and Milind G. Parikh3, 2
(1)
University of Florida, College of Medicine, Gainesville, FL, USA
(2)
Private Practice:, Orlando, FL, USA
(3)
Department of Internal Medicine/Cardiology, University of Central Florida College of Medicine, Orlando, FL, USA
Keywords
AcnePsoriasisDermatitisHairPhotodermatosesPregnancy dermatosesRosaceaLichen planusAllergensANA3.1 Acne and Related Conditions
A. Acne Vulgaris and Special Forms (Tables 3-1, 3-2)
Acne Vulgaris
Inflammation of the pilosebaceous unit (PSU) causing comedones, papulopustules and nodules
Four key pathogenic factors
Abnormal follicular keratinization
↑ Corneocyte cohesiveness and proliferation
Propionibacterium acnes ( P. acnes ) in sebum
Gram + anaerobic rod, resident flora in follicle but acne patients with higher concentration
Naturally produces porphyrins (coproporphyrin III), which is the target of light-based acne therapy
Secretes lipases which cleave lipids in sebum into pro-inflammatory free fatty acids (FFAs), which are both comedogenic and chemotactic
Binds/activate toll-like receptor 2 (TLR2)
Inflammation
↑ IL-1, IL-8, and TNF-α through TLR-2 pathway
Hormonal effect on sebum due to androgens
↑ Sebum production due to androgen-stimulated sebaceous glands
Androgen receptors present on basal layer of sebaceous gland and ORS of hair follicle; respond to most potent androgen, dihydrotestosterone (DHT), and testosterone (latter produced by gonads and can be converted to DHT via 5α reductase)
Dehydroepiandrosterone sulfate (DHEA-S): weak androgen produced by adrenal glands
Microscopic precursor lesion: microcomedo
May present with non-inflammatory comedones (open/closed), inflammatory papules, pustules, ± nodules
Histology: follicular distension often with ruptured PSU and accompanying brisk inflammatory response, ± foreign body reaction with multinucleated giant cells
Treatment:
Topical therapy | Benzoyl peroxide, retinoids (tretinoin, adapalene, tazarotene), azelaic acid, dapsone, clindamycin, sodium sulfacetamide/sulfur and salicylic acid |
Topical retinoids: comedolytic and downregulate TLR-2 | |
Other therapies | Oral antibiotic, isotretinoin, oral contraceptive pill, spironolactone; photodynamic therapy or blue light alone |
Cannot combine isotretinoin and tetracycline due to risk of pseudotumor cerebri |
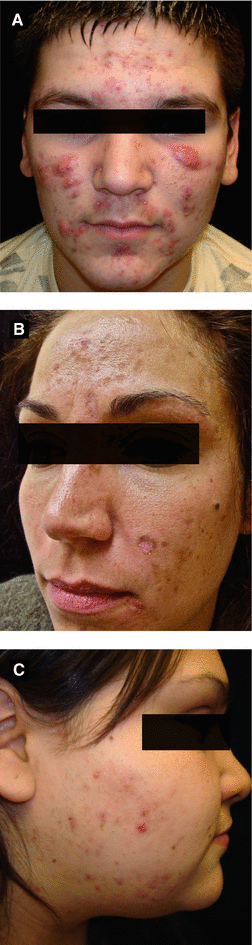
Figure 3.1:
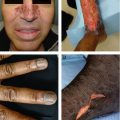
A: Acne conglobataB: Acne excoriéeC: Acne in PCOS
Table 3-1:
Acne Variants
Type | Clinical Features |
|---|---|
Acne fulminans | – Severe form of nodulocystic acne in young males (13-16 years old) – Presents with sudden-onset suppurative nodular acne with ulceration, eschars and systemic symptoms (may include myalgias, arthralgias, fever, ↑ ESR, ↑ WBCs, ± sterile osteolytic bone lesions typically over clavicle or sternum) – Treat with low-dose isotretinoin and prednisone or prednisone alone initially, followed by isotretinoin to prevent flare and formation of granulation tissue |
Acne conglobata (Figure 3.1A) | – Acute-onset nodulocystic acne without systemic manifestations – Part of the follicular occlusion triad (dissecting cellulitis of scalp, pilonidal cyst and hidradenitis suppurativa) |
Acne excoriée (Figure 3.1B) | – Mainly seen in young women with emotional or psychological disorders (such as obsessive-compulsive disorder) who repeatedly pick at lesions – Presents as mild acne with several excoriations, crusted erosions and sometimes ulcerations with subsequent scarring – Antidepressants may be warranted |
Acne with underlying endocrinologic abnormality (Figure 3.1C) | – If acne with accompanying hirsutism ± irregular menses, check lab work for hormonal abnormality (check LH, FSH, DHEA-S, free and total testosterone) – Source of androgens: ovarian androgens: testosterone adrenal androgens: DHEA-S, 17-hydroxyprogesterone Polycystic ovarian syndrome (PCOS): – Seen in 5-10% of women of reproductive age – Androgen excess causing hirsutism, irregular menses, ± polycystic ovaries, ± obesity, insulin resistance, ↑ LH/FSH ratio, ↓ fertility, ↑ testosterone – Acne lesions typically nodular; involve lower ½ of face (especially jawline) – Treatment: oral contraceptive pill (resulting in ↑ SHBG, ↓ free testosterone), spironolactone (off-label, blocks androgen receptor) Late congenital adrenal hyperplasia – ↑ DHEA-S or 17-hydroxyprogesterone due to partial deficiency of adrenal enzymes (commonly 21-hydroxylase or 11-hydroxylase) |
Industrial acne | – Due to exposure to insoluble cutting oils or chlorinated aromatic hydrocarbons (such as chlorinated dioxins and dibenzofurans) – Chloracne (form of industrial acne): presents with comedones, pustules and cysts over malar cheeks, retroauricular region, and scrotum |
Acne mechanica | – Due to repeated obstruction of the pilosebaceous unit through friction/pressure |
Neonatal acne (Cephalic neonatal pustulosis) | – Begins around 2 weeks of age and often resolves by 3rd month of age – Presents with erythematous small papules on cheeks |
Infantile acne | – Typically begins around 3-6 months of age, resolves within 1-2 years |
Drug-induced acne (Acneiform eruption) | – Due to corticosteroid, phenytoin, lithium, isoniazid, iodides, epidermal growth factor receptor inhibitors (EGFRI: cetuximab, erlotinib, gefitinib), anabolic steroids – Presents with abrupt-onset monomorphic-appearing papules and pustules; comedones typically not seen |
Table 3-2:
Syndromes Associated with Acne
Syndrome | Clinical Features |
|---|---|
PAPA syndrome | Pyogenic Arthritis (sterile), Pyoderma gangrenosum, Acne – Inherited (AD), CD2 binding protein 1 (CD2BP1) mutation; CD2BP1 is a pyrin-interacting protein, which is part of inflammatory pathway associated with familial Mediterranean fever, Muckle-Wells syndrome, and familial cold urticaria – Skin changes typically present near or at puberty |
HAIR-AN | HyperAndrogenism, Insulin Resistance, Acanthosis Nigricans |
SAPHO(Chronic recurrent multifocal osteomyelitis) | Synovitis, Acne (conglobata), Pustulosis (palmoplantar), Hyperostosis, Osteitis – Inflammatory bone changes (commonly involving sternoclavicular joint, spine {spondyloarthropathy} and long bones); peripheral arthritis also common – 1st line treatment: bisphosphonates suggested in many case reports and series; other treatments include mainstay therapies for psoriatic arthritis (methotrexate, anti-TNFα agents, etc) |
B. Acneiform Conditions
Gram-Negative Folliculitis
Exacerbation of acne after long-term antibiotic use
Presents with centrofacial pustules, especially perinasal
Treat with isotretinoin if severe or recurrent
Chronic folliculitis of occipital scalp and posterior neck in men of African, Hispanic or Asian descent
Presents initially as inflammatory follicular papules and pustules which over time evolve into firm, dome-shaped papules with fibrosis, ± coalesce into keloidal plaques
Treatment: avoid mechanical irritation; mixture of tretinoin and potent corticosteroid gel (if noninflamed), topical and/or systemic antibiotics, punch excision or intralesional corticosteroid injection for papules/keloids, laser epilation
Pseudofolliculitis Barbae (Figure 3.2B)
Inflammatory condition typically in men of African descent, especially with tightly curled hair
Distal end of curved hair penetrates epidermis and dermis after being shaved → subsequent foreign-body inflammatory reaction
Inflammatory papules, pustules and sometimes abscesses in beard region and anterolateral neck; may form hypertrophic scars and keloids
Treatment: topical or systemic antibiotics, topical corticosteroid, topical clindamycin/benzoyl peroxide; prevention with clippers, chemical depilatories and glycolic acid lotion
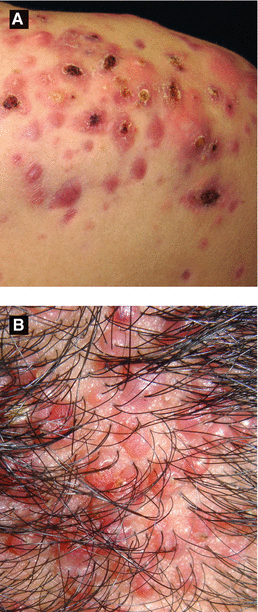
Figure 3.2:
A: Acne conglobata, shoulderB: Acne keloidalis nuchae
Distinctive dermatitis with discrete papules and pustules with erythematous, sometimes scaly, base around the mouth, nose, and possibly periorbital area
Previous use of fluorinated topical steroid may cause or exacerbate condition
Treatment: oral tetracycline, topical metronidazole, azelaic acid, and/or sodium sulfacetamide/sulfur
Chronic inflammatory and scarring disease of apocrine gland-bearing skin sites (axillae and anogenital area)
Initially thought to be due to obstruction of apocrine glands, but now thought to be from occlusion of follicular infundibula with subsequent rupture of the follicle and surrounding inflammation
