bacterial infection Adults with more severe presentation (pneumonia, 10–30% mortality if untreated) All stages of development seen simultaneously |
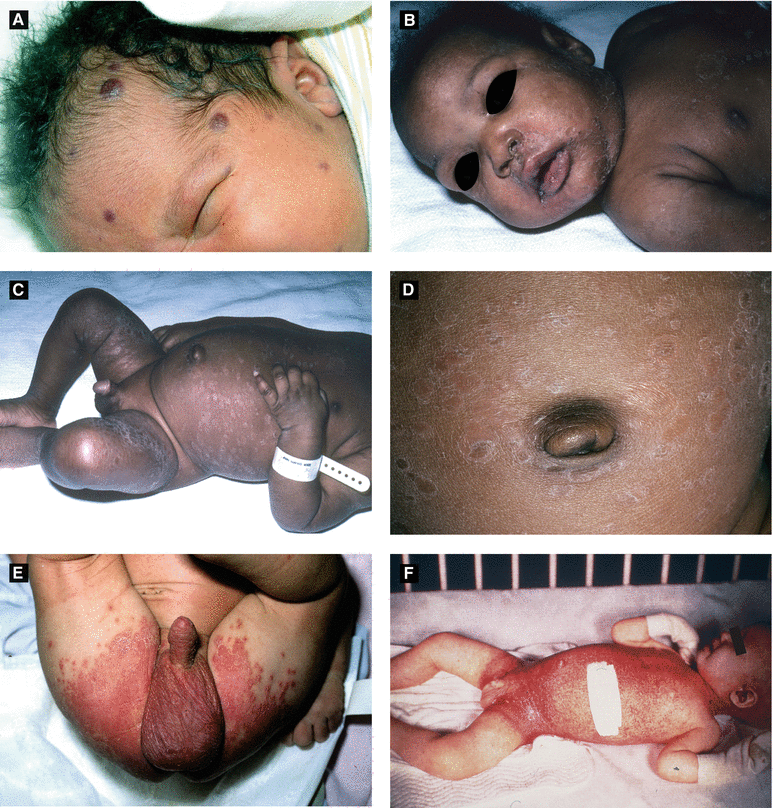
Figure 2.3:
A: Dermal hematopoiesis B: Congenital syphilis(Courtesy of Dr. Vandana Mehta) (Courtesy of Dr. Paul Getz)C: Congenital syphilis D: Congenital syphilis(Courtesy of Dr. Paul Getz) (Courtesy of Dr. Paul Getz)E: Candidiasis F: Langerhans cell histiocytosis(Courtesy of Dr. Paul Getz) (Reprint from Morgan MB, Smoller BR, Somach SC. Deadly Dermatologic Diseases. New York, NY: Springer; 2007.)
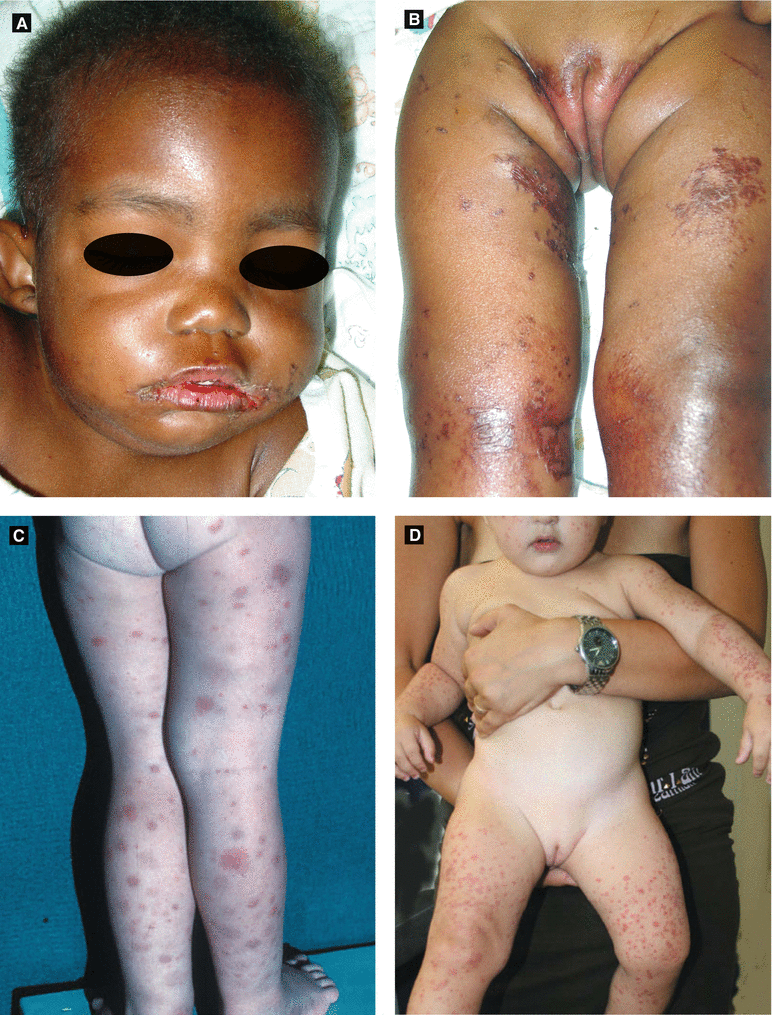
Figure 2.4:
A: Acrodermatitis enteropathica B: Acrodermatitis enteropathica(Courtesy of Dr. Michelle B. Bain) (Courtesy of Dr. Michelle B. Bain)C: Henoch Schonlein purpura D: Gianotti-Crosti syndrome(Reprint from Bonfazi E, (ed). Differential Diagnosis in (Reprint from Bonfazi E, (ed). Differential DiagnosisPediatric Dermatology. Milan, Italy: Springer; 2013.) in Pediatric Dermatology. Milan, Italy: Springer; 2013.)
