(1)
Department of Dermatology, Freiburg University Medical Center, Freiburg, Germany
12.1.1 Incontinentia Pigmenti
12.1.2 Focal Dermal Hypoplasia
12.1.4 MIDAS Syndrome
12.1.8 Menkes Syndrome
12.1.9 IFAP Syndrome
12.1.10 Aicardi Syndrome
12.1.13 X-linked Dominant Hypertrichosis
12.2.1.1 Syringocystadenoma Papilliferum
12.2.1.2 Maffucci Syndrome
12.2.1.3 Happle-Tinschert Syndrome
12.2.3 Other Autosomal Non-Nevi
12.2.3.3 “Basal Cell Nevus”
12.2.3.4 “Blue Rubber Bleb Nevus”
12.3.1 Lichen Striatus
12.3.3 Lichen Aureus
12.3.4 Linear Grover Disease
12.3.7.1 Acquired Inflammatory Disorders
12.3.7.2 Vitiligo
12.3.7.3 Cherry Angiomas
Abstract
Abstract This group includes X-linked skin disorders, some autosomal non-nevi, and mosaic manifestations of common, acquired skin disorders with a polygenic background Among the X-linked genodermatoses, some phenotyopes exclusively occur in women because the underlying gene is acting as a lethal factor for male embryos. Affected female embryos survive by the mechanism of lyonization. This group of disorders includes incontinentia pigmenti, focal dermal hypoplasia, Conradi-Hünermann-Happle syndrome, MIDAS syndrome, and oral-facial digital syndrome type I. Another X-linked, male-lethal trait is Aicardi syndrome that cannot be categorized as a genodermatosis because constant cutaneous features are lacking, although a unilateral hairy bridge between eyebrow and scalp hair (“trichogephyrosis”) was noted in one case. – In X-linked genodermatoses caused by non-lethal mutations, male patients are diffusely and severely affected, whereas female carriers usually show a Blaschko-linear pattern of lyonization. Such phenotypes include Christ-Siemens-Touraine syndrome, X-linked dyskeratosis congenita, Menkes syndrome, IFAP syndrome, and the reticulate pigmentary disorder of Partington. By contrast, women heterozygous for X-linked generalized hypertrichosis tend to show a checkerboard pattern of lyonization. In the X-linked albinism-deafness syndrome, the arrangement of depigmented areas as noted in affected females is so far unclassifiable, whereas their irides show a definite sectorial pattern of hypopigmentation. Autosomal non-nevi: Some skin disorders look like nevi but are definitely no nevi, either because they represent benign neoplastic lesions or because someone has called them nevi without reason. The lesions of syringocystadenoma papilliferum tend to be arranged in a linear, nevus-like pattern, but they are benign neoplasias, which is why they do not fulfil the criteria of a nevus. Similarly, the hemangiomas and enchondromas of Maffucci syndrome are distributed in a mosaic arrangement, but they are likewise neoplastic lesions. The same holds for the segmentally arranged basaloid follicular hamartomas of Happle-Tinschert syndrome, and for the subcutaneous lipomas of hemihyperplasia-multiple lipomata syndrome. – The salmon patch (“Unna’s nevus”, “median nevus flammeus”) is no nevus because the criterion of mosaicism is lacking. – “White sponge nevus of the oral mucosa” is an absurd term. The symmetric lesions of this autosomal dominant trait may better be called “white sponge hyperplasia of the mucosa”. – The term “basal cell nevus” is incorrect because it denotes neoplastic skin lesions. Moreover, the term is ambiguous because it has been used to describe quite different disorders such as the disseminated basal cell carcinomas of Gorlin syndrome, or a mosaic manifestation of Gorlin syndrome, or hereditary nonsyndromic basal cell carcinomas, or the basaloid follicular hamartomas of Happle-Tinschert syndrome. – Finally, “blue rubber bleb nevus syndrome” is a misnomer because these vascular lesions are benign neoplasias. Hence, the term “blue rubber bleb angiomatosis” is more appropriate. Acquired skin disorders: Some of these diseases occur in a mosaic arrangement but do not fulfill the criteria of a nevus because they tend to resolve spontaneously after some time. Examples are lichen striatus (including “blaschkitis”), lichen aureus, linear Grover disease, and linear juvenile xanthogranuloma. – On the other hand, numerous common skin disorders with a polygenic background sometimes show a linear or otherwise segmental distribution, either as an isolated nevoid manifestation or being superimposed on the nonsegmental phenotype. The segmental lesions would originate either from an early event of allelic loss at one of the predisposing loci, or from a postzygotic new mutation at an additional locus predisposing to the disorder. Clinical examples suggesting a superimposed segmental involvement include psoriasis vulgaris, pustular psoriasis, atopic dermatitis, lichen planus, lichen planopilaris, lichen nitidus, acne vulgaris, discoid lupus erythematosis, lupus erythematosus profundus, subacute lupus erythematosus, systemic lupus erythematosus, dermatomyositis, pemphigus vulgaris, bullous pemphigoid, graft-versus-host disease, morphea, granuloma annulare, erythema multiforme, common drug eruption, fixed drug eruption, lateralized exanthema of childhood, vitiligo, and cherry angiomas.
The group of nevoid conditions includes all skin disorders that are reminiscent of a nevus, but do not fulfill the criteria of a true nevus [83] (see Chap. 7). On the other hand, this group comprises some X-linked “true nevi” such as the cutaneous lesions of focal dermal hypoplasia that, by convention, are categorized as “nevoid.”
12.1 Cutaneous Lesions Reflecting Functional X-Chromosome Mosaicism
The skin lesions reflecting lyonization in women with X-linked traits are taken, by convention, as nevoid conditions although they fulfill, in principle, the criteria of true nevi [83]. There is, however, one exception from this rule. The CHILD nevus reflects functional X-chromosome mosaicism but is categorized as a true nevus (see Sect. 7.3.1.9 and Table 7.1).
12.1.1 Incontinentia Pigmenti
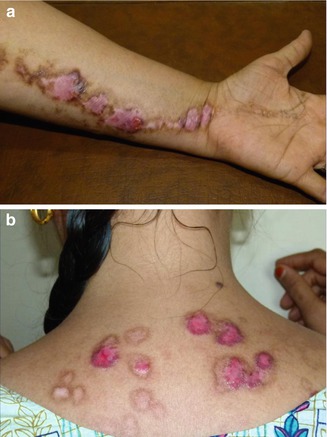
This X-linked multisystem birth defect is caused by NEMO mutations [227]. The disorder is characterized by linear skin lesions that develop in four stages. During the first months of life, an episode of inflammation and blistering of the skin occurs, being arranged along Blaschko’s lines (Fig. 12.1). Later, these lesions are replaced by hyperkeratotic verrucous areas, whereas in older girls a linear or patchy hyperpigmentation is noted (Figs. 3.14 and 12.1b). In adult women, this hyperpigmentation often fades away and gives rise to hypopigmented linear lesions that are slightly atrophic and hairless. This fourth stage tends to be most conspicuous on the calves.