Presents with double-ended comedones, tender nodules and sterile abscesses in groin, axillae, perianal and/or inframammary region; sequelae include sinus tracts, chronic drainage, and scarring (hypertrophic scars, rope-like elevation of skin, dermal contractures)
Histology: follicular hyperkeratosis, rupture of follicular epithelium, heavy inflammatory infiltrate (lymphocytes, neutrophils, plasma cells) around hair follicles ± sweat glands (sometimes extending to apocrine glands), abscess formation, foreign body-type granulomas, fibrosis in late stages
Treatment: weight reduction, smoking cessation, antiseptic soap, absorbent powder, topical aluminum chloride, intralesional corticosteroid injection into early inflammatory lesions, adalimumab, systemic corticosteroids (but typically flare when discontinued), isotretinoin (results often disappointing), acitretin, surgical excision and grafting; avoid incision and drainage (can result in scarring and chronic sinus tract formation)
Fox-Fordyce Disease (Figure 3.3C)
Chronic pruritic disorder of the apocrine glands due to obstruction and dilation
Intensely pruritic, dome-shaped flesh-colored follicular papules in axillae, ± anogenital and periareolar skin
Treatment (difficult): topical and intralesional corticosteroids, topical tretinoin, cleocin lotion
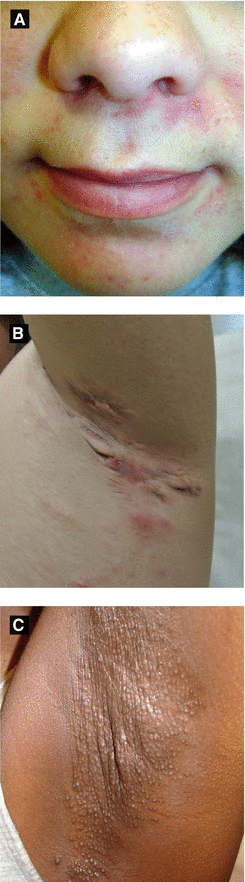
Figure 3.3:
A: Perioral dermatitisB: Hidradenitis suppurativa(Courtesy of Dr. Sima Jain)C: Fox-Fordyce disease(Courtesy of Dr. Sophie M. Worobec)
C. Rosacea (Figure 3.4A-C)
Chronic inflammatory condition of facial pilosebaceous units with increased vascular hyperreactivity; common in fair-skinned patients with peak in the 3rd to 5th decade
Presents with easy flushing and gradual reddening of complexion; exacerbating factors may include particular foods (especially spicy), alcoholic beverages, UV exposure, hot weather, warm beverages, and exercise
Type | Clinical Findings |
|---|---|
Erythematotelangiectatic (Type 1) | Prolonged flushing (>10min), persistent central facial erythema, ± telangiectasias, ± burning/stinging sensation, easy irritation |
Papulopustular (Type 2) | Persistent central facial erythema with acneiform pustules and papules (no comedones) |
Phymatous (Type 3) | Indurated erythematous to yellow-brown papules/nodules with persistent edema; almost exclusively in men; rhinophyma (subtype) over distal half of nose; less common sites include forehead, chin, philtrum, ears and eyelids |
Ocular (Type 4) | Xerophthalmia, tearing, pain, blurry vision, blepharitis, conjunctivitis, recurrent chalazion, keratitis, iritis, scleritis |
Steroid rosacea: use of oral/topical corticosteroid results in exacerbation of disease after initial improvement
Granulomatous rosacea: red-brown papules/nodules with underlying granulomatous inflammation
Histology: early lesions with dilated blood and lymphatic vessels; later lesions show lymphectasia, perivascular and perifollicular lymphohistiocytic infiltrate, ± poorly organized granulomas, dermal fibrosis, sebaceous gland hyperplasia, ± Demodex folliculorum mites within infundibula
Treatment:
Sun protection and avoidance of triggers, green-tinted makeup (conceals redness)
Topical therapy: metronidazole or azelaic acid best for inflammatory lesions, sodium sulfacetamide/sulfur, topical brimonidine (best for erythema)
Oral therapy: tetracyclines, macrolides, isotretinoin
Other: pulsed-dye laser or intense pulsed light for erythema/telangiectasias, CO2 laser for phymatous rosacea
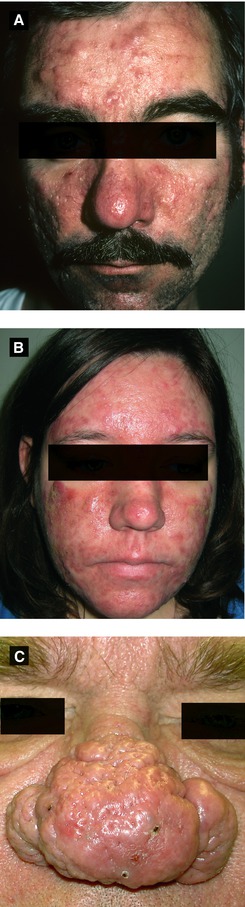
Figure 3.4:
A: Rosacea, papulopustular(Courtesy of Dr. Paul Getz)B: Rosacea, granulomatous(Courtesy of Dr. Iris K. Aronson)C: Rhinophyma
D. Rosacea Variants
Lupus Miliaris Disseminatus Faciei (Figure 3.5A)
Yellow-brown to red small monomorphic smooth papules on malar cheeks and periorifically
Lack history of flushing and lack telangiectasias
Histology: prominent small granulomas, ± central necrosis or caseation
Treatment: long term therapy with minocycline or isotretinoin
Mainly seen in postadolescent females; may be rare variant of rosacea
Presents with acute onset of erythematous papules, pustules, nodules and abscesses in centrofacial region with background of dull cyanotic erythema, ± draining sinuses; ± mild systemic symptoms (myalgias, fever,↑ ESR, ↑ WBC)
Treatment: initial use of oral corticosteroid followed by low-dose isotretinoin and slow taper of corticosteroid
Morbihan’s Disease (Solid Facial Edema)
Presents with woody, nonscaling edema involving midline face and cheeks
Treatment: isotretinoin ± ketotifen × 4-5 months
3.2 Papulosquamous, Lichenoid and Eczematous Dermatoses
A. Papulosquamous Dermatoses
Polygenic inflammatory disease with chronic, recurrent course; affects 2% of population
Bimodal onset with 3rd or 6th decade; earlier onset associated with more severe disease
Pathogenetic factors
Abnormal T cell activation
Thought to be mediated by new distinct type of T helper cell, Th17 cells; T cell-mediated autoimmunity toward poorly defined antigens
Cytokine profile:
↑ IL-2, IFNγ (Th1 cytokines)
↑ IL-1, IL-6, TNFα (innate cytokines)
↑ IL-12 (stimulates Th1 cells)
↑ IL-23 (stimulates Th17 cells)
↑ IL-22, IL-17, TNFα (Th17 cytokines)
IL-22 correlates with disease severity
T cell disease supported by response to T cell inhibitors like cyclosporine; worsening through IFNγ
IL-12 and IL-23 have common subunit p40: target of ustekinumab
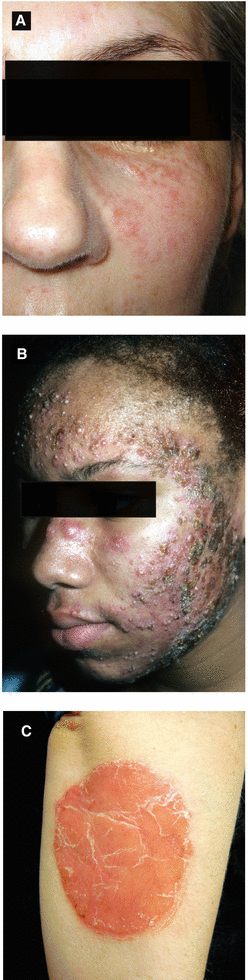
Figure 3.5:
A: Lupus miliaris disseminata(Courtesy of Dr. Iris K. Aronson)B: Pyoderma faciale(Courtesy of Dr. Paul Getz)C: Plaque psoriasis
Abnormal keratinocytes
↑ Mitotic activity and leukocyte recruitment; keratinocytes move to upper layer in 3–5 days (vs. normal 28 days)
↑ Human β-defensin-2 (hBD2), secretory leukocyte protease inhibitor (SLPI), and skin-derived anti-leukoproteinase (SKALP): antimicrobial peptides resulting in ↓ risk of secondary infection
↑ Involucrin in all layers (except basal)
Shed without loss of nucleus (parakeratotic scales)
Epidermal expression of STAT-3, which induces upregulation of TGFα and ICAM-1
Secrete IL-1, TNFα, IL-6, IL-8 (chemoattractant)
Genetics (polygenic and multifactorial)
HLA associations:
Cw6 (strongest) associated with early onset
B13, DR7, B17, B57 associated with earlier onset
B27 associated with psoriatic arthritis
A2 and Cw2 (weaker association)
B13, B17, Cw6: guttate psoriasis
In moderate to severe psoriasis, ↑ risk of cardiovascular disease; other comorbidities may include hypertension, obesity, dyslipidemia, diabetes mellitus; may also have ↑ risk of developing cancer particularly nonmelanoma skin cancer, lymphoma and lung cancer (according to recent study); common genetic factors (B27) may also cause ↑ incidence of inflammatory bowel disease
Triggering factors: physical trauma (Köebner phenomenon), infection (especially streptococcal pharyngitis), stress and medications (lithium, β-blocker, antimalarial, IFN, ACEI, NSAID, withdrawal of systemic corticosteroid, G-CSF)
Chronic plaque psoriasis presents with erythematous, scaly sharply-bordered macules, papules and plaques covered with silvery scale over extensor surfaces (knees, elbows), scalp, gluteal cleft, ± genitalia, ± intertriginous areas; chronic course with flares
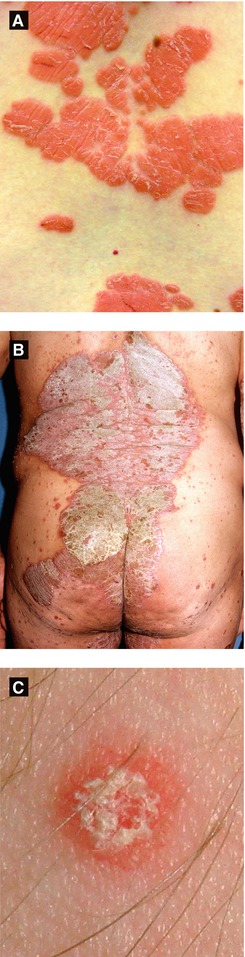
Figure 3.6:
A: Plaque psoriasisB: Plaque psoriasis(Courtesy of Dr. Paul Getz)C: Guttate psoriasis
Guttate psoriasis (Figure 3.7A) presents with acute-onset rain drop-like scaly papules on the trunk and extremities often in young patients; frequently preceded by streptococcal infection and eruption usually resolves completely within few months (may turn into chronic plaque psoriasis in adults)
Special locations:
Palms and soles (palmoplantar psoriasis)
Intertriginous areas (inverse psoriasis)
Psoriatic nails (Figure 3.7BC): may affect nail matrix, bed or plate; may be sole involvement or part of extensive disease
Pitting (nail matrix → focal parakeratosis in nail plate)
Leukonychia (nail matrix)
Onychodystrophy (nail matrix ± bed → subungual hyperkeratosis)
Oil spots (nail bed → yellow brown spots)
Onycholysis (nail bed → separation of plate from bed)
Splinter hemorrhages (↑ capillary fragility in nail bed)
Erythroderma (Figure 3.7C)