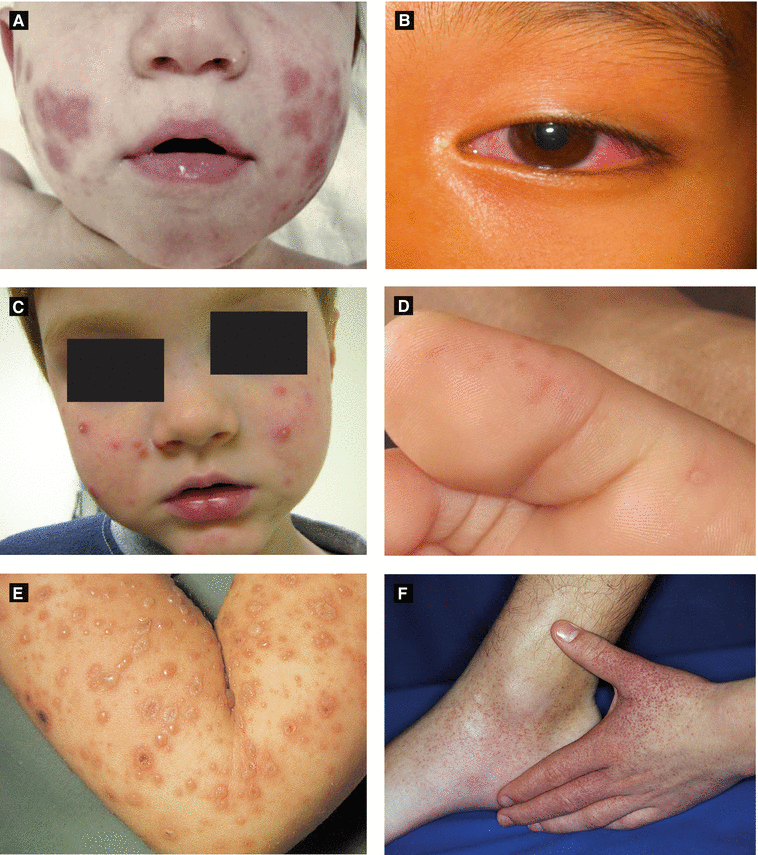
Figure 2.5:
A: Acute hemorrhagic edema of infancy B: Kawasaki disease(Photograph courtesy of SpringerImages Database, (Reprint from Buka B, Uliasz A, Krishnamurthy K (eds). Buka’s Springer Publishing Company) emergencies in dermatology. New York, NY: Springer, 2013)C: Gianotti-Crosti syndrome D: Hand-foot-and-mouth disease(Courtesy of Dr. Michelle B. Bain) (Courtesy of Dr. Sima Jain)E: Varicella F: Papular purpuric gloves and socks syndrome(Reprint from Abdel-Halim AW. Passing (Reprint from Burgdorf WH, Plewig G, Wolff HH, Landthalerthe USMLE. New York, NY: Springer, 2009.) M, eds. Braun-Falco’s Dermatology. 3rd ed. Heidelberg: Springer; 2009)
2.3 Papulosquamous and Eczematous Dermatoses
Psoriasis
Approximately 25% patients will have presentation before age 15
Presents as erythematous well-demarcated plaques with micaceous scale
Guttate psoriasis more common in children; presents with rain drop-like papules in an eruptive pattern; common triggers include strep infection, viral infection, stress and trauma
Pityriasis Lichenoides (Figure 2.6A)
Two diseases forming spectrum: pityriasis lichenoides et varioliformis acuta (PLEVA) and pityriasis lichenoides chronica (PLC)
PLEVA: abrupt onset of erythematous papules and vesicles with crusted or necrotic centers, often involuting within weeks to months; treat with oral erythromycin, phototherapy, and/or topical corticosteroid
PLC: reddish-brown papules with adherent scale, heals with dyschromia; more chronic course lasting months to years
Acropustulosis of Infancy (Figure 2.6B)
Onset from 6 months to 2 years; resolves by age 3
Presents with recurrent crops of pruritic pustules on palms, soles, distal extremities (may mimic scabies infection so prudent to perform mineral oil scraping)
Treatment: topical corticosteroid
Three juvenile forms in addition to two adult forms (I/II)
Classic juvenile form (III) | Resembles classic adult form but with early onset (1st 2 years of life); most resolve within 3 years; 10% cases |
Circumscribed juvenile form (IV) | Lesions on extensor surfaces and present in prepubertal years; 25% cases (50% persist into adulthood) |
Atypical juvenile form (V) | Similar to type III + scleroderma-like changes of hands/feet, familial basis; presents in early childhood with unrelenting course; 5% cases |
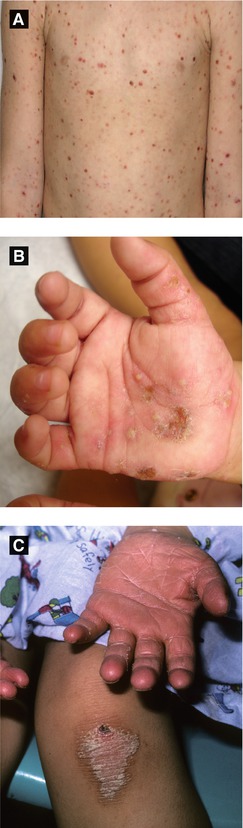
Figure 2.6:
A: PLEVA(Reprint from Tom WL (ed). Severe skin diseases in children: Beyond Topical Therapy. Heidelberg, Germany: Springer; 2013.)B: Acropustulosis of infancyC: Pityriasis rubra pilaris(Courtesy of Dr. Paul Getz)
Pityriasis Rosea (PR)
Self-limited papulosquamous eruption; likely viral pathogen (human herpesvirus 7, less likely HHV 6)
Presents with initial herald patch (precedes eruption by 1-2 weeks) followed by salmon-colored oval patches and plaques with inner scale along long axis of Langer’s lines of cleavage (‘Christmas tree’ pattern on posterior trunk); variants include inverse pattern (flexural accentuation) and papular PR (young children and darker-skinned patients)
Lichen Striatus (Figure 2.7A-B)
Self-limited, linear inflammatory condition in children
Presents with small erythematous scaly papules forming linear band → spreads down extremity or trunk and typically follows lines of Blaschko, ± nail involvement
Hypopigmentation may persist for months to years after lesions resolve and points to diagnosis