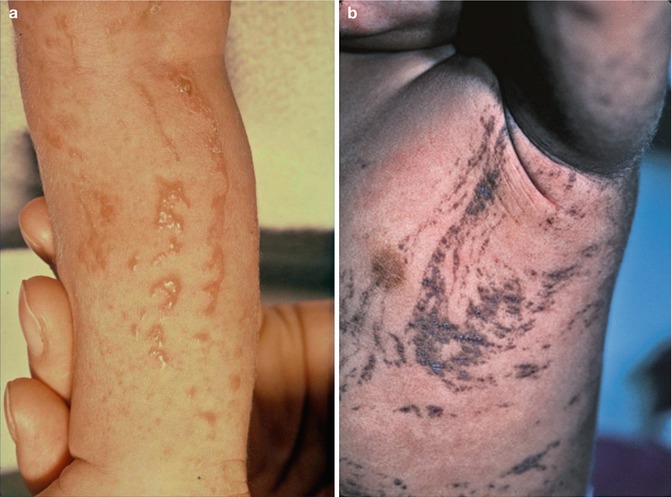
Fig. 12.1
Incontinentia pigmenti. (a) First stage of in the form of linear blistering; (b) third stage characterized by linear hyperpigmentation [19] (Reprinted with permission from John Wiley & Sons, USA)
The early inflammatory stage apparently reflects an attempt of the functionally normal cells to eliminate the functionally aberrant clone. This elimination process, however, is not always complete because exceptional eruptions reiterating a stage 1 may occur in older children [6, 50, 197] or even in adult women [14].
Hypomorphic NEMO alleles may cause a quite different phenotype in the form of ectodermal dysplasia of Zonana (see Chap. 4).
12.1.2 Focal Dermal Hypoplasia
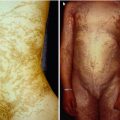
Focal dermal hypoplasia (Goltz syndrome) is an X-linked dominant, male-lethal disorder caused by PORCN mutations [77]. The syndrome includes multiple congenital defects of the mesoderm and the ectoderm, reflecting lyonization. A cutaneous hallmark are linear lesions of thinned or absent dermis (Fig. 12.2), giving rise to herniation of the underlying fatty tissue [82]. Longitudinal striation of the long bones as noted on x-rays is likewise very characteristic (Fig. 12.3) [97].
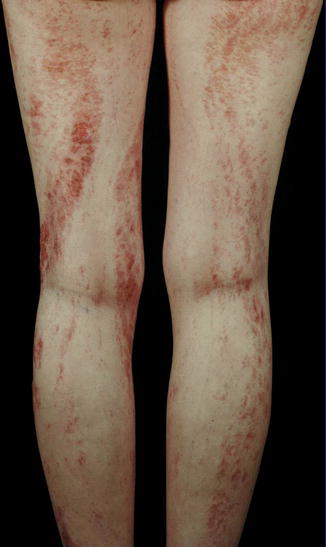
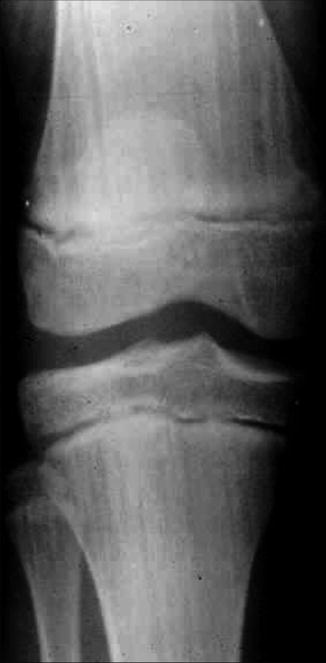
Fig. 12.2
Focal dermal hypoplasia showing a systematized linear arrangement (Courtesy of Dr. Regina Föster-Holst, Kiel, Germany)
Fig. 12.3
Longitudinal striation of long bones reflecting functional X-chromosome mosaicism in a patient with focal dermal hypoplasia
12.1.3 Conradi-Hünermann-Happle Syndrome
This X-linked dominant, male-lethal phenotype is caused by EBP mutations [102]. The syndrome is also called X-linked dominant chondrodysplasia punctata and includes asymmetric bone defects, sectorial cataracts, and skin lesions following Blaschko’s lines. Epiphyseal stippling is found on x-rays taken shortly after birth. During the first weeks of life, the skin shows linear areas of inflammation and hyperkeratosis (Fig. 12.4). This stage is followed by linear areas of atrophy and hypopigmentation with rather mild scaling. Lesions of follicular atrophoderma are predominantly found on the scalp, the wrists, and the dorsal and lateral aspects of the hands (Fig. 12.5).
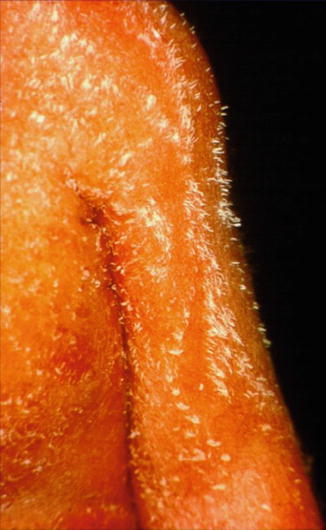
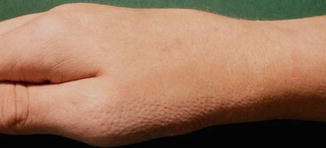
Fig. 12.4
Characteristic feather-like hyperkeratoses showing a linear arrangement in a newborn girl with Conradi-Hünermann-Happle syndrome (Courtesy of Dr. Elzo Folkers, Zaandam, The Netherlands)
Fig. 12.5
Linear arrangement of follicular atrophoderma in an adolescent girl with Conradi-Hünermann-Happle syndrome
A girl with an EBP mutation and classical features of the syndrome had a mother who was phenotypically healthy with the exception of a very small patch of erythematous and slightly hyperkeratotic skin present on one of her legs. The mutation was absent in the mother’s blood lymphocytes but present in DNA obtained from her tiny skin lesion, providing evidence that she carried an EBP mutation in the form of postzygotic mosaicism [165].
12.1.4 MIDAS Syndrome
MIDAS is an acronym for microphthalmia, dermal aplasia, and sclerocornea [36, 92]. The disorder is also called MLS (microphthalmia with linear skin defects) syndrome [255]. It is caused by male-lethal mutations in one of the X-linked genes, holocytochrome c-type synthetase [198] or COX7B [113]. Oncocytic cardiomyopathy may cause early death [21]. For unknown reasons, the cutaneous lesions tend to involve almost exclusively the face and the neck (Fig. 12.6) [179, 263].
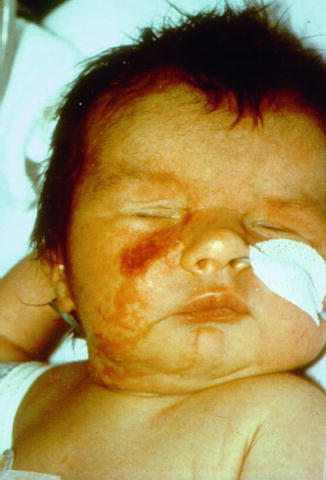
Fig. 12.6
MIDAS syndrome in a newborn girl affected with bilateral microphthalmia, blepharophimosis, saddle nose, and unilateral linear areas of dermal aplasia [92] (Reprinted with permission from John Wiley & Sons, USA)
12.1.5 Oral-Facial-Digital Syndrome Type 1
This X-linked, male-lethal trait is caused by CXORF5 mutations. It includes gingival frenula and multilobulated tongue (Fig. 12.7), median cleft of upper lip, asymmetric cleft palate, malformed digits, and mild mental deficiency. Multiple milia involve the face and the ears but disappear after infancy. On the scalp a spiral pattern of congenital alopecia, reflecting lyonization, is noted (Fig. 12.7) [25, 95].