Generalized erythema and scaling (>90% skin surface affected), ± leukocytosis, ↑ ESR, lymphadenopathy; may develop suddenly from guttate or stable psoriasis but typically due to inappropriate treatment of disease
Histology: confluent parakeratosis (focal in guttate), hyperkeratosis, collection of neutrophils in s. corneum (Munro microabscesses) and spinous layer (spongiform pustules of Kogoj), hypogranulosis, acanthosis with clubbed rete ridges, suprapillary thinning, dilated blood vessels in papillary dermis, perivascular lymphocytes
Treatment:
Topicals
Vitamin D3 analogues (ie. calcipotriol, calcipotriene)
Corticosteroids
Calcineurin inhibitors (pimecrolimus/tacrolimus for intertriginous areas)
Tazarotene
Coal tar
Anthralin
Fixed combination betamethasone dipropionate/calcipotriol
Oral
Apremilast
Acitretin
Methotrexate
Cyclosporine
Other:
TNFα inhibitors, IL-17 inhibitor and p40 (IL-12/IL-23) inhibitor
Phototherapy (PUVA, NBUVB)
Excimer laser
Topical calcipotriene may be inactivated in acidic environment (ie. salicylic acid)
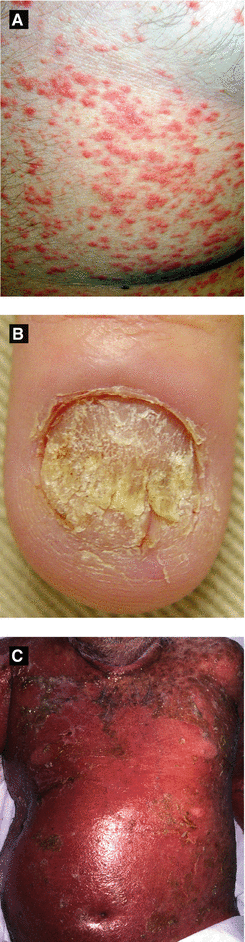
Figure 3.7:
A: Guttate psoriasisB: Psoriasis involving nailC: Erythrodermic psoriasis(Courtesy of Dr. Paul Getz)
Pustular Psoriasis (Figure 3.8A-C)
Distinct from psoriasis vulgaris in both features and clinical course
↑ HLA-B27 incidence
Two types: generalized and localized (palmoplantar pustulosis, acrodermatitis continua suppurativa)
Generalized (von Zumbusch)
Presents initially with malaise and fever, subsequent onset of erythematous macules studded with sterile pustules; initially in intertriginous areas but quickly spreads to trunk, extremities and nails (skin feels painful), ↑ risk for infection
Risk factors: tapering oral corticosteroid, infection, hypocalcemia, pregnancy (impetigo herpetiformis)
Labs: leukocytosis, hypoalbuminemia
Treatment: correct electrolyte and protein imbalance, methotrexate or cyclosporine (avoid systemic corticosteroid), later treatment can include photherapy or biologic treatment
Palmoplantar pustulosis
Tense, sterile pustules over palmoplantar surface with yellow-brown macules; may be associated with SAPHO syndrome (so prudent to inquire about any sternoclavicular tenderness and/or back pain)
Treatment: acitretin, topical corticosteroid
Acrodermatitis continua of Hallopeau
Variant of pustular psoriasis limited to finger tip or digit; HLA-B27 association
Presents with sterile pustules on erythematous base at tip of finger (less likely on toe) forming lakes of pus, associated pain and impaired use of digit; if pustules within nail bed, nail will typically be shed; may have loss of bony structures
Treatment: topical calcipotriene, topical corticosteroid, acitretin
Psoriatic Arthritis (Table 3-3)
Up to 30% of patients with psoriasis have arthritis; associated with moderate to severe psoriasis and typically occurs several years after appearance of skin lesions
Rheumatoid factor negative (seronegative) arthritis; HLA-B27 association
Tendons and ligaments often involved (enthesopathy or enthesitis) in addition to bone and cartilage
↑ TNFα level in synovium and serum in patients with psoriasis and psoriatic arthritis
Almost all patients with psoriatic arthritis have nail changes (up to 20% may have no skin findings)
Common features include pain at tendon insertion sites, digital involvement and sacroiliac disease, asymmetric joint involvement, negative rheumatoid factor, morning stiffness lasting more than 1 hour
Treatment: TNFα antagonists, IL-17 inhibitor, p40 (IL-12/IL-23) inhibitor, oral apremilast, methotrexate, NSAIDs, cyclosporine, sulfasalazine
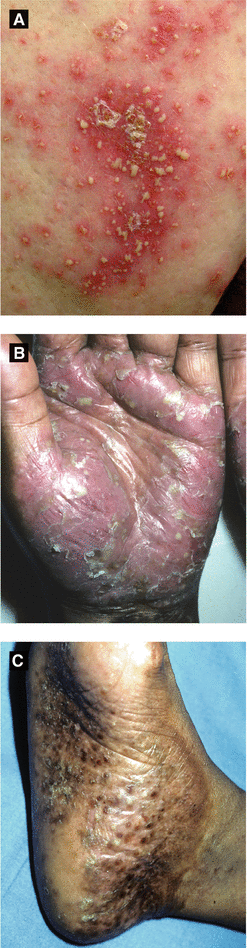
Figure 3.8:
A: Pustular psoriasisB: Palmoplantar psoriasis(Courtesy of Dr. Paul Getz)C: Palmoplantar psoriasis(Courtesy of Dr. Paul Getz)
Table 3-3:
Forms of Psoriatic Arthritis
Type of Arthritis | % | Salient Features |
|---|---|---|
Asymmetric oligoarthritis | 70% | Single or multiple distal joints in hands or feet involved; synovitis and joint swelling, ± swelling of digit (dactylitis or ‘sausage finger’); knees, ankles and sometimes axial involvement (if HLA-B27 positive) may also occur |
Asymmetrical DIP arthritis | 5-10% | Single or multiple DIP joint involvement; periarticular swelling with concomitant nail involvement |
Symmetrical polyarthritis (RA-like) | 15% | Involvement of small and medium-sized joints (PIP, MCP, wrists, elbows); difficult to distinguish from rheumatoid arthritis (RA); usually seronegative |
Spondylitis and sacroiliitis | 5% | Typically in men and resembles ankylosing spondylitis, with addition of knee and sacroiliac involvement, ± peripheral joint involvement, ± inflammatory bowel disease or uveitis, often positive for HLA-B27 |
Arthritis mutilans | 5% | Digits become shorter, wider, softer due to osteolysis of phalanges and metacarpals; results in telescoping motion of digits |
X-ray findings: ‘pencil in cup’ deformity (distal head of bone apearing sharpened like a point) fusiform tissue swelling (‘sausage digit’), tuft resorption, eccentric erosions
Reactive Arthritis (Reiter’s Disease) (Figure 3.9)
Autoimmune condition that develops in response to infection in another part of the body (cross reactivity)
Linked to two factors
Genetic factor: HLA-B27
Exposure to pathogen
May follow urethritis after exposure to GU pathogens (likely Chlamydia trachomatis)
May follow GI infection after exposure to enteric pathogens such as Campylobacter spp., Shigella flexneri, Ureaplasma urealyticum, Salmonella spp., or Yersinia spp.
Bacterial antigen mimics portion of HLA molecule with subsequent dysregulation of immune control mechanism
More common and severe in HIV patients; may be presenting sign of HIV
Presentation
Peripheral arthritis > 1 month duration, with
Associated urethritis or cervicitis
Other findings: urethritis, conjunctivitis, fever weakness, weight loss, erythema nodosum
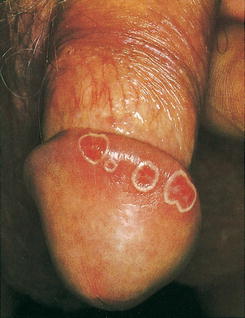
Figure 3.9:
Circinate balanitis(Reprint from Burgdorf WH, Plewig G, Wolff HH, Landthaler M, eds. Braun-Falco’s Dermatology. 3rd ed. Heidelberg: Springer; 2009.)
Skin findings in 5% patients: psoriasiform lesions similar to psoriasis
Keratoderma blenorrhagicum: thick plaques with pustules and erythema on plantar surfaces
Circinate balanitis: circinate erythematous lesions on glans penis (almost pathognomonic)
Classic triad: urethritis, arthritis, conjunctivitis
Treatment: treatment of any triggering infection (doxycycline 100mg bid × 14 days); arthritic symptoms may treat with biologic agent, methotrexate, cyclosporine, acitretin or NSAID; cutaneous lesions with high-potency topical corticosteroid
Pityriasis Rubra Pilaris (PRP)
Disorder of keratinization with bimodal distribution involving children and adults, nearly always acquired (occasional familial cases described)
Presents with follicular hyperkeratotic papules on erythematous base, which coalesce into large orange-red to red patches with characteristic ‘islands of sparing’; palms and soles with waxy orange-red keratoderma; may rapidly evolve into erythroderma; nail changes include subungual hyperkeratosis and nail plate thickening
5 types: Type I and II in adults, Type III-V in children (see Chapter 2 for pediatric types)
Type I (classic): over half of all cases; sudden onset of symptoms with duration of 2-5 years
Type II (atypical): about 5% cases, slow onset with alopecia, localized lesions and chronic course
Histology: acanthosis, thickened granular layer, ‘checkerboard parakeratosis’ (orthokeratosis alternating with parakeratosis both vertically and horizontally), ‘shoulder parakeratosis’ adjacent to follicular plugs, perivascular lymphocytic infiltrate
Treatment: high potency topical corticosteroid, systemic retinoid, methotrexate, ± phototherapy, azathioprine, infliximab
B. Lichenoid Dermatoses
Pruritic papular disease of skin and mucous membranes
Due to cell-mediated autoimmune reaction toward basal layer keratinocytes; may be idiopathic, drug-related or infection-related (HCV)
4P’s: papular, pruritic, polygonal, purple
Often lasts 1-2 years (except oral and hypertrophic forms, which typically have protracted courses)
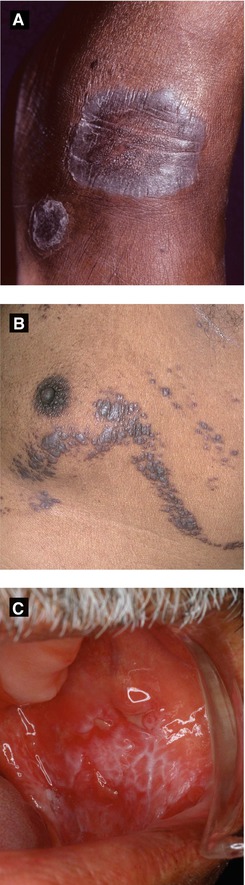
Figure 3.10:
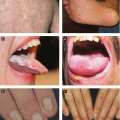
A: Annular lichen planus(Courtesy of Dr. Paul Getz)B: Blaschkoid lichen planus(Courtesy of Dr. Iris K. Aronson)C: Oral lichen planus(Reprint from Norman R, ed. Diagnosis of aging skin diseases. London: Springer; 2008.)
