(1)
Department of Dermatology, Freiburg University Medical Center, Freiburg, Germany
7.2 Pigmentary Nevi
7.2.1 Melanocytic Nevi
7.2.1.1 Common Small Melanocytic Nevus
7.2.1.4 Spitz Nevus
7.2.1.5 Cellular Blue Nevus
7.2.1.6 Papular Nevus Spilus
7.2.1.7 Macular Nevus Spilus
7.2.1.8 Linear Lentiginous Nevus
7.2.2.6 Phylloid Hypomelanosis
7.2.2.7 Phylloid Hypermelanosis
7.3 Epidermal Nevi
7.3.1 Keratinocytic Nevi
7.3.1.3 SASKIA Nevus (Segmentally Arranged Seborrheic Keratoses with Impending Atypia): A New Skin Disorder?
7.3.1.7 Epidermolytic Epidermal Nevus
7.3.1.9 CHILD Nevus
7.3.1.10 Nevus Corniculatus
7.3.1.11 Nevus Kerinokeratoticus
7.3.1.12 Other Keratinocytic Nevi
7.3.2 Organoid Epidermal Nevi
7.3.2.1 Nevus Sebaceus
7.3.2.2 Nevus Comedonicus
7.3.2.3 Linear Epidermolytic Comedones
7.3.2.8 Nevus Trichilemmocysticus
7.3.2.9 Acne Nevus of Munro
7.4 Vascular Nevi
7.4.1 Capillary Nevi
7.4.1.1 Nevus Flammeus
7.4.1.2 Nevus Roseus
7.4.1.6 Angiokeratoma Circumscriptum
7.4.1.8 Nevus Anemicus
7.4.1.9 Nevus Vascularis Mixtus
7.4.2 Venous Nevi
7.4.2.1 Large Venous Nevus
7.5.2 Linear Collagen Nevus
7.5.3 Elastin-Rich Nevus
7.6.2 Nevus Psiloliparus
Abstract
Mosaic skin disorders can be divided into three major groups: i) nevi; ii) nevoid skin disorders that do not fulfill the criteria of true nevi; and iii) benign or malignant neoplastic skin lesions. According to present knowledge, nevi are visible, circumscribed, long-lasting lesions of the skin or the neighboring mucosa, reflecting mosaicism. With the exception of melanocytic nevi, they do not show neoplastic growth. They never show malignant neoplasia. (Obviously, some nevi may show secondary malignant growth, but such tumors do no longer represent nevi.) Most nevi reflect the action of a lethal gene that can only survive in an admixture with wild-type cells. There are, however, many important exceptions from this rule, such as the nonlethal postzygotic mutations causing the epidermal nevus of the epidermolytic type. Pigmentary nevi: This group includes melanocytic nevi and other pigmentary lesions reflecting mosaicism. Common small melanocytic nevi represent a polygenic trait. Molecular analysis has revealed postzygotic allelic loss involving various genes such as BRAF, NRAS, BRCA1, MC1R or IRF4, thus corroborating the concept of mosaicism.– In “atypical” melanocytic nevi (formerly called “dysplastic nevi”) the number of such events of LOH is rather high. Spitz nevi appear to be genetically different because BRAF mutations are absent, whereas HRAS mutations are frequently noted. By way of exception, all of these small nevi may show a segmental arrangement. Large or giant melanocytic nevi may sometimes represent a superimposed mosaic manifestation of a polygenic trait consisting of multiple small melanocytic nevi. – Papular nevus spilus is an entity that can clearly be distinguished from macular nevus spilus. Both types are arranged in a checkerboard pattern. – The linear lentiginous nevus is characterized by complete absence of any hypo- or hyperpigmented background . – Nevus cesius (segmental dermal melanocytosis) has also been called nevus fuscucoeruleus, or “aberrant Mongolian spot” which is an obsolete name. – Linear hypermelanosis in broad bands is a characteristic feature of McCune-Albright syndrome. – The linear lesions of “hypomelanosis of Ito” are no nosological entity but merely a cutaneous marker of many different states of mosaicism. The same is true for so-called “linear and whorled nevoid hypermelanosis” and for phylloid hypermelanosis, whereas phylloid hypomelanosis represents a distinct entity reflecting a mosaic trisomy 13q. – A checkerboard arrangement of hyper- or hypomelanosis arranged has rarely been reported. Epidermal nevi can be divided into two large groups, keratinocytic nevi and organoid nevi. Most of the keratinocytic nevi have today been elucidated at the molecular level. Types of so far unknown etiology include inflammatory linear verrucous epidermal nevus, nevus corniculatus, nevus kerinokeratoticus, and the hystrix-lke epidermal nevus of NEVADA syndrome. Keratinocytic nevus syndromes with a known molecular basis include Proteus syndrome, CLOVES syndrome, type 2 segmental PTEN hamartoma syndrome, and CHILD syndrome. Organoid epidermal nevus syndromes with a known molecular cause comprise Schimmelpenning syndrome (including phacomatosis pigmentikeratotica) and the “porokeratotic eccrine nevus syndrome” representing a mosaic manifestation of KID syndrome. Organoid nevus syndromes of unknown etiology include angora hair nevus syndrome (Schauder syndrome), nevus comedonicus syndrome, Becker nevus syndrome, and