Keratosis Pilaris (KP)
Excessive keratinization causing horny follicular plugs on upper arms, thighs and cheeks; associated with atopy
KP Atrophicans (Figure 2.7C)
Group of disorders in children with faulty follicular keratinization followed by atrophy and scarring
KP atrophican faciei: erythema with follicular spiny papules of eyebrows, cheeks and scalp; involute and leave pitted atrophic scars; term ulerythema ophyrogenes if limited to lateral 1/3 of eyebrows, associated with Noonan syndrome
Atrophoderma vermiculata: pit-like atrophic scarring of follicles on face (‘honeycomb’ atrophy), associated with Rombo syndrome, Nicolau-Balus syndrome, and Down syndrome
Rombo syndrome: milia, atrophoderma vermiculata, acral cyanosis, trichoepitheliomas, multiple BCCS, hypotrichosis, alopecia
Nicolau-Balus syndrome: milia, atrophoderma vermiculata, eruptive generalized syringomas (DO NOT confuse with Blau syndrome, which has granulomatous arthritis, uveitis and skin granulomas)
Atopic Dermatitis (AD)
Occurs in 10–15% children, often presenting at 2-3 months of age; multifactorial pathogenesis but includes ↑ secretion of TH2 cytokines (IL-4, IL-5)
Triad of atopy: AD, allergic rhinitis, asthma
Few may have allergy to specific foods, which may exacerbate AD (eggs, milk, soybeans, fish, wheat, peanuts)
Atopic patients with ↓ amount of innate antimicrobial peptides: human β-defensins (HBD) and canthelicidins (LL37)
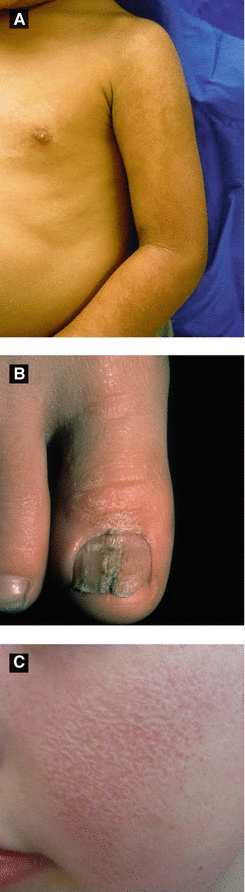
Figure 2.7:
A: Lichen striatus(Courtesy of Dr. Michelle B. Bain)B: Lichen striatus(Courtesy of Dr. Paul Getz)C: Atrophoderma vermiculata(Reprint from Jafari R, Tronnier M. Honigwabenartige Narben an den Wangen Welche Verdachtsdiagnose stellen Sie? pädiatrie: Kinder- und Jugendmedizin hautnah. 2015. © Urban & Vogel 2015)
Presents with eczematous lesions, xerosis and lichenification
Distribution varies with age
Infants: face, scalp, and extensors
Children: antecubital/popliteal fossae, neck, ankles
Adults: typically hands (chronic hand eczema)
Pityriasis alba: hypopigmented patches with minimal scale; may be only manifestation of AD (Figure 2.8A)
Complications: keratoconus (conical deformity of cornea), eyelid dermatitis, ↑ risk of infection (impetigo, eczema herpeticum, molluscum contagiosum)
Treatment: topical corticosteroid, topical calcineurin inhibitor, oral corticosteroid (short course), oral antihistamine, phototherapy; if recalcitrant and severe, consider oral immunosuppressant
Juvenile Plantar Dermatosis (Figure 2.8B)
Typically in children with an atopic diathesis; related to increased humidity from impermeable material in shoes
Presents with dry, scaly glazed patches with fissures involving forefoot plantar surface
Chronic but typically self-limited
2.4 Pigmented Lesions
Café Au Lait Macule (CALM)
Presents as a light to dark brown macule or patch
Single lesion in 10-20% of normal population; multiple lesions ± associated with different genodermatoses (McCune-Albright syndrome, neurofibromatosis)
Lentigines
Presents as brown macules with increased number of melanocytes; no relationship to sunlight
Multiple lentigines ± associated with following: (See Figure 2.10B)
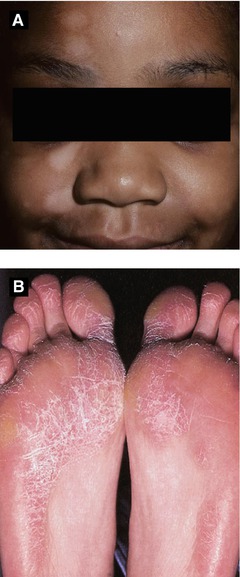
Figure 2.8:
A: Pityriasis alba (Courtesy of Dr. Paul Getz)B: Juvenile plantar dermatosis (Reprint from Johansen JD, Frosch PJ, Lepoittevin J-P (eds). Contact Dermatitis. Heidelberg, Germany: Springer; 2011.)