Table 3-4:
Types of Lichen Planus
Type | Description |
|---|---|
Acute LP | Eruptive lichenoid papules with wide distribution, heals with hyperpigmentation; self-limited (typically resolves within 9 months) |
Actinic LP | Photosensitive variant of LP with melasma-like appearance or lichenoid papules over face, neck and dorsal hands; typically in children and young adults with spring or summer onset (some consider entity as lichenoid form of PMLE) |
Annular LP | Annular papules and plaques with central clearing (commonly over penis) |
Atrophic LP | Lichenoid papules replaced with depressed atrophic areas typically over lower legs, ± residual hyperpigmentation; resembles lichen sclerosus clinically |
Bullous LP | Vesicles and bullae arising within existing LP lesions (intense inflammatory reaction at dermoepidermal junction causes subepidermal bullae) |
Drug-induced LP | Distribution typically generalized or sun-exposed sites; Wickham’s striae uncommon; ± eosinophils and parakeratosis on histology; medication typically taken for several months before eruption appears Common meds: β-blockers, captopril, penicillamine, HCTZ, antimalarials, furosemide, quinidine, NSAID, tetracycline, quinacrine, gold, sulfonylureas, hydroxyurea, methyldopa |
Erosive or ulcerative LP | Painful, chronic, recalcitrant erosive lesions especially on oral mucosa and palmoplantar surface; small ↑ risk of SCC within longstanding lesions; erosive oral LP associated with liver disease (HCV) |
Genital LP | Seen in up to 20% of LP patients; glans penis common site for men (annular lesions, small grouped papules, or larger plaques); vulvar LP commonly erosive and may coexist with gingival involvment (‘vulvovaginal gingival syndrome’) |
Hypertrophic LP | Thick, hyperkeratotic intensely pruritic plaques commonly found over shins or dorsal feet; also known as LP verrucosus |
Inverse LP | LP lesions in groin, axillae and inframammary regions |
Linear LP | Linear groups of lichenoid papules following lines of Blaschko |
Mucosal LP | Up to 50% patients with skin disease may have oral mucosal changes; ranges from reticular, atrophic, erosive, bullous, papular to pigmented; reticular type most common with lacy white hyperkeratosis on buccal mucosa, lips, tongue and gingiva; typically asymptomatic unless erosive; rarely may see esophageal, laryngeal or conjunctival involvement |
Nail LP | 10% of LP patients; may be isolated finding; typically lateral nail thinning, longitudinal ridging, dorsal pterygium, splitting, ± twenty nail dystrophy |
LP/LE overlap | Clinical and histologic features of both LE and LP |
Palmoplantar LP | Painful hyperkeratotic yellow to erythematous plaques on palms and soles (lateral borders and pressure points), ± ulceration, erosions; recalcitrant to therapy |
LP Pemphigoides | DIF: linear IgG/C3 at BMZ IIF: IgG at roof of blister (salt-split skin) Tense vesicles and bullae arise in normal, uninvolved skin; typically blisters occur weeks to months after appearance of typical LP lesions; overlap between bullous pemphigoid and LP; + IgG antibody to BP180 (NC16A) |
LP Pigmentosus | Gray-brown macules in sun-exposed areas ± flexural folds in darker-skinned patients; similar to erythema dyschromicum perstans |
Lichen planopilaris | Keratotic follicular papules with violaceous rim leading to cicatricial alopecia |
Graham-Little-Piccardi-Lasseur syndrome : typical skin/mucous membrane LP, scarring alopecia of scalp, nonscarring loss of pubic/axillary hairs |
Presents with intense pruritus and violaceous, smooth flat-topped papules and plaques with fine scale (Wickham’s striae) over flexor wrists, forearms, legs, presacrum and other areas, + Köebner phenomenon
No conclusive evidence to support association with autoimmune disease (per Bolognia); difficult to determine if true HLA association
Histology
Hyperkeratosis (without parakeratosis)
↑ Granular layer
Partially effaced rete ridges with widened papillae (‘sawtooth’ appearance)
Vacuolar change of basal layer with colloid bodies
Band-like lymphocytic infiltrate at dermoepidemal junction
Dermal melanophages
Treatment: superpotent topical corticosteroids, topical calcineurin inhibitors, intralesional or systemic corticosteroid, phototherapy, methotrexate, acitretin, metronidazole (latter for erosive oral form)
Lichenoid Keratosis
Likely due to inflammation of lentigo, actinic keratosis or seborrheic keratosis
Brown to red scaly papule on sun-exposed extremity
Solitary lesion mimicking lichen planus histologically
Clinical syndrome resulting from the transfer of immunologically competent cells to an immunosuppressed host
Donor lymphocytes recognize the recipient as ‘foreign’ and mount an immunological attack primarily against the skin, mucosa, gastrointestinal tract and liver
Histocompatibility (both major and minor complexes) between the donor and host is the most important factor in the development of GVHD
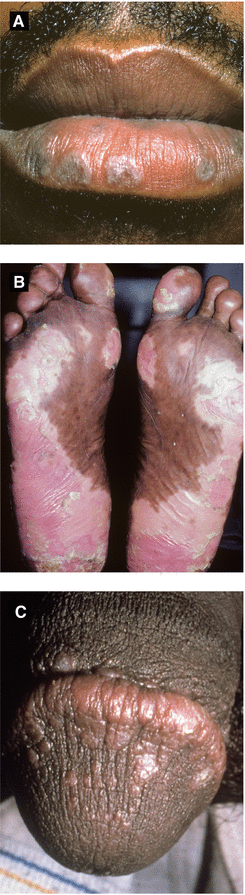
Figure 3.11:
A: Lichen planus, lips*B: Lichen planus, palmoplantar*C: Lichen planus, genital* *Courtesy of Dr. Paul Getz
One of the major complications of allogeneic hematopoietic stem cell transplantation (can also occur after transfusion of unirradiated blood products or donor lymphocytes in setting of solid organ transplantation)
Two forms (acute and chronic GVHD) based on time of presentation since transplant date (part of same spectrum)
Acute GVHD:
Occurs within first 100 days following transplantation (typically within 1-3 weeks after transplantation)
Presents with a maculopapular eruption which may coalesce into confluent erythema (± evolve into erythroderma or bullae resembling toxic epidermal necrolysis); acral erythema with violaceous discoloration of pinna of ear suggestive
Triad of dermatitis, enteritis with diarrhea, and hepatitis with abnormal LFTs, ± high fever, conjunctival erythema
Histology: vacuolization of basal layer, necrotic keratinocytes, sparse perivascular or interface dermatitis, ± complete epidermal necrosis (severe cases)
Course: 30-50% of patients with moderate to severe acute GVHD die
Treatment: systemic corticosteroid
Chronic GVHD:
Occurs after mean of 4 months (as early as 40 days)
Evolves from acute GVHD in approximately 50% surviving patients, otherwise de novo
Divided into early lichenoid and late sclerodermoid
Lichenoid GVHD: violaceous to erythematous lichenoid papules over dorsal hands, forearms, trunk and may become widespread, ± mucous membrane involvement (lacy white plaques or erosions in mouth, salivary gland involvement with Sjögren-like syndrome)
Sclerodermoid GVHD: sclerotic plaques similar to morphea, ± hyperpigmentation, ± sicca symptoms, may also involve the gastrointestinal tract, lungs, liver and musculoskeletal system
Main cause of death of chronic GVHD: infection due to immuno-suppression
Histology: chronic lichenoid GVHD similar to lichen planus; sclerodermoid GVHD with epidermal atrophy and dermal fibrosis
Treatment: topical calcineurin inhibitor, PUVA, UVB, prednisone, hydroxychloroquine, cyclosporine, mycophenolate mofetil, azathioprine, photopheresis
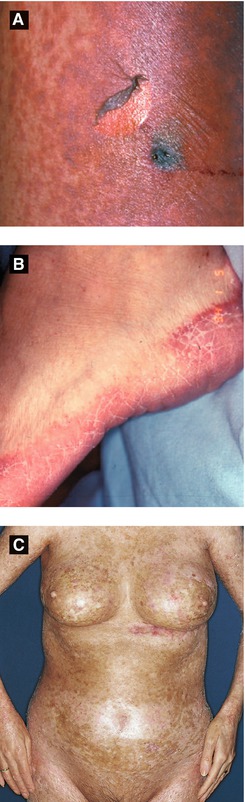
Figure 3.12:
A: GVHD, acute*B: GVHD, acute* *Reprint from Morgan MB, Smoller BR, Somach SC, Deadly Dermatologic Diseases. New York, NY: Springer; 2007.C: GVHD, chronic(Reprint from Burgdorf WH, Plewig G, Landthaler M, Wolff HH, eds. Braun-Falco’s Dermatology. 3rd ed. Heidelberg: Springer; 2009.)