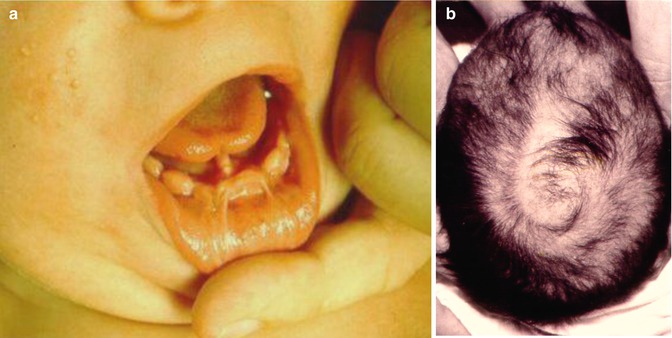
Fig. 12.7
Oral-facial-digital syndrome type 1. (a) Note gingival frenula and lobulated tongue; (b) spiral pattern of hairlessness reflecting lyonization [95]
12.1.6 Christ-Siemens-Touraine Syndrome
With few exceptions, the full-blown phenotype of hypohidrosis is observed in male patients only, whereas heterozygous female individuals tend to be mildly affected. Sweat testing of carrier females reveals hypohidrotic areas arranged along Blaschko’s lines (Fig. 12.8) [15, 94].
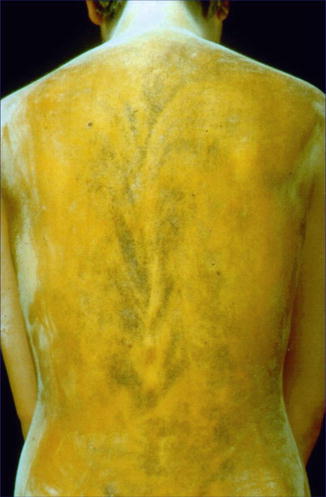
Fig. 12.8
Minor’s sweat testing in a female carrier of Christ-Siemens-Touraine syndrome [94] (Reprinted with permission from John Wiley & Sons, USA)
12.1.7 X-Linked Dyskeratosis Congenita
Male patients with X-linked dyskeratosis congenita show a diffusely arranged reticular hyperpigmentation and atrophy of skin, dystrophic nails, mucosal leukoplakia, progressive failure of bone marrow, and increased proneness to develop malignancies. Heterozygous women are, as a rule, only mildly affected, and their skin lesions follow Blaschko’s lines (Fig. 12.9) [54].
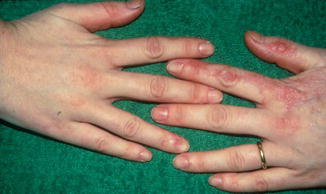
Fig. 12.9
Linear involvement of left hand in a female carrier of dyskeratosis congenita [54] (Courtesy of Dr. Hugo Degreef, Leuven, Belgium)
12.1.8 Menkes Syndrome
This neurodegenerative disorder is caused by mutations in the gene encoding Cu(2+)-transporting ATPase, alpha polypeptide (ATP7A). Major features include twisting and kinking of scalp hair, mental deficiency, seizures, bone defects, and hypothermia. Affected boys usually die during early infancy. Heterozygous women may show a linear or otherwise segmental pattern of cutaneous hypopigmentation (Fig. 12.10) [147, 253] and a mixture of anomalous and normal scalp hair [40, 49].
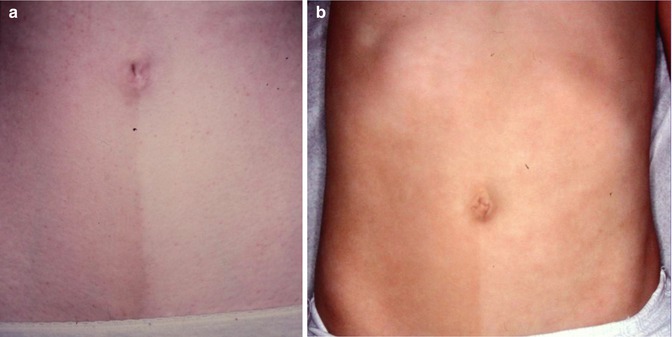
Fig. 12.10
(a, b) Unilateral arrangement of abdominal hypopigmentation in two sisters of a boy with Menkes syndrome [147] (Courtesy of Dr. Gérard Lorette, Tours, France)
12.1.9 IFAP Syndrome
IFAP (ichthyosis follicularis, atrichia, photophobia) syndrome is caused by mutations in MBTPS2, an intramembrane zinc metalloprotease controlling cholesterol homeostasis and stress response of the endoplasmatic reticulum [178]. Boys are diffusely and severely affected (Fig. 12.11a), whereas female carriers show a linear pattern of follicular ichthyosis, atrophoderma, asymmetric hairlessness (Fig. 12.11b), and diminished sweating [56, 137]. Hyperkeratotic streaks involving the feet have likewise been reported [240]. Unilateral acne lesions are another mosaic manifestation as noted in a 14-year-old heterozygous girl (Fig. 12.11c).
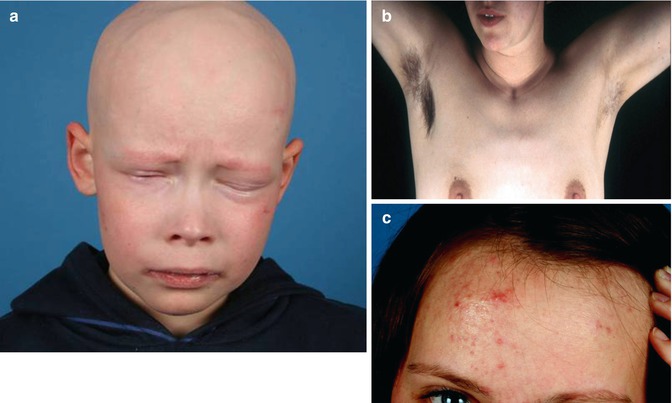
Fig. 12.11
IFAP syndrome. (a) A boy with complete baldness and pronounced photophobia [137]; (b) unilateral axillary hypotrichosis in his heterozygous mother; (c) asymmetrical acne lesions in his heterozygous sister
12.1.10 Aicardi Syndrome
This X-linked dominant, male-lethal disorder is characterized by chorioretinal lacunae, agenesis of corpus callosum, and infantile spasms [209]. Associated features include microphthalmia, coloboma, hypoplasia of the optic nerve, and various cerebral defects [231]. A 3-month-old blind girl with Aicardi syndrome had a “trichogephyrosis” in the form of hypertrichosis bridging the gap between the right eyebrow and scalp hair (Fig. 12.12) [96]. Additional clinical reports are needed to determine whether the unilateral involvement reflects lyonization.