Bannayan-Riley-Ruvalcaba syndrome | AD, PTEN gene, penile > vulvar lentigines, lipomas, hemangiomas |
Carney complex (LAMB or NAME syndrome) | AD, PRKAR1A gene, psammomatous melanotic schwannomas, cardiac/cutaneous myxomas, blue nevi, endocrine overactivity |
Laugier-Hunziker syndrome | Mucocutaneous lentigines, longitudinal melanonychia, genital melanosis |
LEOPARD syndrome | AD, PTPN11 gene*, café-noir macules, EKG changes, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, growth retardation, deafness |
Peutz-Jeghers syndrome | AD, STK11 gene (serine threonine kinase), mucocutaneous (oral/acral) lentigines, intestinal polyposis, ± intussusception, various malignancies |
Ephelides (Freckles)
Present as light brown macules in sun-exposed areas; more prominent in children with fair skin and during summer time; onset typically within first 3 years of age
Can be a marker for UV-induced damage if acquired
Histology: normal number of melanocytes, increased pigment in keratinocytes
Onset at birth or first year typically; 1–2% of population
Categorized as small (<1.5 cm), medium (1.5-20cm), and large (>20cm or 10% BSA)
Slight ↑ risk of melanoma (highest in large CNs); 3–12% of giant (large) CNs may develop melanoma (studies show varying percentages); axial nevi with greatest risk
If large nevus over scalp, rule out neurocutaneous melanosis with MRI
Neurocutaneous melanosis: ↑ intracranial pressure, leptomeningeal melanoma, spinal cord compression
Presents as dome-shaped red-brown or tan-pink smooth surfaced papule; typically occurs within first 2 decades
Pigmented, congenital and agminated variants reported
Histology: Kamino bodies (PAS+ globules)
Characteristic starburst dermoscopic finding in pigmented Spitz nevi
Halo Nevus (Sutton’s Nevus)
Melanocytic nevus with surrounding hypopigmented halo in which central nevus either persists or involutes
Typically appears in adolescence; may appear in setting of vitiligo; prudent to rule out concomitant melanoma (rare) by performing full skin exam
Presents as tan, regularly-bordered patch with darker macules within lesion
Melanoma rarely arises within nevus component
Associated with phakomatosis pigmentovascularis and pigmentokeratotica (latter with organoid nevus + hemiatrophy + neurologic defects)
Melanoma
0.3-0.4% of melanomas in prepubertal children
↑ Risk with fair skin, blue eyes, blonde/red hair, CDKN2A or p16 mutation, xeroderma pigmentosum, dysplastic nevus syndrome, large congenital nevus or neurocutaneous melanosis
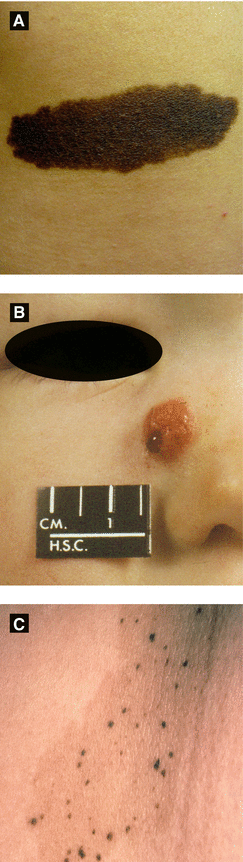
Figure 2.9:
A: Congenital nevusB: Spitz nevus(Reprint from Laxer RM, ed. The Hospital for Sick Children: Atlas of Pediatrics. Philadelphia, PA: Current Medicine; 2005.)C: Nevus spilus(Courtesy of Dr. Paul Getz)
Acquired unilateral lesion found in adolescent males (second or third decade) typically on shoulder, upper chest or back
Presents as hyperpigmented hypertrichotic patch or plaque associated with underlying smooth muscle hamartoma (arrector pili)
Histology: ↑ melanin in epidermis, often smooth muscle hamartoma present in dermis
Blue Nevus (Figure 2.11A)
Congenital or acquired (typically early childhood)
Different types: common, cellular and combined
Multiple blue nevi associated with Carney complex (LAMB/NAME syndrome)
Histology: normal epidermis, many elongated dendritic melanocytes within dermis, large amounts of melanin often seen within melanocytes
Similar presentation to nevus of Ota but typically occurs in shoulder region (supraclavicular, scapular and deltoid)
Nevus of Ota (Nevus Fuscoceruleus Ophthalmomaxillaris, Oculodermal Melanocytosis) (Figure 2.10D, 2.11B)
Onset either near birth or during puberty
Most common in Asian population, mainly women
Presents as unilateral, blue-gray macules typically involving V1 and V2 distribution of trigeminal nerve
Most common extracutaneous sites: sclera > tympanum > nasal mucosa > pharynx > palate
Hori’s Nevus (Acquired Nevus of Ota-like Macules)
Onset in late adolescence, mainly in Asian women
Bilateral nevus of Ota-like macules of the zygomatic region; may be misdiagnosed as melasma
Congenital Dermal Melanocytosis (Mongolian Spot) (Figure 2.11C)
Common in infants with pigmented skin
Presents with blue-gray macules or patches typically over lumbosacral skin or buttocks
If extensive, consider phakomatosis pigmentovascularis
Histology: dendritic melanocytes situated in lower half of dermis, cells arranged parallel to epidermis
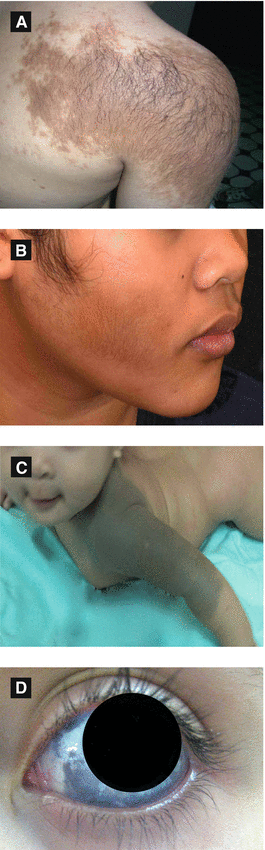
Figure 2.10:
A: Becker’s nevus*B: Becker’s nevus**C: Nevus of Ito**D: Nevus of Ota* *Reprint from Baykal C, Yazganoğlu KD (eds). Clinical Atlas of Skin Tumors. Heidelberg, Germany. Springer; 2014. **Reprint from Silverberg NB, Durán-McKinster C, Tay Y-K (eds). Pediatric Skin of color. New York, NY. Springer; 2015.