C. Eczematous Dermatoses
Acute or chronic inflammatory reaction to a substance in contact with the skin; divided into irritant and allergic
Irritant contact dermatitis (ICD)
Accounts for 80% of CD
Due to direct local cytotoxic effect of irritant on skin
Acute ICD: acute exposure to toxic agent; presents with pruritus and sharply-localized erythema with vesicles, edematous papules or hemorrhagic blisters, ± scaling or crusting; no distant spread
Chronic ICD: repeated exposure to mild irritants (low-grade irritation); presents with diffuse or localized but ill-defined scaly patches and plaques
Allergic contact dermatitis (ACD)
Accounts for 20% of all CD
Type IV delayed hypersensitivity to contactant (to which already sensitized), onset may be delayed as long as 24 to 96 hours after allergen exposure
Nickel and poison ivy most common
Patch testing (Figure 3.13C) is the gold standard for diagnosing ACD; grading system:
Reaction | Description |
+ | Weak reaction with erythema, infiltration, ± papules |
++ | Strong reaction: vesicles, erythema, infiltration, papules |
+++ | Spreading bullous reaction |
Poral reaction: non-allergic reaction of erythem- atous dots (cobalt residing in acrosyringium)
Acute ACD: typically presents 24-48 hours after exposure and presents with pruritus, vesicles, weeping and erythema, ± dissemination of lesions
Subacute ACD: presents with eczematous scaly plaques or lichenification correlating to areas of contact with allergen
Histology: spongiosis, intraepidermal vesicles and superficial perivascular infiltrate in acute setting; acanthosis, hyperkeratosis and mild superficial infiltrate in chronic setting
Treatment: avoid exposure irritants/allergens; topical corticosteroid, patch testing (for ACD), if severe may use short-term systemic corticosteroid
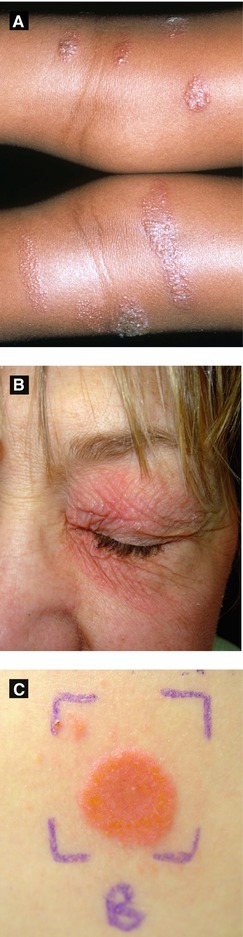
Figure 3.13:
A: Allergic contact dermatitis(Courtesy of Dr. Paul Getz)B: Subacute contact dermatitisC: Patch testing, 2+ reaction(Courtesy of Dr. Sophie M. Worobec)
Factors | ACD | ICD |
|---|---|---|
Previous exposure required | Yes | No |
Immunologic reaction | Yes | No |
Distant spread | Yes | No |
Dose-related response | No | Yes |
Similar reaction in others w/ exposure | No | Yes |
Table 3-5:
Table of Contact Allergens
Formaldehyde-Releasing Preservatives | |
|---|---|
Quaternium-15 | Most common cosmetic preservative to cause ACD; personal care products |
2-Bromo-2-nitropropane- 1,3-diol (Bronopol) | Formaldehyde-releasing preservative in personal care products and variety of industrial applications |
Diazolidinyl urea (Germall II) | Formaldehyde-releasing preservative; personal care products especially bubble baths, baby wipes and household detergents |
Imidazolidinyl urea (Germall 115) | Formaldehyde-releasing antimicrobial preservative used in cosmetics |
DMDM hydantoin | Formaldehyde-releasing antimicrobial preservative used in cosmetics like shampoo, hair conditioners and skin care products |
Rubber accelerators | |
Thiuram | Thiuram cross reacts w/ disulfiram Rubber in shoes, rubber gloves, elastic, fungicides, paints, barrier contraceptives |
Mercaptobenzothiazole (MBT) | Most common cause of allergic shoe dermatitis Rubber products, tires, antifreeze, anticorrosive agents, cutting oils, electrical cords, fungicides, rubber shoes (sneakers, tennis shoes, etc) |
Carba mix | Leather shoes, tires, fungicides, cosmetic applicators, gloves, adhesives, elastic, barrier contraceptives; may cross react with thiuram derivatives |
Mercapto mix | Tires, elastic, rubber gloves, electrical cords, rubber soles of shoes, etc |
Black rubber mix | Heavy use rubber products such as tires, hoses, cables and belts |
Mixed dialkyl thioureas | Neoprene rubber (wetsuit), goggles, elastic, paint remover, shoe insoles |
Textiles | |
Disperse blue 106 | Clothing dye (bed linens, blue dye, clothing, tights, garment lining) |
Ethylene urea melamine formaldehyde mix | Permanent press clothing (wrinkle-resistant) |
Textile dermatitis typically occurs where clothing fits tightly (posterior neck, upper back, lateral thorax, axillae, waistband, flexor surfaces); ± purpuric contact dermatitis | |
Adhesives | |
Epoxy resin (bisphenol A) | Glues, plastics, electrical insulation, paint and primer |
Colophony (rosin, abietic acid) | Paper, cosmetics, paint, adhesives, waxes, chewing gum, used in baseball/ballet/musical instruments to ↑ friction |
Cyanoacrylate (methyl or ethyl) | Fast-acting adhesive (Super or Krazy Glue), liquid bandages, Dermabond |
Ethyl acrylate | Artificial nail (adhesive) |
Methacrylate (methyl or ethyl) | Adhesive, artificial nails, dental fillings/sealants, hearing aids, hard contact lenses, glue (bone cement) for artificial joints, acrylic denture material |
p-tert-butylphenol formaldehyde resin | Leather/rubber adhesive |
Sunscreen components | |
Benzophenone-3 | PABA ± cross react w/ sulfonamides, azo dyes, benzocaine, PPD Sunscreens, rubber products, cosmetics |
PABA (Padimate O) | Sunscreen (UVB) |
Oxybenzone | Oxybenzone: most common sunscreen agent to cause photoallergic contact dermatitis Sunscreen (UVA) |
Corticosteroids | |
Tixocortol-21-pivalate | Group A (hydrocortisone acetate, prednisone, methylprednisolone) |
Budesonide | Budesonide also marker for some Gr. D steroids Group B (triamcinolone acetonide, desonide, fluocinolone acetonide, fluocinonide, halcinonide) |
Hydrocortisone-17-butyrate | Group D (hydrocortisone-17-valerate/butyrate, clobetasone-17-butyrate clobetasol proprionate, betamethasone valerate/diproprionate) |
Hair preparations | |
Paraphenylenediamine (PPD) | Permanent hair dye, ‘black henna’ (not natural henna from plant), photographic developer, printing inks, black rubber, temporary tattoos; ACD typically on eyelids/ear helices/hairline or hands May cross react with: PABA, sulfonamides (including thiazide and furosemide), para-aminosalicylic acid, benzocaine and procaine (ester anesthetics), azo dyes (temporary/semi-permanent hair dye, pen ink, coloring agent) |
Monothioglycolate | Permanent wave preparations; typically in glycerol monothioglycolate |
Ammonium persulfate | Hair bleach, bleaching agent in flour |
Others | |
Bacitracin | Topical antibiotic ointment, otic and ophthalmic preparations |
Balsam of Peru | Naturally occurring fragrance material, found in topical medications |
Benzalkonium chloride | Skin disinfectant (ophthalmic solutions), cosmetics |
Benzocaine, tetracaine | May cross-react with PABA, PPD, and sulfonamides Local anesthetic (ester) |
Cobalt | Added to other metals to ↑ hardness; found in jewelry, buttons, cosmetics, hair dye, joint replacements, ceramics, enamel, cement, paint, and pottery |
Dimethyl fumarate | Furniture, shoes, and car seats; added for antifungal property to prevent mold growth |
Cocamidopropyl betaine | Hair and bath products like shampoo (surfactant) |
Ethylenediamine | Medical creams, antifreeze, paint |
Cross-reacts with hydroxyzine and aminophylline | |
Euxyl K400 | Cosmetic and household products (preservative) |
Fragrance mix | Several components (cinnamic aldehyde), detects 70% fragrance allergies |
Formaldehyde | Textile resins (wrinkle-free clothing), cosmetics, tissue fixative, embalming solution, paints, disinfectants, and medications |
Gold | Jewelry, dentistry, electronics, treatment of certain diseases (RA, SLE, etc) |
Glutaraldehyde | Cold sterilization (medical/dental equipment), disinfectant, tan shoe leather |
Lanolin (wool alcohol) | Cosmetics and some topical creams |
Methylchloroiso-thiazolinone (Kathon CG) | Preservative in cosmetics and shampoos (antibacterial properties) |
Neomycin sulfate | Antibiotic ointment, hemorrhoidal cream, otic and ophthalmic preparations Co-sensitivity often between neomycin and bacitracin |
Nickel sulfate | Costume jewelry, buckles, and snaps; dimethylglyoxime test detects nickel (positive if turns pink); ± co-sensitivity seen with nickel and cobalt |
Paraben mix | Cosmetics, topical medications, food, textiles, antiperspirants |
Potassium dichromate | Cement, leather (footwear), plaster, wood finishes, green felt of card tables |
Propylene glycol | Cosmetics, topical medications (vehicle), antifreeze |
Thimerosal | Preservative in contact lens solution, vaccines, otic/ophthalmic solution, antiseptic |
↑ photosensitivity with piroxicam if patient with positive reaction to thimerosal | |
Tocopherol acetate | Vitamin E |
Toluene sulfonamide | Nail polish (typically appears eyelid dermatitis or periungual dermatitis) |
Table 3-6:
Plant Allergens
Allergen | Plant (common name) | Scientific name |
|---|---|---|
Urushiol (includes pentadecacatechol in oleoresin) | Poison ivy, poison sumac, poison oak | Family: Anacardiaceae Genus: Toxicodendron; Species: Rhus May cross react with Japanese lacquer tree (sap), cashew tree, mango tree, Indian marking nut (black juice), Brazilian Pepper tree (sap), gingko (seed pulp) |
Sesquiterpene lactone | Dermatitis may be airborne (face, neck) or direct contact (hands) Chrysanthemum, ragweed, sunflower, artichoke, arnica, daisy, marigold, arnica | May cross react with permethrin Family: Asteraceae or Compositae |
Primin | Primrose | Family: Primulaceae Species: Primula obconica |
Diallyl disulfide, allylpropyl disulfide | Garlic, onion, chives | Family: Alliaceae Genus: Allium |
Allicin | Garlic, onion, chives | Family: Alliaceae Genus: Allium |