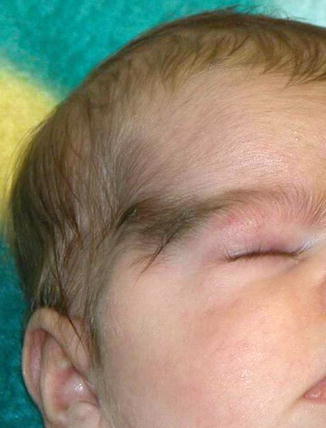
Fig. 12.12
Unilateral nevoid hypertrichosis in the form of “trichogephyrosis” in an infant girl with Aicardi syndrome [96] (Reprinted with permission from John Libbey Eurotext, Montrouge, France)
12.1.11 Reticulate Pigmentary Disorder of Partington
The full-blown syndrome occurs in male individuals who show diffuse reticulate dyspigmentation (Fig. 12.13a), hypohidrosis, coarse hair, failure to thrive, recurrent pneumonia, colitis, and corneal dyskeratosis with photophobia. Female carriers have linear lesions of dyspigmentation (Fig. 12.13b) [9, 131, 190, 191]. The underlying gene has been assigned to Xp22–p21 but is so far unknown [117].
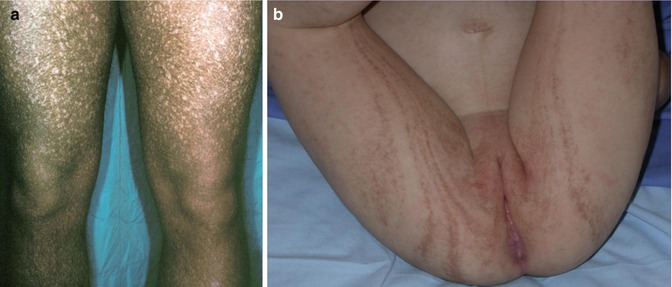
Fig. 12.13
Reticulate pigmentary disorder of Partington. (a) Diffuse involvement in a man; (b) linear lesions in his daughter (Courtesy of Dr. Eulalia Baselga, Barcelona, Spain)
12.1.12 X-linked Albinism-Deafness Syndrome
In 1962, an unusual X-linked form of albinism with deafness was described in two reports on the same family [152, 261]. Initially this was taken as an X-linked recessive trait, but female members of the family were later found to be likewise affected [66]. The trait has been assigned to Xq26.3–q27.1 [223]. Male patients are deaf and blue eyed and show peculiar patches of hyperpigmentation being diffusely distributed within the albinotic skin (Fig. 12.14a), whereas affected women may have difficulties of hearing or may show segmental depigmentated areas involving the skin (Fig. 12.14b) or the irides (Fig. 12.14c). Remarkably, the cutaneous pattern of lyonization does not follow Blaschko’s lines.
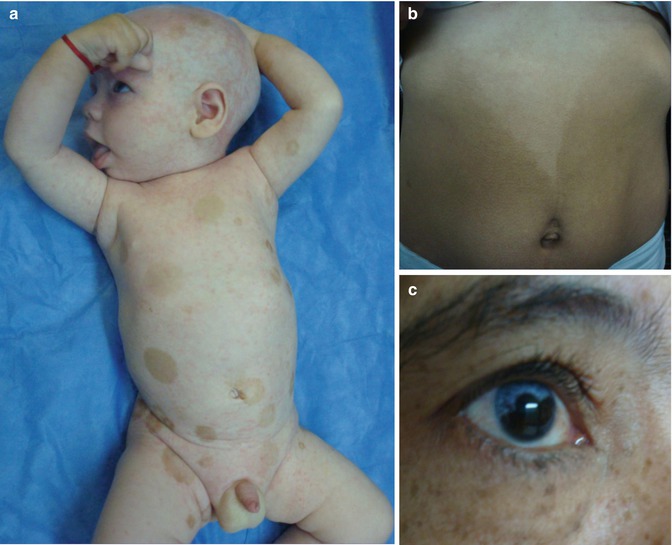
Fig. 12.14
X-linked albinism-deafness syndrome. (a) Diffuse involvement in a boy; (b) segmental hypopigmentation in his mother who suffers from hearing loss; (c) sectorial depigmentation of the iris in the boy’s grandmother who has no hearing loss (Courtesy of Dr. Silvina Sartori, Santa Fe, Argentina)
12.1.13 X-linked Dominant Hypertrichosis
Affected males have a severe and diffusely distributed hypertrichosis, whereas heterozygous females show asymmetrical patches of increased hairiness arranged in a checkerboard pattern, reflecting lyonization (Fig. 12.15) [63, 150]. In a large Chinese family, Zhu et al. [260] confirmed that heterozygous women tend to be more mildly affected. The authors assumed that a palindrome-mediated insertion near SOX3 may be involved in the etiology. A syndromic form of this X-linked hair disorder has likewise been documented [233].
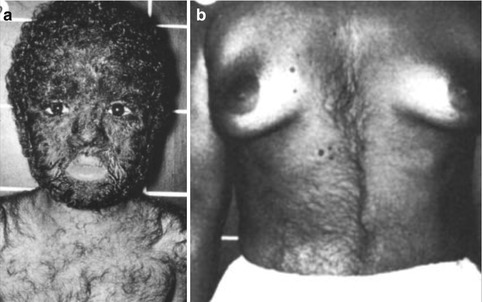
Fig. 12.15
X-linked dominant hypertrichosis. (a) Diffuse involvement in a boy; (b) segmental pattern of lyonization in a woman [63] (Reprinted with permission from Nature Publishing Group)
12.2 Congenital Autosomal Disorders Representing Non-Nevi
The following skin disorders are either autosomal sporadic tumor syndromes or other non-nevi that have so far been mistaken as nevi either because they look like nevi or simply because someone has erroneously categorized them as nevi.
12.2.1 Benign Skin Tumors Reflecting Lethal Autosomal Mutations Surviving by Mosaicism
The following sporadic phenotypes cannot be categorized among the group of nevi because they are characterized by the presence benign neoplasias.
12.2.1.1 Syringocystadenoma Papilliferum
Multiple tumors of this type show rather frequently an arrangement along Blaschko’s lines (Fig. 12.16) [139, 154, 174, 187, 194, 256, 258]. All cases so far reported were sporadic.
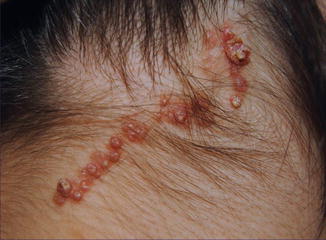
Fig. 12.16
Syringocystadenoma papilliferum showing a linear arrangement [154] (Reprinted with permission from Creative Commons)
12.2.1.2 Maffucci Syndrome
In this syndrome multiple enchondromas, most frequently affecting the fingers and toes, are found to be associated with multiple hemangiomas that predominantly involve the skin (Fig. 12.17) [120, 126] but may also be found in the oral cavity, esophagus, or intestinal tract [222]. The syndrome always occurs sporadically and the lesions show an asymmetrical arrangement. Therefore, the phenotype was proposed to be caused by a lethal mutation surviving by mosaicism [84]. This etiological concept was corroborated in recent molecular studies. Pansuriya et al. [188] found postzygotic heterozygous mutations in IDH1 or IDH2 within the lesional tissue of patients with Maffucci syndrome and a related disorder, Ollier disease.