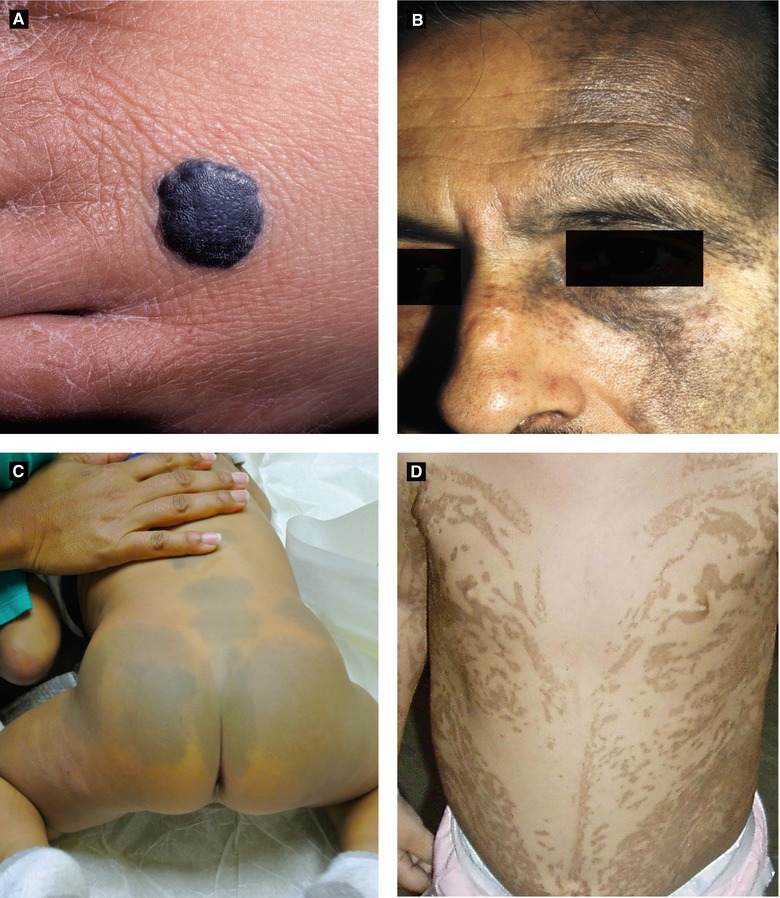
Figure 2.11:
A: Blue nevus B: Nevus of Ota(Courtesy of Dr. Paul Getz) (Reprint from Baykal C, Yazganoğlu KD (eds). Clinical Atlas of Skin Tumors. Heidelberg, Germany. Springer; 2014.)C: Dermal melanocytosis D: Linear and whorled nevoid hypermelanosis(Courtesy of Dr. Sima Jain) (Reprint from Silverberg NB, Durán-McKinster C, Tay Y-K. (eds). Pediatric Skin of Color. New York, NY. Springer; 2015.)
Linear and Whorled Nevoid Hypermelanosis (LWNH) (Figure 2.11D)
Appears within early infancy; progressive for 1-2 years before stabilization; persists indefinitely
Reticular hyperpigmented macules and patches in streaky configuration following lines of Blaschko
2.5 Hypopigmented Lesions
Table 2-4:
Hypopigmented/Depigmented Lesions in Children
Hypopigmented Lesions | |
|---|---|
Nevus depigmentosus (Fig 2.12A) | Congenital (at birth or within first few years), non-progressive hypopigmented macule or patch; most common site – trunk |
Nevus anemicus (Fig 2.12B-C) | Hypopigmented patch due to persistent vasoconstriction of papillary vessels; rubbing only reddens peripheral uninvolved skin; under diascopy, peripheral skin same as involved skin |
Hypomelanosis of Ito (Pigmentary mosaicism) | Descriptive term (not diagnosis) of hypopigmentation along lines of Blaschko; ± extracutaneous involvement (includes CNS, musculoskeletal, dental, cardiac and ocular abnormalities); thought to be due to chromosomal mosaicism and sporadic mutations; also known as nevoid hypopigmentation ± systemic involvement |
“Ash leaf” spots (Fig 2.12D) | Ovoid or ash leaf-shaped hypopigmented macules or patches at birth or early infancy; 3 or more lesions suggest tuberous sclerosis |
Postinflammatory hypopigmentation | Hypopigmentation due to preceding inflammatory skin disorder |
Depigmented Lesions (see Section 2.9 ) | |
Piebaldism | Leukoderma and white forelock (poliosis); depigmented patches with islands of normal to hyperpigmented macules within affected areas; typically involving forehead, central chest, abdomen, upper arms and lower legs |
Waardenburg syndrome | White forelock and depigmented patches (similar to piebaldism); 4 genetic subtypes; heterochromia irides, dystopia canthorum, ± hearing loss |
Vitiligo | Depigmented macules and/or patches; may present during infantile period but not birth |
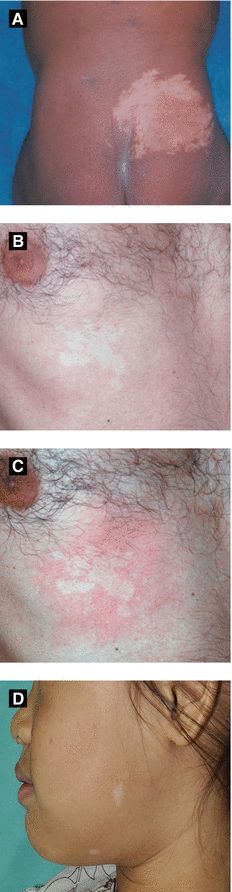
Figure 2.12:
A: Nevus depigmentosus*B: Nevus anemicus*C: Nevus anemicus, after rubbing*D: “Ash leaf” spots** *Reprint from Bonifazi E (ed). Differential Diagnosis in Pediatric Dermatology. Milan, Italy: Springer; 2013.**Reprint from Silverberg NB, Durán-McKinster C, Tay Y-K (eds). Pediatric Skin of Color. New York, NY. Springer; 2015.