Tuliposide A | Peruvian lily | Family: Alstroemeriaceae |
Tulip, hyacinth | Family: Liliaceae | |
d-usnic acid | Lichens | Several genera including Parmelia |
Colophony (Abietic acid, rosin) | Pine tree (resin) | Family: Pinaceae Species: Pinus species |
Tea tree oil (Limonene) | Ti or tea tree | Family: Myrtace ae Species: Melaleuca alternifolia |
3-carene | Turpentine | Family: Pinaceae |
Table 3-7:
Plant irritants (ICD)
Irritant | plant |
|---|---|
Bromelin | Pineapple (Ananas comosus) |
Calcium oxalate | Dumb cane (Araceae), daffodils (Narcissus spp.), hyacinth (Liliaceae), pineapple |
Phorbol esters(in milky latex) | Poinsettias, spurges, crotons (Euphorbiaceae) May cause temporary blindness if latex contacts eye |
Capsaicin | Chili peppers (Solanaceae) |
Thiocyanates (Allyl isothiocyanate) | Garlic (Alliaceae) Black mustard and radish (Brassicaceae) Ranunculin converts to protoanemonin after plant injury |
Protoanemonin(Ranunculin) | Buttercups (Ranunculaceae) Causes intense linear vesiculation |
Table 3-8:
Pigment Reactions from Tattoos
Tattoo Color | Chemical |
|---|---|
Black | Carbon, iron oxide |
Blue | Cobalt |
Brown | Ferric oxide |
Green | Chromic oxide Most common allergic reaction seen from red tattoo pigment |
Purple | Manganese |
Red | Cinnabar (mercury sulfide), cadmium red |
White | Titanium dioxide |
Yellow | Cadmium sulfide |
3.3 Granulomatous, Metabolic and Depositional Diseases
A. Granulomatous Diseases
Asymptomatic, benign and self-limited granulomatous disease of the dermis seen in both adults and children
Unknown etiology; may include trauma and sun exposure
Presents as skin-colored to pink non-scaly papules coalescing into annular or arciform plaque typically over dorsal hand or foot; variants listed below:
GA Variant | Description |
|---|---|
Patch GA (Macular) | Patches of erythema typically over extremities ± trunk |
Generalized GA (Disseminated) | Flesh-colored pink papules over trunk/extremities; poor response to treatment; association with diabetes |
Perforating GA | Small papules with central umbilication and crusting (discharging necrotic collagen) typically over dorsal hands |
Subcutaneous GA | Deep dermal nodules similar to rheumatoid nodules; typically asymptomatic |
Histology: necrobiotic foci in dermis surrounded by histiocytes (palisading granulomas) or histiocytes splayed between collagen bundles (interstitial type), mucin accumulation, ± perivascular lymphocytes/eosinophils
Treatment: clinical observation as typically self-limited (50-75% in 2 years) in localized GA, potent topical corticosteroid, topical calcineurin inhibitor or intralesional corticosteroid; systemic therapy can be considered for disseminated GA but none consistently proven effective
Uncommon necrobiotic disease associated with diabetes mellitus (DM): 30-40% patients with NLD have DM, but only 0.03-3% of patients with DM manifest with NLD
Presents with yellow to red-brown atrophic to indurated plaques typically over pretibial areas; prominent telangiectasias, ± ulceration
Histology: normal to atrophic epidermis, histiocytes often encircling necrobiotic collagen in dermis in layered fashion (tier-like, parallel to epidermis), ± sclerosis, interstitial lymphocytes, plasma cells, histiocytes, and multinucleated giant cells (granulomatous inflammation)
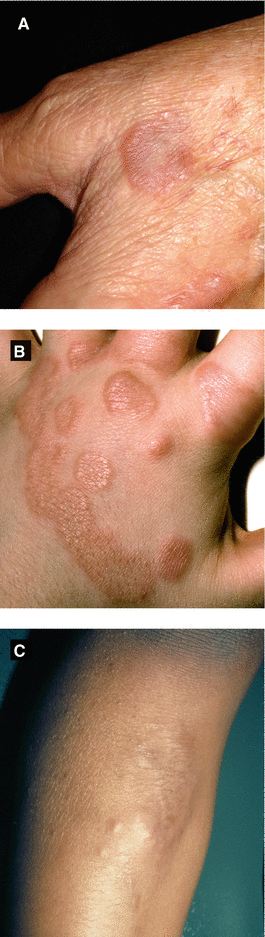
Figure 3.14:
a: Granuloma annulareb: Granuloma annulare(Courtesy of Dr. Paul Getz)c: Subcutaneous GA(Courtesy of Dr. Paul Getz)
Treatment: high potency topical corticosteroid (1st line) or IL injection into active border; aspirin + dipyridamole (to ↓ plt aggregation), niacinamide, and if severe refractory ulcerations consider excision with graft
Annular Elastolytic Giant Cell Granuloma (Actinic Granuloma)
Unclear if distinct disease or variant of GA
± Related to inflammation triggered by actinic damage
Presents as annular erythematous plaque with slightly atrophic center in sun-exposed areas of older adults; tendency for slow spread
Histology: solar elastosis, absence of elastic fibers in center of lesion, elastic fibers adjacent to/within giant cells (elastophagocytosis), granulomatous inflammation
Treatment: topical corticosteroids (inconsistent response)
Rare, multisystem histiocytic disease
Presents as red-orange to yellow papules, nodules or plaques on face (periorbital), tendency toward ulceration
80% with IgG monoclonal gammopathy (↑ risk of plasma cell dyscrasias and lymphoproliferative disorders)
Histology: mid-dermal palisading xanthogranuloma (mid-dermis extending to fat consisting of histiocytes, foamy cells, plasma cells, giant cells (Touton and bizarre foreign body), cholesterol clefts in areas of necrobiosis
Cutaneous Crohn’s Disease
Skin changes related to Crohn’s disease
Genital: labial or scrotal erythema with swelling
Nongenital: erythematous indurated plaque or ulcerations with drainage (typically perianal)
Oral: cobblestoning (buccal mucosa), small aphthae-like ulcers or linear ulcers, pyostomatitis vegetans, cheilitis glandularis, indurated fissuring of lower lip
Histology: non-caseating granulomas (sarcoidal or diffuse)
Treatment: topical/IL/oral corticosteroid, oral antibiotic (ie. metronidazole), azathioprine, sulfasalazine, adalimumab; of note, treatment of intestinal manifestations usually improves skin lesions
Immune response to exogenous or endogenous material that has wounded skin
Histology: ranges from sarcoid-like granulomatous reaction, necrobiotic reaction, suppurative reaction to reaction consisting of mild fibrosis
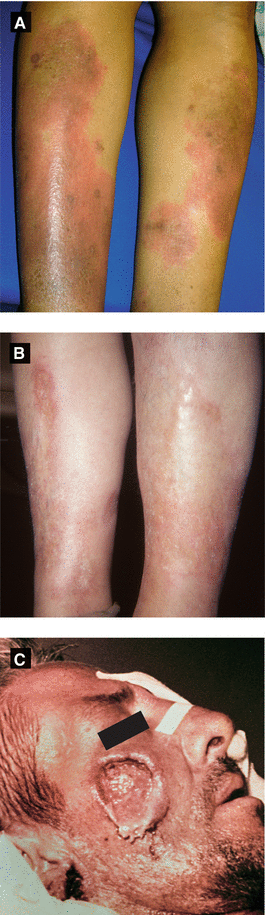
Figure 3.15:
a: Necrobiosis lipoidica(Courtesy of Dr. Sophie M. Worobec)b: Necrobiosis lipoidica(Courtesy of Dr. Paul Getz)c: Necrobiotic xanthogranuloma (Reprint from Morgan MB, Smoller BR, Somach SC. Deadly Dermatologic Diseases. New York, NY: Springer; 2007.)
Table 3-9:
Properties of Foreign Bodies
Doubly refractile with polarized light (Birefringent) | PAS Positive | |
|---|---|---|
Silica (dirt or glass) | Talc (deodorant/powdered gloves) | Starch |
Wood splinter | Sutures (nylon) | Plant material |
Starch | Keratin (hair shafts) | |
Spines of sea urchins | Cholesterol esters | |
Table 3-10:
Common Foreign Body Reactions
Foreign Body | Clinical Findings | Histological Reaction |
|---|---|---|
Intralesional corticosteroid | Yellowish to skin-colored nodule at site of injection | Foreign body granuloma with pale blue (mucin-like) acellular material in center |
Keratin | Erythematous follicular papules (ie. acne keloidalis nuchae) | Foreign body granuloma, ± fragments of keratin (section of hair shafts, birefringent) |
Suture material | Erythematous papule or papules | Birefringent basophilic suture fibers in dermis with surrounding foreign body granuloma |
Tattoo pigment | Lichenoid papules, eczematous dermatitis, erythema or induration | Extracellular pigment (typically black) with foreign body reaction (only if allergic reaction) |
Wood splinter | Induration, erythema or papule | Brownish color with prominent cell walls (honeycomb appearance) surrounded by granulomatous inflammation; birefringent |
Silica, zirconium, beryllium | Erythema, induration or papule | Foreign body granuloma, sarcoidal granuloma or caseating granuloma; doubly refractile spicules |
Silicone | Nodule, indurated plaque or ulceration | ‘Swiss cheese’ appearance due to presence of silicone filled cavities surrounded by histiocytes (may be multinucleated or foamy) |
Hyaluronic acid | Erythema, induration, papule or nodule | Basophlic amorphous material, stains with Alcian blue |
Paraffin | Firm indurated nodule or plaque, ± ulceration | ‘Swiss cheese’ appearance (stains with oil red O) |
Starch | Indurated nodule typically | Maltese cross configuration with polarized light |
Monsel’s solution | Clinically consistent with nevus or tattoo | Brown black deposits (containing iron) in dermis due to ferrous subsulfate |
Sarcoidosis (Figure 3.16A-D)
Chronic multisystem inflammatory disease characterized by non-caseating granulomas of unknown etiology
Related to ↑ activity of cell mediated immune system
↑ Frequency and severity in African American patients
Presents with cutaneous findings in approximately 30-40% patients, may be sole or initial manifestation
Presents typically with non-scaly, skin-colored to red-brown circinate or annular infiltrated papules/plaques on face, lips, neck, trunk, extremities; cutaneous sarcoidosis may develop within pre-existing scars
Hypopigmented lesions not uncommon in African-American patients
Sarcoidal plaques may appear psoriasiform
Less common presentations include ichthyosis over lower legs, hypopigmentation, scarring alopecia, and ulcerations
Variants (listed below)
Histology: superficial and deep sharply defined naked epithelioid granulomas with minimal lymphocytes, giant cells, eosinophilic stellate inclusion bodies (asteroid bodies) or round basophilic laminated inclusions (Schaumann bodies) seen in giant cells
Diagnosis (of exclusion): ↑ ACE, ↑ calcium, ↑ ESR
Treatment: topical, IL or systemic corticosteroid, hydroxychloroquine, methotrexate or other immunosuppressant
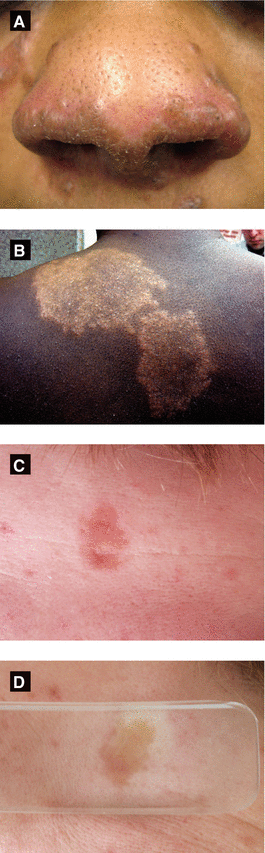
Figure 3.16:
A: SarcoidosisB: Sarcoidosis, hypopigmented (Courtesy of Dr. Iris K. Aronson)C: Sarcoidosis*D: Sarcoidosis**Reprint from Lipsker D. Clinical examination and differential diagnosis of skin lesions. Paris, France: Springer;2013.