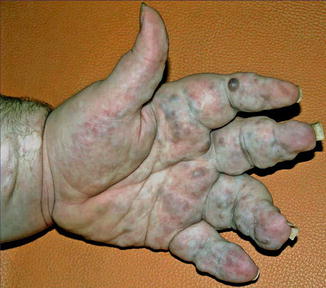
Fig. 12.17
Maffucci syndrome [205] (Reprinted with permission from the American Society of Clinical Oncology)
12.2.1.3 Happle-Tinschert Syndrome
This birth defect includes multiple, segmentally arranged basaloid follicular hamartomas (Fig. 12.18a), linear atrophoderma (Fig. 12.18b), and osseous, dental, ocular, or cerebral anomalies [22, 99, 116]. A case described by Bedi [17] as “nevus unius lateris” is another typical example of this disorder.
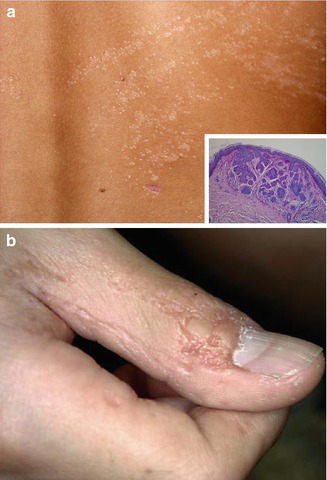
Fig. 12.18
Happle-Tinschert syndrome. (a) Linear arrangement of multiple follicular basaloid hamartomas in a 7-year-old boy; (b) ipsilateral linear atrophoderma involving the thumb [116] (Reprinted with permission from S. Karger AG, Basel, Switzerland)
It is unlikely that the phenotype represents a type 2 segmental manifestation of hereditary multiple basaloid follicular hamartoma because so far this autosomal dominant trait has neither been observed in patients with Happle-Tinschert syndrome nor in their relatives [99]. Admittedly, however, it is so far difficult to determine whether cases of multiple congenital, segmentally arranged and tightly packed basaloid follicular hamartomas [8, 100, 110] may represent a minimal form of this syndrome or a superimposed manifestation of autosomal dominant multiple basaloid follicular hamartoma (see Sect. 10.1.4).
12.2.2 Hemihyperplasia-Multiple Lipomata Syndrome: A Nevoid Disorder of Unknown Origin
This phenotype was delineated in 1998 by Biesecker et al. [20]. It is characterized by a rather stable overgrowth of one half of the body and ipsilateral development of multiple lipomas (Fig. 12.19) [30, 215]. The term “multiple lipomatosis” [20] is redundant because the word lipomatosis implies multiplicity of the tumors. The syndrome has always occurred sporadically. Its molecular basis is so far unknown. Possible mechanisms are discussed in OMIM under the entry 235000 (“hemihyperplasia, isolated”) [182].
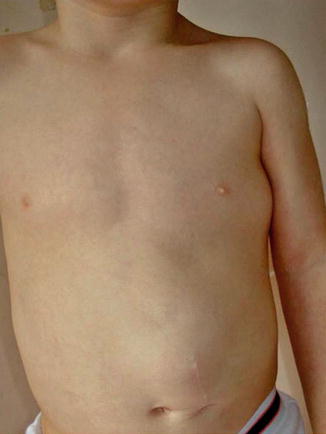
Fig. 12.19
Hemihyperplasia-multiple lipomata syndrome in a 3-year-old boy. Note strictly unilateral arrangement of the lipomas [30] (Reprinted with permission from Elsevier Limited, Oxford, UK)
Practical aspect: Liposuction appears to be a suitable therapeutic option to alleviate the burden of large lipomas [144].
12.2.3 Other Autosomal Non-Nevi
Some other skin lesions are called nevi without being nevi. The following disorders refer to such historical misnomers.
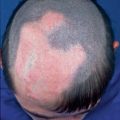
12.2.3.1 Salmon Patch (“Unna’s Nevus,” “Median Nevus Flammeus,” “Nevus Simplex”)
Congenital telangiectatic macules involving the glabella (“angels kiss”) or the occipital or nuchal region (“stork bite”) (Fig. 12.20) or the sacral area have so far been categorized as median vascular nevi [122, 193]. These lesions do not fulfill, however, the criteria of a nevus because they do not reflect mosaicism. Frontal salmon patches are noted in 30–40 % of newborns, whereas the prevalence of the nuchal erythema is even higher [250]. If we would include such median vascular lesions into the group of nevi, this term would lose any specific meaning.
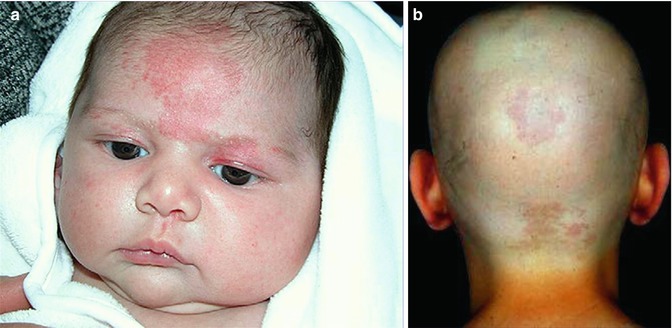
Fig. 12.20
Salmon patch, a frequently occurring non-nevus. (a) Rather extensive glabellar involvement [122]; (b) occipital involvement in a adult patient with alopecia totalis (a: Reprinted with permission from Elsevier Limited, Oxford, UK)
12.2.3.2 White Sponge Hyperplasia of the Mucosa (“White Sponge Nevus”)
This autosomal dominant trait is caused by mutations of keratin 4 or 13 and has so far been called “white sponge nevus” (Fig. 12.21) [146]. This name, however, is an absurdity. With the same right one could categorize palmoplantar keratoderma as a “hyperkeratotic nevus.” The symmetric oral leukoplakia does not look like a nevus, nor does it reflect mosaicism.
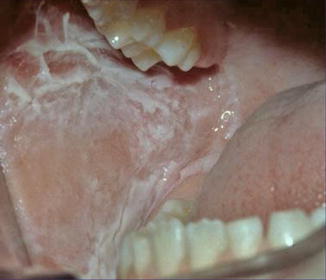
Fig. 12.21
White sponge hyperplasia of the mucosa [146] (Reprinted with permission from John Wiley & Sons, USA)
12.2.3.3 “Basal Cell Nevus”
This term should no longer be used since it is both incorrect and ambiguous [99]. It is incorrect because it refers to non-melanocytic neoplasias and thus not to nevi. It is ambiguous because it has been used in the past to describe Gorlin syndrome [32] and Happle-Tinschert syndrome [7, 116] and mosaic manifestations of either Gorlin syndrome [221] or nonsyndromic basal cell carcinomas [31] or nonsyndromic basaloid follicular hamartomas [44].
12.2.3.4 “Blue Rubber Bleb Nevus”
Blue rubber bleb “nevi” are not nevi but represent true angiomas. Hence, the so-called blue rubber bleb nevus syndrome is in fact a blue rubber bleb angiomatosis (see Sect. 10.1.11).
12.3 Nevoid Arrangement of Acquired Skin Disorders
This group includes some disorders that tend to occur exclusively in a segmental arrangement, and mosaic manifestations of many common skin disorders with a polygenic background.
12.3.1 Lichen Striatus
This transient disorder is preponderantly noted in children and consists of one or more linear areas showing inflammatory papular lesions (Fig. 12.22). The distribution follows Blaschko’s lines. Histopathologically, a lichenoid infiltrate with lymphocytic exocytosis and spongiosis is noted [229]. Spontaneous resolution tends to occur after some weeks or months. The cause of the disorder is so far unknown. Some cases of lichen striatus being associated with atopic dermatitis [55, 192, 243] may in fact represent examples of superimposed linear atopic dermatitis [245].