2.6 Bullous Diseases
Table 2-5:
Epidermolysis Bullosa
EB Subtype | Inh | Gene | Clinical Features |
|---|---|---|---|
EB SIMPLEX (EBS) Split: Epidermal Basal Layer | |||
Dowling-Meara (EBS Herpetiformis) | AD | K5/K14 | Onset at birth, grouped or herpetiform blisters (figurate), significant mucosal membrane and laryngeal/esophageal involvement (± hoarseness), nail dystrophy, confluent PPK, scarring, early death EM: clumped tonofilaments in basal keratinocytes |
Weber-Cockayne (Localized) | AD | K5/K14 | Onset typically childhood/adolescence, palmoplantar bullae/erosions, heal without scarring |
Koebner (Generalized) | AD | K5/K14 | Generalized bullae at birth, PPK, nail dystrophy, mucosal erosions, heals without scarring |
EBS Muscular Dystrophy | AR | Plectin | Widespread bullae at birth, muscular dystrophy, scarring, hair/nail/tooth/oral disease, early death |
EBS Mottled Digmentation | Resembles localized and generalized EBS + reticulated hyperpigmentation over trunk | ||
JUNCTIONAL EB (JEB) Split: Basement Membrane (Lamina Lucida) | |||
Herlitz (EB Lethalis) Premature termination codon | AR | Laminin 5 (laminin-332) | Severe, widespread bullae, nonhealing exuberant granulation tissue (perioral, axillae, neck), enamel defects, absent nails, mucosal involvement (respiratory/GI tract with hoarseness), early death |
Non-Herlitz (Generalized Atrophic Benign EB or GABEB) | AR | Laminin 5 or BPAG2 (BP180) | Widespread bullae at birth, heal with atrophic scars, mild oral involvement, scarring alopecia, nail dystrophy, improves with time |
JEB with Pyloric Atresia | AR | α6β4 integrin | Severe congenital blistering, hydronephrosis, pyloric atresia, mucosal erosions |
DYSTROPHIC EB (DEB) Split: Dermal (Sublamina Densa) | |||
Hallopeau-Siemens Recessive DEB (RDEB-HS) Premature termination codon | AR | Type VII collagen | Severe widespread bullae at birth, heals with atrophic scarring (hands/feet → ‘mitten deformity’), milia, mucosal strictures, nail dystrophy, ↑skin, oral, esophageal SCCs |
Non-Hallopeau-Siemens (RDEB-nHS) | AR | Type VII collagen | Skin changes localized to acral bony prominences, Hallopeau-Siemens symptoms but less severe |
Cockayne-Touraine (DDEB-CT) | AD | Type VII collagen | Bullae mainly over extremities, heal with milia/atrophic scars/keloids, nail dystrophy |
Pasini Variant (DDEB-P) | AD | Type VII collagen | Similar to Cockayne subtype + albopapuloid lesions (white perifollicular papules, slowly enlarge) |
Transient Bullous Dermolysis of the Newborn | AD/AR | Type VII collagen | Uncommon variant of dystrophic EB; benign, self-limited disease without scarring in newborn period |
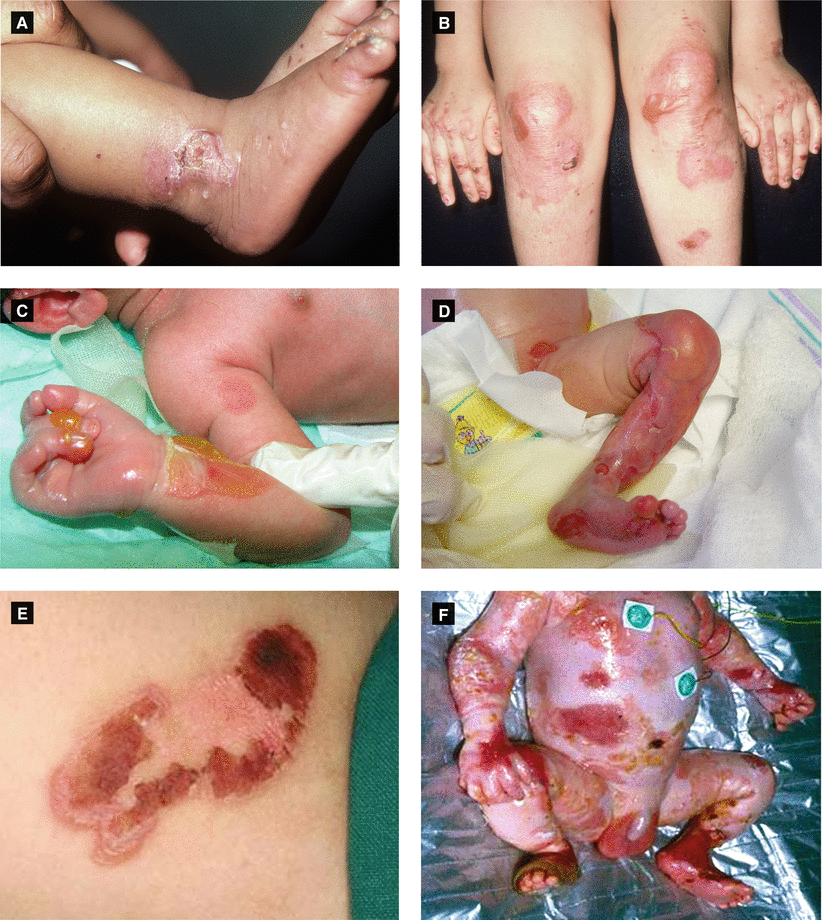
Figure 2.13:
A: EB simplex (Weber-Cockayne) B: Dominant dystrophic EB (Cockayne-Touraine)(Courtesy of Dr. Paul Getz) (Courtesy of Dr. Paul Getz)C: Recessive dystrophic EB D: Recessive dystrophic EBE: EB simplex (Dowling-Meara) F: EB simplex (Dowling-Meara)(Reprint from Laimer M et al. Epidermolysis (Reprint from Has C et al. Hereditare Blasen bildendebullosa hereditaria. Monatsschrift Kinderheilkunde Hauterkrankungen. Der Hautarzt. 2004:55(10);920-30.)Zeitschrift für Kinder und Jugendmedizin.2008:156(2);110-21.)