Sarcoidosis Variant | Clinical Findings |
|---|---|
Lupus pernio | Violaceous doughy infiltration on nose, cheeks or earlobes; often associated with chronic sarcoidosis of lungs, chronic uveitis and bone cysts; chronic course |
Darier-Roussy disease | Also known as sarcoidal panniculitis; painless subcutaneous mobile nodules without epidermal change |
Löfgren’s syndrome | Acute sarcoidosis; erythema nodosum, hilar adenopathy, acute iritis, migrating polyarthritis, and fever |
Mikulicz syndrome | Complex of symptoms caused by a variety of systemic disorders (ie. Sjögren syndrome, lymphoma and sometimes sarcoidosis) Parotid and lacrimal enlargement with swelling, ± sicca symptoms |
Parinaud oculoglandular syndrome | Conjunctivitis with ipsilateral lymphadenopathy Also caused by infection (cat-scratch fever and tularemia) |
Heerfordt’s syndrome | ‘Uveoparotid fever’; fever, parotid gland enlargement, anterior uveitis, facial nerve palsy |
Erythema nodosum | Seen in acute or subacute sarcoidosis; good prognostic sign, associated with transient sarcoidosis that resolves spontaneously |
Oral sarcoidosis | May involve mucosa, tongue, major salivary glands, hard palate and gingival tissue |
Ocular sarcoidosis | Seen in 15-25%: anterior uveitis (common), lacrimal gland involvement, chronic uveitis leading to adhesions, glaucoma, and blindness |
Non-mucocutaneous findings | Lung disease (alveolitis, fibrosis, hilar adenopathy), liver, spleen, bone, kidney, heart, GI involvement; hypercalcemia |
B. Metabolic and Depositional Diseases
Amyloidosis (Figure 3.17)
Refers to several diseases sharing common feature of abnormal deposition of eosinophilic amyloid protein in various tissues
Amyloid properties: insoluble fibril protein aggregates with β-pleated sheet configuration
Classified into systemic and organ-limited amyloidosis, with the former being associated with ↑ morbidity and mortality (unlike the cutaneous counterpart)
Histology: deposits of eosinophilic, homogenous and amorphous material limited to papillary dermis with melanin incontinence in lichen/macular amyloidosis; waxy eosinophilic fissured nodules involving dermis in nodular amyloidosis; characteristic staining pattern showing green birefringence under polarized light with Congo red stain; other stains include methyl violet, crystal violet, PAS + (diastase resistant), Sirius red, pagoda red 9, scarlet red (RIT), and thioflavin T
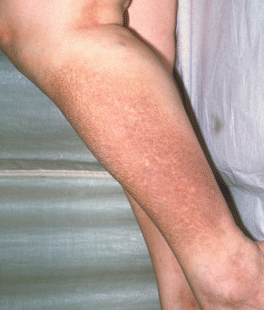
Figure 3.17:
Lichen amyloidosis(Courtesy of Dr. Paul Getz)
Table 3-11:
Types of Cutaneous Amyloidosis
Type | Description | Protein |
|---|---|---|
Macular amyloidosis | Presents with hyperpigmented small firm papules in rippled appearance coalescing into thin plaque typically over interscapular region; asymptomatic or moderately pruritic; ± associated notalgia paresthetica Treatment: potent topical corticosteroid, topical capsaicin | Keratinocyte-derived |
Seen in MEN type 2A | ||
Lichen amyloidosis (Figure 3.17) | Presents with small, flat-topped shiny papules typically over shins, highly pruritic Treatment: reduce friction, potent topical corticosteroid ± occlusion or IL corticosteroid, phototherapy | Keratinocyte-derived |
Seen in MEN type 2A | ||
Nodular amyloidosis (Figure 3.18c) | Presents with single or multiple waxy nodules ± purpura on limbs or trunk Can progress to systemic involvement in about 7% cases → long term follow up needed Treatment: excision or laser ablation if few lesions | AL (immunoglobulin light chains, typically λ) |
Secondary amyloidosis | Amyloid deposits seen both in benign and malignant cutaneous tumors | Keratinocyte-derived |
Multiple endocrine neoplasia (MEN) type 2A (Sipple syndrome): RET gene, AD
Medullary thyroid carcinoma, pheochromocytoma, hyperparathyroidism, ± lichen or macular amyloidosis
Of note, MEN type 1 (Wermer syndrome) associated with facial angiofibromas, collagenomas and lipomas
Type 2B (aka type 3) associated with mucosal neuromas
Table 3-12:
Types of Systemic Amyloidosis
TTR transports thyroxine & retinol
Type | Description/treatment | Protein |
|---|---|---|
Primary systemic amyloidosis | Usually associated with underlying plasma cell dyscrasia; up to 50% may have mucocutaneous lesions including macroglossia (± indentation of teeth), difficulty swallowing, ecchymosis and ‘pinch’ purpura due to vessel fragility from perivascular amyloid deposition (‘raccoon eyes’), waxy nodules and plaques, bullous lesions (especially hemorrhagic); hoarseness; other non-cutaneous involvement include carpal tunnel syndrome, RA-like arthropathy, shoulder pad sign (amyloid infiltration around periarticular soft tissue), cardiac arrhythmias, heart failure, restrictive cardiomyopathy; may be associated with multiple myeloma; confirmation of diagnosis in absence of cutaneous findings aspiration of abdominal fat to detect amyloid deposits | AL (light chain) |
Secondary systemic amyloidosis | Amyloid deposition in organs due to underlying chronic inflammatory or infectious process (ie. rheumatoid arthritis, tuberculosis, chronic abscess, periodic fever syndromes such as familial Mediterranean fever, TRAPS and Muckle-Wells syndrome – see below); skin typically not involved | AA (non-immunoglobulin protein: amyloid-associated) |
Hemodialysis-associated amyloidosis | Due to ↓ excretion of β2-microglobulin in patients with long-term hemodialysis; deposition in synovial membranes resulting in carpal tunnel syndrome and spondyloarthropathy | Aβ 2 M (β2-microglobulin) |
Familial amyloidosis | Includes familial amyloidotic polyneuropathy, AD inheritance; findings include peripheral and autonomic neuropathy; treatment: orthotopic liver transplantation (remove major source of TTR) | ATTR (TTR or transthyretin) |
Senile systemic amyloidosis | Late-onset disease seen in elderly due to deposition of TTR-derived amyloid fibrils in heart causing CHF, cardiomyopathy | ATTR (transthyretin or TTR) |
Muckle-Wells syndrome (MWS): CIAS1 mutation (encodes cryopyrin), AD → urticaria, deafness, renal amyloidosis, acute attacks of fever, abdominal pain, myalgias, arthralgias, and conjunctivitis; treat w/ glucocorticoids or anakinra (recombinant human IL-1 receptor antagonist)
Familial Mediterranean fever (FMF): MEFV mutation (encodes pyrin, also known as marenostrin), AR → recurrent episodes of polyserositis, fever, erysipelas-like erythema (legs); treat w/ colchicine (prophylaxis)
TNF receptor associated periodic syndrome (TRAPS): TNFR1 mutation, AD → high fever, erythematous annular or serpiginous patches/plaques on extremities, abdominal pain, arthralgias/myalgias; treat w/ TNF inhibitors or glucocorticoids
Mucinoses
Heterogenous group of skin disorders involving abnormal accumulation of mucin
Mucin
Mixture of acid glycosaminoglycans normally produced in small amounts by fibroblasts
Routine H&E shows blue-staining material between collagen bundles or empty space
Special stains for mucin: Alcian blue, colloidal iron or toluidine blue
Table 3-13:
Forms of mucinoses
Type | Description/treatment |
|---|---|
Scleromyxedema (Figure 3.18D) | Presents with generalized symmetric eruption of several waxy firm papules accompanied by induration and thickening of the skin (sclerodermoid) with ↓ mobility; typically involves hands, forearms, face (‘leonine facies’), neck, thighs and upper trunk; associated with monoclonal gammopathy (paraproteinemia) IgG λ (lambda light chain); due to fibroblast proliferation and mucin deposition in dermis; non-cutaneous manifestations include myopathy, arthropathy, neuropathy, dysphagia, lung and renal disease; poor prognosis Treatment: disappointing; stem cell transplant, oral immunosuppressants (including thalidomide), electron beam therapy; of note, monthly melphalan used in past but associated with ↑ mortality |