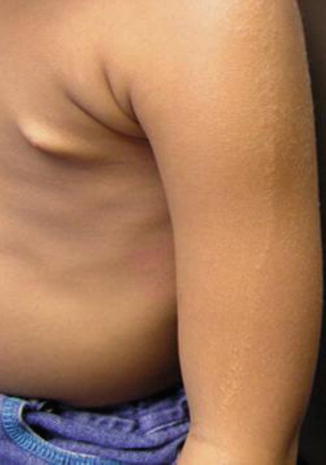
Fig. 12.22
Lichen striatus in a 3-year-old girl [200] (Reprinted with permission from John Wiley & Sons, USA)
12.3.2 “Blaschkitis”: No Entity, but Either a Variant of Lichen Striatus or an Umbrella Term Including the Linear Manifestation of Various Acquired Inflammatory Skin Disorders
This term was proposed by Grosshans and Marot [76] for a transient linear skin disorder that resembled lichen striatus but showed a more pronounced inflammation and was preponderantly noted in adults (Fig. 12.23). A clear-cut separation from lichen striatus, however, appears to be rather difficult [161, 235], and “pediatric blaschkitis” was also reported [53, 130]. Many authors believe that blaschkitis is merely a variant of lichen striatus [79, 108, 163, 169, 201, 241]. In 2006, Grosshans himself agreed that “blaschkitis probably belongs to the spectrum of lichen striatus” [236].
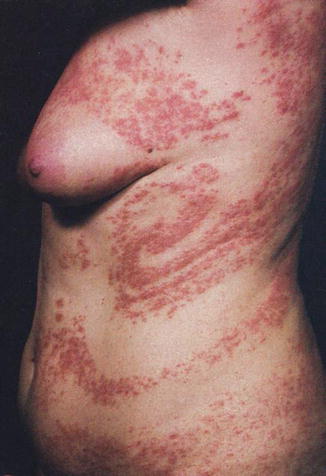
Fig. 12.23
Unilateral systematized “Blaschko dermatitis” in a 44-year-old woman [158] (Reprinted with permission from Elsevier Limited, Oxford, UK)
In a broad sense, the term “blaschkitis” includes the isolated or superimposed linear manifestation of many acquired inflammatory skin disorders such as atopic dermatitis, lichen planus, various types of lupus erythematosus, dermatomyositis, or drug eruptions (see Sect. 12.3.7.1).
12.3.3 Lichen Aureus
Lichen aureus is usually arranged in a segmental pattern (Fig. 12.24) [142, 162] and can, therefore, be categorized as a mosaic disorder. Synonyms include unilateral linear capillaritis [149], unilateral linear capillaropathy [259], and linear progressive pigmentary purpura [106]. It is a controversial issue whether there is a relationship between this disorder and the nonsegmental lesions of Schamberg disease. As a rule of thumb, a nonsegmental involvement that usually occurs in adults tends to be called “Schamberg disease,” whereas the name “lichen aureus” is preferred for the segmental lesions that are preponderantly noted in children [119]. Remarkably, however, cases of superimposed segmental involvement have so far not been reported.
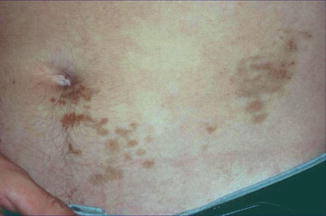
Fig. 12.24
Lichen aureus showing a characteristic segmental distribution [162] (Reprinted with permission from the Australasian College of Dermatologists © 2010 the Australasian College of Dermatologists)
12.3.4 Linear Grover Disease
Grover disease is an acquired disorder characterized by multiple disseminated papular lesions resembling those of Darier disease both clinically and histopathologically [189]. Contrasting with Darier disease, however, the disorder is rather pruritic [12]. It tends to develop later in life and usually disappears spontaneously after some months. A systematized linear arrangement of the disorder (Fig. 12.25) was reported by Asahina et al. [12].
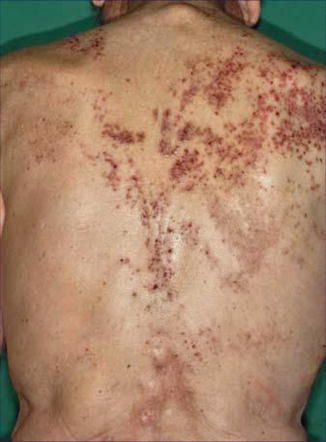
Fig. 12.25
Grover disease arranged along Blaschko’s lines [12] (Reprinted with permission from S. Karger AG, Basel, Switzerland)
12.3.5 Linear Juvenile Xanthogranuloma
The small yellowish plaques of juvenile xanthogranuloma are a rather common disorder of infancy. On rare occasions, a pronounced linear arrangement may occur [68, 133]. This segmental involvement may include large nodules that even may show ulceration (Fig 12.26) [176]. Such cases may herald the presence of one or more genes predisposing to juvenile xanthogranuloma.
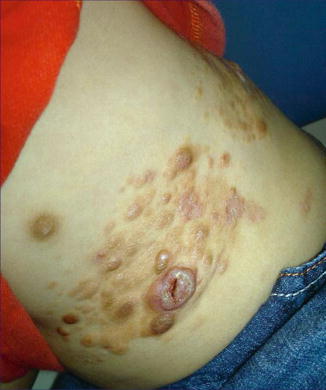
Fig. 12.26
Segmentally arranged juvenile xanthogranuloma [176] (Reprinted with permission from John Wiley & Sons, USA)
12.3.6 Linear Atrophoderma of Moulin
This acquired disorder consists of atrophic and hyperpigmented bands arranged along Blaschko’s lines (Fig. 12.27) [16, 166]. As a rule, no initial inflammatory stage is reported, and sclerotic skin changes are absent [208]. The disorder may reflect an early postzygotic mutation giving rise to mosaicism [48].
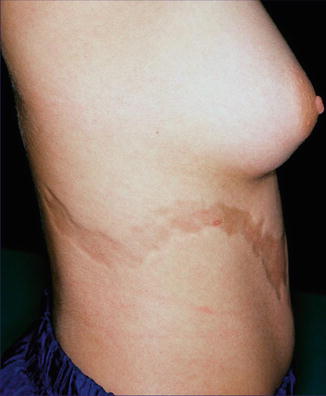
Fig. 12.27
Linear atrophoderma of Moulin [208] (Reprinted with permission from John Libbey Eurotext, Montrouge, France)
12.3.7 Superimposed Segmental Manifestation of Common Polygenic Skin Disorders
Numerous acquired skin disorders such as psoriasis or atopic dermatitis have a polygenic background. Such diseases sometimes show a linear or otherwise segmental arrangement that may either occur as an isolated manifestation or be superimposed on the ordinary nonsegmental phenotype [86, 88] (see Sect. 3.1.3). The following paragraphs deal with both types of mosaicism but shall focus on the superimposed segmental manifestation of polygenic skin disorders.
12.3.7.1 Acquired Inflammatory Disorders
In common inflammatory skin disorders, many examples of mosaic involvement have been reported.