Blistering disorder with onset typically before age 5
Target antigen: 97 kDa Ag (LAD-1 or LABD97): cleaved ectodomain of BPAG2
Presents with annular and herpetiform bullae favoring extensor surfaces/groin (‘crown of jewels’ configuration)
Histology: subepidermal bullae with neutrophils in dermal papillae (similar to dermatitis herpetiformis)
Treat with dapsone or sulfapyridine
Neonatal Pemphigus
Presents in infants whose mothers have pemphigus vulgaris; due to passive transfer of maternal IgG to fetus
Self-limited; resolves within few weeks of birth
AD, ATP2C1 gene (encodes Golgi-associated Ca2+ ATPase hSPCA1), results in abnormal intracellular calcium signaling; onset typically 2nd to 3rd decade
Presents with flaccid vesicles initially on erythematous base over intertriginous areas, ruptures easily and gives rise to macerated or crusted erosions
Histology: extensive epidermal acantholysis ‘dilapidated brick wall’
Think of ‘Hailey’s Comet’ to remember ATP2 C 1
2.7 Epidermal, Appendageal and Dermal Tumors
Hamartoma of epidermis and papillary dermis; onset typically at birth (± adolescence, rare in adulthood)
Presents as hyperpigmented papillomatous papules and plaques along lines of Blaschko, usually stops at midline
ILVEN (inflammatory linear verrucous epidermal nevus): erythematous scaly plaque along lines of Blaschko; not associated with any neurologic defects
Ichthyosis hystrix (generalized epidermal nevus): extensive bilateral systematized lesions
Nevus comedonicus (NC); if associated ipsilateral cataract and skeletal/CNS defects, think NC syndrome
Epidermal nevus syndrome (Schimmelpenning syndrome): sporadic; widespread EN, underlying CNS, ocular, cardiac and skeletal defects, biopsy to r/o EHK
Of note, if biopsy of EN shows epidermolytic hyperkeratosis, the patient may be at risk for offspring with full-blown EHK
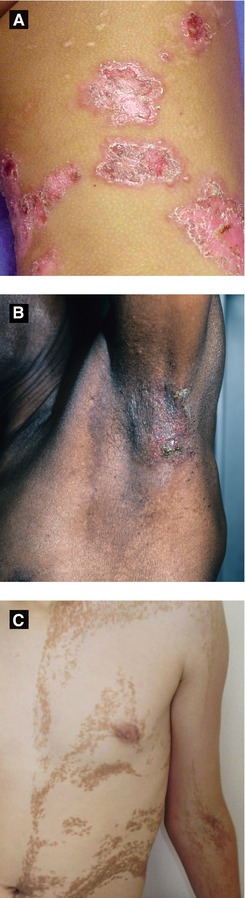
Figure 2.14:
A: Chronic bullous disease of childhood(Courtesy of Dr. Michelle B. Bain)B: Hailey-Hailey disease (Courtesy of Dr. Paul Getz)C: Unilateral epidermal nevus(Reprint from Baykal C, Yazganoğlu KD (eds). Clinical Atlas of Skin Tumors. Heidelberg, Germany. Springer; 2014.)
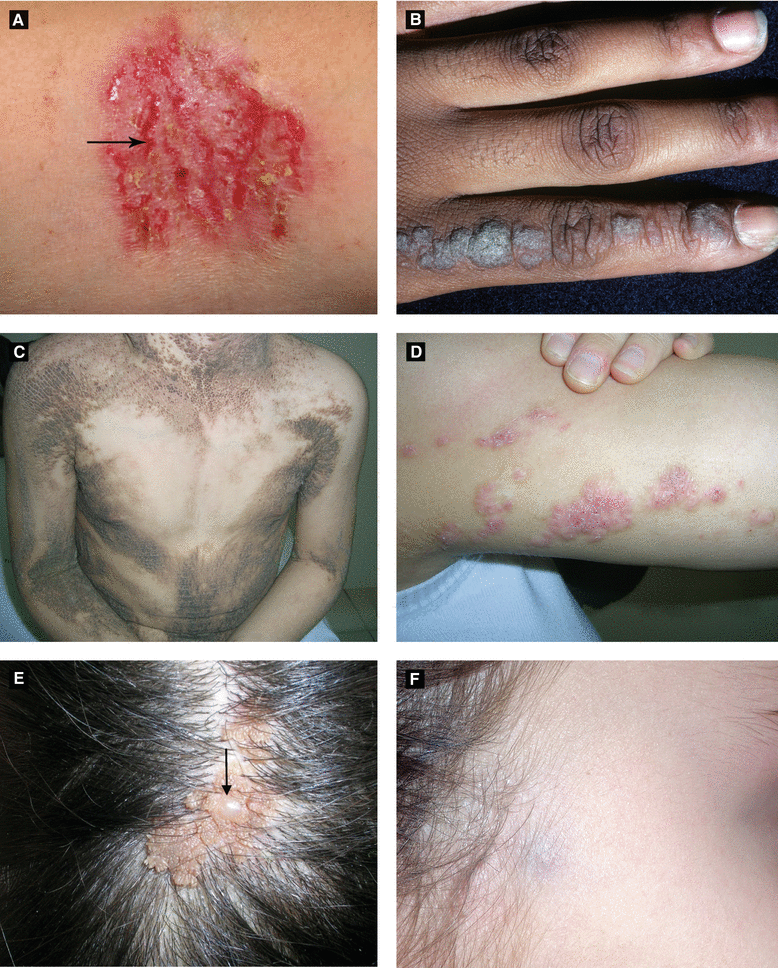
Figure 2.15:
A: Hailey-Hailey disease with linear erosions (arrow)* B: Inflamed linear verrucous epidermal nevus (ILVEN)**C: Bilateral epidermal nevus*** D: ILVEN***E: Nevus sebaceous with trichoblastoma (arrow)*** F: Pilomatricoma ****Reprint from Lipsker D. Clinical Examination and Differential Diagnosis of Skin Lesions. Paris, France: Springer;2013.**Courtesy of Dr. Paul Getz ***Reprint from Baykal C, Yazganoğlu KD (eds). Clinical Atlas of Skin Tumors. Heidelberg, Germany. Springer; 2014.