Lichen myxedematosus (Papular mucinosis) | Localized form of scleromyxedema (spectrum) with small, flat-topped shiny papules mainly over extensor extremities; does not progress to scleromyxedema but shows little tendency for spontaneous resolution Treatment: observation or topical corticosteroid |
Scleredema (Scleredema of Buschke) (Scleredema diabeticorum) (Figure 3.18E) | Three forms: – Infection-related: preceding fever, malaise and infection (typically streptococcal) in children and women; presents with induration of cervicofacial area with extension to proximal extremities and trunk; typically self-limited – Gammopathy-related: similar to above but with insidious onset and without preceding infection; typically associated with monoclonal gammopathy – Diabetes-related: subtle onset erythema and induration of neck and back(± peau d’orange appearance) in obese men with IDDM; persistent involvement All three may have some form of systemic involvement: dysphagia, cardiac abnormalities, serositis Treatment (for latter 2 types): phothotherapy, cyclophosphamide, oral glucocorticoid, cyclosporine |
Reticular erythematous mucinosis (REM) | Erythematous macules and papules in reticulated pattern over midline chest and back; may be induced by UV light Treatment: antimalarials, sun protection |
Porphyria (Figure 3.18D)
Inherited or acquired disorders due to enzyme deficiency causing ↑ production of porphyrins (photosensitizing) or their precursors during heme synthesis
Porphyrins (uroporphyrin, coproporphyrin, protoporphyrin) absorb light intensely in the Soret band (400-410 nm) → reactive oxygen species with subsequent damage to skin, liver, and/or rbcs
Table 3-14:
Types of Porphyria
Inheritance/Defect | Labs | Description | Treatment |
|---|---|---|---|
Congenital Erythropoietic Porphyria (CEP) (Gunther’s disease) | |||
AR Uroporphyrinogen III cosynthase | Urine: ++ uro/coproStool: ++ copro RBC: + uroPlasma: + fluoresce CEP: Colorless (anemia), Erythrodontia, PhotosensitivityUTC (uro three cosynthase): Ur Teeth r Colored | Extreme photosensitivity (bullae with subsequent mutilated scarring), hypertrichosis, erythrodontia (red fluorescent teeth), hemolysis, red urine (stains diapers), ↑ risk skin CA | Light avoidance, transfusions for anemia, ± bone marrow transplantation (BMT), ± splenectomy |
Erythropoietic Protoporphyria (EPP) | |||
AD Ferrochelatase EPP: enzyme starts w/F | Urine: normal levelsStool: ++ proto RBC: ++ protoPlasma: ± fluoresce | Photosensitivity with burning, heals with waxy scars, porphyrin gallstones, hepatic damage | Light avoidance, oral β-carotene, monitor liver |
Porphyria Cutanea Tarda (PCT) | |||
AD (familial form) or acquired Uroporphyrinogen decarboxylase (UD) PCT – UD: Urine Dazzles pink (fluoresce with Wood’s light) | Urine: ++ uro >copro Stool: + isocopro RBC: normal levels | Tense bullae, erosions, milia and scarring on sun-exposed skin, hypertrichosis (temples), iron overload, scleroderma-like changes, facial hyperpigmentation | Phlebotomy every 2 weeks, oral antimalarial agent Triggers: alcohol, HCV, estrogen, polychlorinated hydrocarbons iron overload (hemochromatosis, C282Y gene), HIV |
Acute Intermittent Porphyria (AIP) | |||
AD Porphobilinogen deaminase (PBD) AIP-PBD: Abdomen Is Painful, Please Barbiturates D/C | Urine: + ALA, PBG, Stool/RBC/plasma: all normal levels | NO skin findings; neurologic and psychiatric findings w/↑ abdominal pain | Remove trigger, glucose loading, hematin infusion Triggers: drugs (barbiturates, sulfonamides, griseofulvin), stress, fasting, alcohol, hormonal changes (progesterone) and infection |
Variegate Porphyria (VP) | |||
AD Protoporphyrinogen oxidase (PPO) | Urine: + ALA/PBG Stool: + proto, copro Plasma: + fluoresce | Overlap between AIP and PCT | Same treatment for AIP during acute attacks VP-PPO: ViPs have Pink Plasma Optimized at 626 nm (fluoresces) |
Hereditary Coproporphyria | |||
AD Coproporphyrinogen oxidase (CPO) | Urine: ALA, PBG, Stool: + copro, proto RBC: normal level | Acute attacks similar to mild version of AIP, may have skin findings (mimics PCT) | Same a variegate prophyria |
Hepatoerythropoietic Porphyria | |||
AR Uroporphyrinogen decarboxylase | Urine: + uro Stool: + uro, coproRBC: + proto | Overlap between PCT and CEP | Photoprotection (phlebotomy NOT effective) |
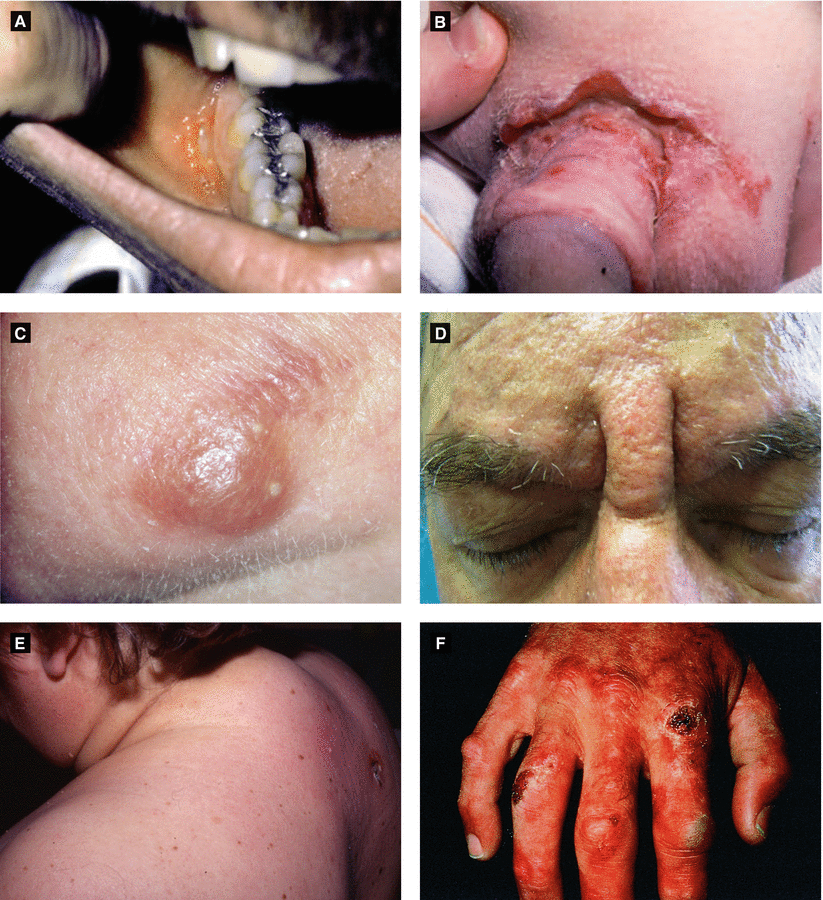
Figure 3.18:
A: Cutaneous Crohn’s disease* B: Cutaneous Crohn’s disease*C: Nodular amyloidosis** D: Scleromyxedema***E: Scleredema*** F: Porphyria cutanea tarda*****Reprint from Wu, Selsky, Grant-Kels (eds). Atlas of Dermatological Manifestations of Gastrointestinal Disease. New York, NY: Springer; 2013.**Reprint from Lipsker D. Clinical Examination and Differential Diagnosis of Skin Lesions. Paris, France: Springer; 2013.***Reprint from Smoller BR, Rongioletti F (eds).Clinical and Pathological Aspects of Skin Diseases in Endocrine, Metabolic, Nutritional and Deposition Disease. New York, NY: Springer; 2010.****Reprint from Maddrey WC (ed). Atlas of the Liver. New York, NY: Springer; 2004.
Rare condition of systemic calcification involving small and medium-sized vessels with ischemic necrosis of the skin and soft tissue; high mortality
Presents initially as painful violaceous mottling (reticulated) typically on lower extremities → stellate purpura with central necrosis, ± central bulla, followed by necrosis and ulceration; death due to gangrene and sepsis
Most commonly in patients with end-stage renal disease on hemodialysis or after renal transplant with elevated calcium-phosphate product
Treatment
Normalization of calcium-phosphate product (by low calcium dialysis)
Sodium thiosulfate (↑ solubility of calcium deposits)
Bisphosphonates (pamidronate, etidronate)
Calcimimetics (cinacalcet)
Parathyroidectomy
Wound therapy
Table 3-15:
Types of Familial Hyperlipidemia
Type | Defect | ↑ CAD Risk | Serum | Serum levels | Clinical findings |
|---|---|---|---|---|---|
Type I Familial LPL deficiencyFamilial hyperchylomicronemia | ↓ Lipoprotein lipase (LPL) or apoprotein CII defect | No | Creamy top layer | ↑↑ TG (chylomicrons) | Eruptive xanthomas, acute pancreatitis abdominal pain, lipemia retinalis |
Type IIa Familial hypercholesterolemiaFamilial defective apo B100 | LDL receptor defect (mutation of LDLR or apo B) | Yes | Clear | ↑↑ Cholesterol (LDL) | Tendinous and tuberous xanthomas, xanthelasma |
Type IIb Familial combined hypercholesterolemia | LDL receptor defect | Yes | Clear or cloudy | ↑ Cholesterol ↑ TG (LDL, VLDL) | Tendinous and tuberous xanthomas, xanthelasma |
Type III Familial dysbetalipoproteinemia | Apoprotein E defect | Yes | Turbid | ↑ Cholesterol ↑ TG (IDL, VLDL) | Xanthoma striatum palmare, tuberous xanthomas pathognomonic |
Type IV Familial hypertriglyceridemia | ↑ Production of VLDL | ± | Turbid | ↑ TG (VLDL, IDL) | Eruptive xanthomas; associated with DM, obesity, alcoholism |
Type V | Apolipoprotein C-II defect | ± | Creamy top layer | ↑↑ TG ↑ Cholesterol (VLDL, chylo) | Eruptive xanthomas, acute pancreatitis abdominal pain |
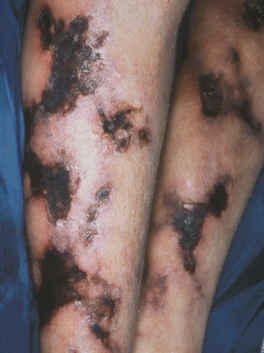
Figure 3.19:
Calciphylaxis(Reprint from Morgan MB, Smoller BR, Somach SC. Deadly Dermatologic Diseases. New York, NY: Springer; 2007.)
Type of Xanthoma | Description
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
|---|