Psoriasis Vulgaris
Linear psoriasis may occur as an isolated disorder [33, 38, 226]. The similarities between, and the distinguishing criteria of, linear psoriasis and ILVEN have fervently been debated [3, 73, 109, 202]. In my view, the two disorders can usually be distinguished by application of clinical and histochemical criteria [85].
Historical cases of superimposed linear psoriasis [90] have been reviewed elsewhere [86, 88]. More recently published reports (Fig. 12.28) [11, 41, 134, 217, 219] corroborate the rule that the superimposed linear lesions tend to appear much earlier than the nonsegmental psoriatic plaques (Table 12.1). A novel phenomenon is the unmasking of linear involvement by effective treatment of nonlinear psoriasis with biologicals [11, 41, 183, 219].
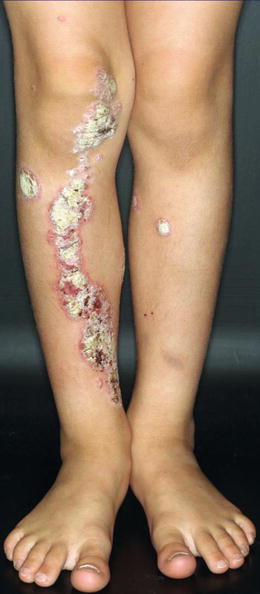
Fig. 12.28
Superimposed linear psoriasis in a 7-year-old boy. Note additional nonsegmental lesions on both legs [167]
Table 12.1
Recently published reports on superimposed linear psoriasis
Authors | Age | Age at onset of segmental lesions | Age at onset of nonsegmental lesions | Remarks |
|---|---|---|---|---|
Seitz et al. [217] | 19 | Birth | Adolescence | One grandfather had psoriasis vulgaris |
Linear lesions were resistant to topical treatment with steroids and dithranol | ||||
Sfia et al. [219] | 29 | ? | 12 years | Linear lesions were unmasked by treatment with infliximab |
Arnold et al. [11] | 50 | ? | Adolescence | Linear lesions were unmasked by treatment with adalimumab |
Kira and Katayama [134] | 18 | 3 years | 17 years | Linear lesion showed epidermal expression of involucrin, thus excluding “ILVEN” |
It was resistant to topical treatment with calcipotriol and clobetasol | ||||
Colombo et al. [41] | 67 | 65 | Prior to the onset of linear lesions (!) | Linear lesions were less responsive to treatment with etanercept |
Özdemir et al. [184] | 28 | 3 | Later in life | Linear lesions described as “ILVEN” were unresponsive to treatment with etanercept |
Özdemir et al. [183] | 42 | 7 | Later in life | Linear lesions described as “ILVEN” were less responsive to treatment with adalimumab |
Linear psoriasis is not arranged in a “zosteriform” pattern. It should be distinguished from true zosteriform psoriasis occurring as a Köbner phenomenon in a dermatome that had previously been involved by herpes zoster.
Psoriasis Pustulosa
There are four reports on linear psoriasis pustulosa being superimposed on the ordinary nonsegmental disorder (Fig. 12.29) [124, 151, 185, 211]. In three of the patients, the pustular psoriasis was preceded by plaques of psoriasis vulgaris. In one patient the linear lesions were unmasked by oral etretinate treatment [124]. Such cases indicate that generalized pustular psoriasis is not always inherited as a Mendelian trait caused by a homozygous IL36RN mutation [153].
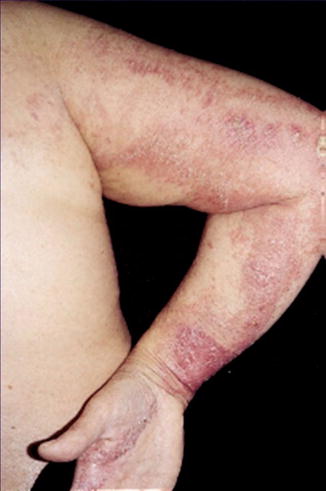
Fig. 12.29
Linear pattern of pustular psoriasis in a 57-year-old woman who had previously developed nonsegmental lesions of both psoriasis vulgaris and pustular psoriasis [185] (Reprinted with permission from Elsevier Limited, Oxford, UK)
Atopic Dermatitis
Some convincing examples of linear atopic dermatitis superimposed on nonlinear eczematous lesions have been documented (Fig. 12.30) [27, 86, 107, 234, 245]. These cases indicate that keratinocytic epitopes play a primary role in the pathogenesis of atopic dermatitis. In another report, an 18-year-old woman had itchy papules in a linear distribution down the inner aspect of her right leg [248]. Nonsegmental skin lesions were absent, but she had atopic features in the form of hay fever: positive prick tests to house dust mite, feathers, and grass; and raised IgE. Removal of split skin in an itchy area did not yield lasting relief, whereas excision of full thickness skin with subsequent grafting of the defect by split skin from an unaffected donor area resulted in permanent relief.
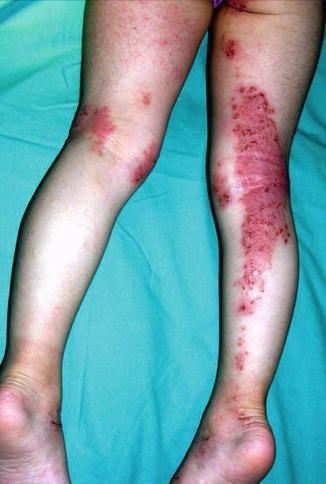
Fig. 12.30
Superimposed linear atopic dermatitis. Note nonlinear involvement of the left leg (Courtesy of Dr. Antonio Torrelo, Madrid, Spain)
Chronic Prurigo
Kawachi et al. [128] described a 65-year-old man with a 20-year history of chronic prurigo and a 3-year history of a pronounced linear pruritic lesion involving his left leg (Fig. 12.31). It is not clear whether the authors wanted to separate the disorder from atopic dermatitis, but by all means this case represents an impressive example of superimposed segmental manifestation of a chronic pruriginous disorder.
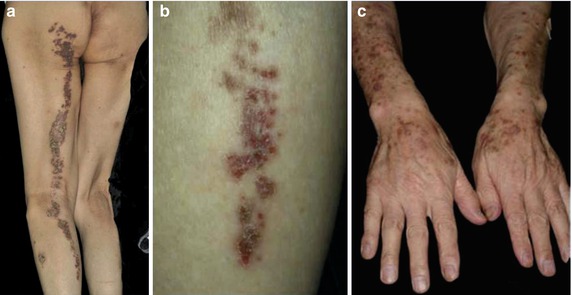
Fig. 12.31
Superimposed linear pruriginous dermatitis in a 65-year-old man. (a, b) Segmental involvement present for 3 years; (c) nonsegmental prurigo present for 20 years [128] (Reprinted with permission from John Libbey Eurotext, Montrouge, France)
Lichen Planus
Isolated forms of linear involvement are often described [28, 132, 145, 214, 218, 230, 249]. By contrast, cases of superimposed linear lichen planus (Fig. 12.32) are more rarely published [86]. Sometimes their arrangement is erroneously described as being “zosteriform” [195]. From the reports documenting the sequence of events, it is obvious that the linear lesions tend to appear earlier than the nonlinear ones (Table 12.2).