Presents as solitary yellow-orange slightly raised plaque typically on scalp or face; plaque typically thickens and becomes more verrucous or pebbly during childhood
Mutation in PTCH gene has been reported (deletion)
Benign tumors (trichoblastoma, syringocystadenoma papilliferum) and malignant tumors (BCC < 1% cases) can arise within lesion
Basal Cell Carcinoma
Seen in children with xeroderma pigmentosum (XP) and basal cell nevus syndrome (BCNS)
Squamous Cell Carcinoma
Seen in children with XP, dystrophic EB and albinism
Onset typically in childhood
Presents as solitary firm, skin-colored to faint blue papule or cyst on face or upper trunk
Histology: anucleate cornified cells (‘ghost’ or ‘shadow’ cells), calcification seen in late lesions
Multiple pilomatricomas may be associated with myotonic dystrophy (β−catenin defect)
Trichoepithelioma (Figure 2.16C)
Benign adnexal neoplasm usually appearing in childhood
Presents as skin-colored translucent papules (usually multiple) along the nasolabial folds or periorbital regions
Multiple lesions in Brooke-Spiegler syndrome (trichoepitheliomas, cylindromas, spiradenomas)
Angiofibroma (Fibrous Papule)
Skin-colored firm papule on face
Multiple lesions with onset in childhood associated with tuberous sclerosis (once known as adenoma sebaceum)
Neurofibroma (NF)
Presents as skin-colored, soft or rubbery papulonodule with positive ‘buttonhole’ sign (easily invaginated)
Commonly seen as solitary lesion; multiple lesions associated with neurofibromatosis
Plexiform NF considered pathognomonic for NF1, malignant transformation in 2-13%
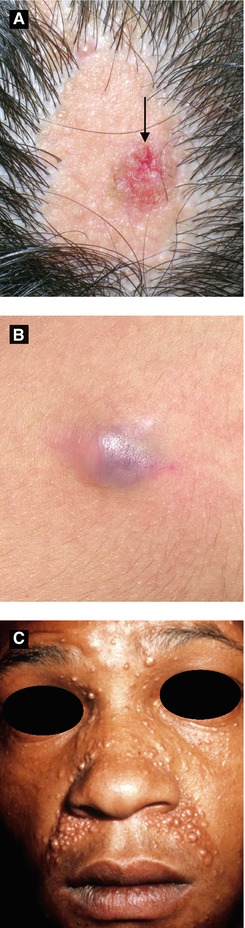
Figure 2.16:
A: Nevus sebaceus with syringocystadenoma papilliferum (arrow)*B: Pilomatricoma*C: Trichoepitheliomas** *Reprint from Baykal C, Yazganoğlu KD (eds). Clinical Atlas of Skin Tumors. Heidelberg, Germany. Springer; 2014.**Courtesy of Dr. Paul Getz
Connective Tissue Nevus (Figure 2.17A-B)
Also known as shagreen patch (tuberous sclerosis) collagenoma, elastoma, or dermatofibrosis leticularis disseminata (latter in Buschke-Ollendorf syndrome)
Onset at birth or early childhood; likely hamartoma
Presents as firm, solitary or multiple skin-colored papules, nodules or plaques
Onset within 1 year of age
Presents as multiple firm, smooth dome-shaped nodules on dorsolateral fingers/toes (spares thumb and great toe)
Benign with spontaneous regression within 2-3 years typically; high local recurrence rate with surgical excision
Histology: eosinophlic intracytoplasmic perinuclear inclusions within spindle cells
Infantile Myofibromatosis (Congenital Generalized Fibromatosis)
Rare, onset at birth or within first two years
Presents as one or more firm, rubbery skin-colored to purple papulonodules on head, neck or trunk
Two types: localized with no visceral involvement, good prognosis; visceral involvement with high mortality
Fibrous Hamartoma of Infancy
Onset at birth or within first year of life
Presents as painless, solitary skin-colored subcutaneous nodule typically involving axilla, shoulder or upper arm (less likely groin area)
Treat with local excision
Fibromatosis Colli
Infiltration of fibrous tissue involving the lower third of the sternocleidomastoid muscle at birth
Typically spontaneous remission within few months
Due to mutation in capillary morphogenesis protein 2
Multiple firm papules and nodules involving the face, extremities, and scalp; hypertrophic gums and disfigurement with flexion contractions
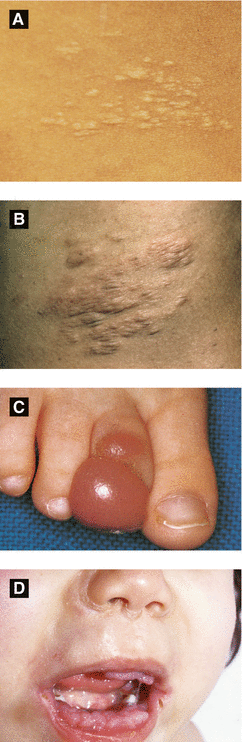
Figure 2.17:
A: Connective tissue nevus(Reprint from Laxer RM, ed. The Hospital for Sick Children: Atlas of Pediatrics. Philadelphia, PA: Current Medicine, 2005.)B: Connective tissue nevus(Courtesy of Dr. Paul Getz)C: Infantile digital fibroma(Reprint from Burgdorf WH, Plewig G, Wolff HH, Landthaler M, eds. Braun-Falco’s Dermatology. 3rd ed. Heidelberg: Springer; 2009.)D: Juvenile hyaline fibromatosis(Reprint from Baykal C, Yazganoğlu KD (eds). Clinical Atlas of Skin Tumors. Heidelberg, Germany. Springer; 2014.)
Non-Langerhans cell histiocytosis with Touton giant cells; onset typically within first year of life
Two types: micronodular (small, multiple) or macronodular (larger size, few in number)
Presents as single or multiple firm, pink-red papulonodules with yellow hue on head/neck > trunk/upper extremities
Regression typically seen in children (not in adults)
0.5% with ocular involvement: (iris most common site): glaucoma, hyphema (may rarely result in blindness)
Association with NF1 and juvenile myelomonocytic leukemia (JMML)
Clonal proliferative disease of Langerhans cells (comma-shaped nuclei, S100+, CD1a+, intracytoplasmic Birbeck granules seen on EM), four overlapping syndromes
Current classification by number of organ systems involved (single vs. multisystem), but historically grouped as follows:
Letterer-Siwe Disease (acute disseminated type) | – Multisystem involvement; onset typically before 2 years of age |
– Small, pink papules, pustules, vesicles with scale/crust/petechiae in seborrheic distribution | |
Hand-Schuller-Christian disease (chronic multifocal) | – Onset between 2-6 years of age |
– Typical triad: diabetes insipidus, bone lesions, exophthalmos | |
– Osteolytic bone lesions (cranium) | |
Eosinophilic Granuloma(chronic unifocal)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|