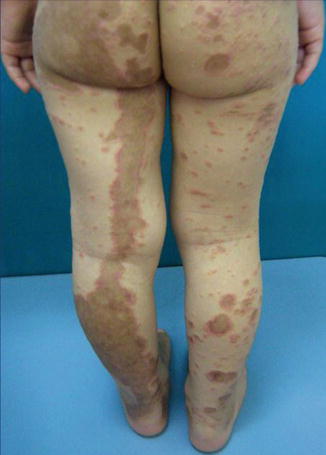
Fig. 12.32
Superimposed linear lichen planus in a 4-year-old girl [181] (Reprinted with permission from John Wiley & Sons, USA)
Table 12.2
Cases of superimposed linear lichen planus preceding the appearance of nonlinear lesions
Authors | Age at onset of linear lesions | Onset ofnonlinear lesions |
|---|---|---|
Davis [52] | 42 years | 45 years |
Irgang [115] | 46 years | 4 months later |
Nagy et al. [172] | 68 years | Some weeks later |
Fink-Pucheset al. [64] | 42 years | “Later” |
Gunning and Turiansky [78] | 54 years | 4 months latera |
Kanwar and De [125] | Childhood | “Subsequently” |
Case 1 | ||
Kanwar and De [125] | Childhood | “Subsequently” |
Case 2 | ||
Hamade et al. [80] | 5 years | Some weeks or months later |
Onder et al. [181] | 4 years | “During the following 3 months” |
In a report on 100 children with lichen planus, Kanwar and De [125] diagnosed a linear type in 12 patients. Subsequently two of these children developed, in addition, nonsegmental lesions. From other studies on lichen planus in childhood, one has the impression that the authors still follow the simple scheme of “classic” versus “linear” involvement, without accepting the possibility of “mixed” cases [81, 148, 173, 177, 220].
Lichen Planopilaris
An isolated mosaic manifestation is rather frequently reported. Cases of linear facial lesions predominate [70, 71, 138, 257], but other regions of the body may likewise be affected [13].
A superimposed linear involvement was described in a 29-year-old man who had itchy papules arranged in a linear distribution on the right side of his scalp, neck, and trunk (Fig. 12.33a–c) [35]. On his abdomen multiple, disseminated, nonsegmental lesions of lichen planopilaris giving rise to hairlessness were noted (Fig. 12.33d).
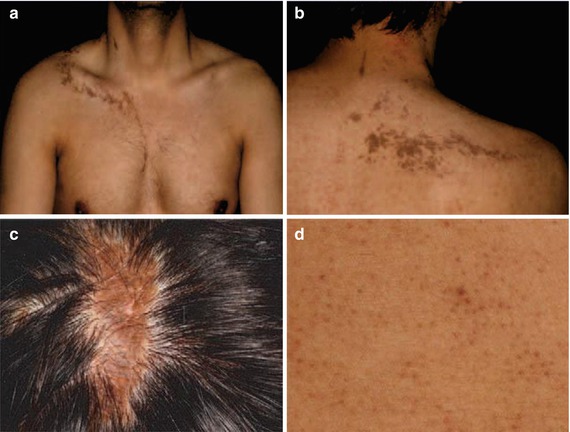
Fig. 12.33
Superimposed linear lichen planopilaris. (a, b) Linear lesions involving the trunk; (c) linear lesion on the scalp; (d) disseminated lesions on the abdomen [35] (Reprinted with permission from John Libbey Eurotext, Montrouge, France)
Lichen Nitidus
A few cases of isolated linear lichen nitidus have been documented [196, 199]. In another report, a 6-year-old boy had pronounced lesions of linear lichen nitidus on his left hand, giving rise to disturbed nail growth [23]. Moreover, the patient developed bilateral disseminated lesions of lichen nitidus (Fig. 12.34). Hence, this appears to be a classical example of superimposed linear lichen nitidus [87].
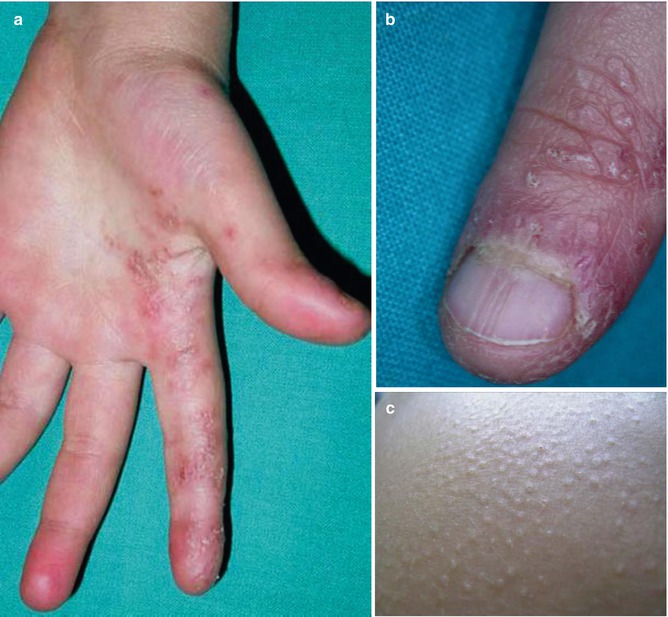
Fig. 12.34
Superimposed linear lichen nitidus. (a) Pronounced linear involvement of the left hand; (b) disturbed growth of left thumb nail; (c) disseminated papules on the abdomen [23] (Reprinted with permission from European Journal of Pediatric Dermatology, Bari, Italy)
Acne Vulgaris
The following case reports can today be categorized as examples of superimposed segmental manifestation of acne vulgaris. Hughes and Cunliffe [111, 112] described a 17-year-old boy who had, since 2 years, bilateral acne lesions involving his face, back, and chest. In addition, a patchy area of severe acne involved the left side of his chest (Fig. 12.35). This “acne nevus” was said to have been present since birth. Similarly, González-Hermosa et al. [72] described a 26-year-old man with a 9-year history of recalcitrant acne lesions involving the right half of the back, whereas only three individual lesions were found on the left side of his body. In a 26-year-old woman with “grade 2 acne on the left side and grade 4 on the right side,” Cooper et al. [42] found that the sebum excretion rate was higher on the right-hand side, and that conversion of dehydroepiandrosterone to androstenedione and 5α-steroids was greater in the biopsy from the right side.
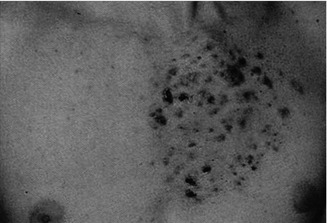
An interesting counterpart of superimposed segmental acne vulgaris was described in the form of an “acne-free nevus” [46]. A 17-year-old boy had mild facial acne and severe papulopustular acne on his back and chest. On the back, four well-demarcated patches being completely free of acne were noted. In these acne-free areas, the sebaceous glands were smaller and produced less sebum when compared to acne-bearing skin. Moreover, the percentage of squalene was lower in the acne-free patches.
Discoid Lupus Erythematosus
An isolated linear involvement is rather frequently reported in children [57, 75, 129, 203], adolescents [47], and adults [2, 4, 29, 67, 123, 216, 242]. The lesions are predominantly noted on the face. Sometimes antinuclear antibodies are found in the blood [1, 75, 212], which would raise the question of a “superimposed” manifestation. A convincing case of superimposed linear manifestation of the disorder (Fig. 12.36) was reported by Verma and Wollina [252].