Castori syndrome. The acne nevus of Munro reflects a postzygotic FGFR2 mutation and thus represents a mosaic manifestation of Apert syndrome. Vascular nevi: The frequently used name “capillary malformation” is an umbrella term being unsuitable to denote any of the different capillary nevi such as nevus flammeus, nevus roseus, rhodoid nevus, cutis marmorata telangiectatica congenita, livedo reticularis congenita, angiokeratoma circumscriptum, segmentally arranged angioma serpiginosum, and nevus anemicus. – Facial nevi flammei are most likely not arranged according to the trigeminal dermatomes. Sturge-Weber syndrome and Klippel-Trenaunay syndrome most likely represent one single entity that may be called Sturge-Weber-Klippel-Trenaunay syndrome. – Nevus roseus is a lateralized birthmark characterized by its pale-pink color. – The rhodoid nevus is a hallmark of the autosomal dominant trait, rhodoid nevus syndrome (including both “capillary malformation-arteriovenous malformation” and “capillary malformation without arteriovenous malformation” (OMIM 608354), being caused by RASA1 mutations. Rhodoid nevi are lighter than nevus flammeus, but darker than nevus roseus. They are often surrounded by an anemic halo. – Cutis marmorata telangiectatica congenita (including van Lohuizen syndrome) should be distinguished from livedo reticularis congenita (including “macrocephaly-capillary malformation syndrome”). – The venous nevi represent a group of different disorders. Large venous nevi occur sporadically and are arranged in a segmental pattern with a strict midline separation. Small venous nevi occur as an autosomal dominant trait being caused by TEK mutations. The venous nevus of the Servelle-Martorell type represents a segmentally arranged, huge phlebectasia with hyperplasia of the involved limb. Connective tissue nevi are often noted in patients with tuberous sclerosis in the form of large collagen nevi that are described under various names such as shagreen patch, forehead patch, cobblestone nevus or fibrous hamartoma of infancy. All of these lesions represent a type 2 segmental manifestation of the disorder. – The linear collagen nevus that has also been described under the name “papulolinear collagenoma” follows the system of Blaschko’s lines. – Multiple, disseminated, small elastin-rich nevi are a characteristic feature of Buschke-Ollendorff syndrome, whereas large segmental lesions (“juvenile elastoma”) represent a type 2 mosaic involvement of the disorder. – Within the group of autosomal dominant Ehlers-Danlos syndromes, examples of both type 1 and type 2 segmental manifestation have been documented. Fatty tissue nevi: Two different types of such nevi can be distinguished. Nevus lipomatosus superficialis is a well known disorder that tends to be arranged on the trunk in a linear pattern. Involvement of the scalp is extremely rare. By contrast, nevus psiloliparus is a less well known skin lesion consisting of a flat and hairless lesion involving the scalp. In the past it has often been confused with nevus sebaceus. Nevus psiloliparus is a hallmark of encephalocraniocutaneous lipomatosis, a sporadically occurring multisystem birth defect. Its molecular basis is so far unknown but the hypothesis of a lethal mutation surviving by mosaicism will most likely be corroborated in the near future.
The concept of mosaicism has helped develop a reasonable definition of the term nevus. In 1995 the following criteria were proposed: “Nevi are visible, circumscribed, long-lasting lesions of the skin or the neighboring mucosa, reflecting mosaicism. With the exception of melanocytic nevi, they do not show neoplastic growth. They never show malignant neoplasia” [82]. Recent molecular research has shown that this is a workable definition.
The statement that nevi “never show malignant neoplasia” needs a clarifying comment. Of course, some nevi may show secondary malignant growth, but these superimposed tumors do no longer represent nevi.
The new criteria were developed from an earlier tentative definition as proposed by Hermann Pinkus in 1965: “The definition of the two forms of nevi can approximately be given in the following way: (1) The nevus cell nevus, or cellular nevus, is a benign neoplasia of the skin, consisting of specific tumor cells, the nevus cells. (2) Otherwise we can designate as nevi all those circumscribed malformations of the skin that are characterized by a surplus (or, occasionally, a deficit) of one or several mature tissue components, and are relatively stable” [181]. A disadvantage of this definition was that it could be applied to all developmental anomalies of the skin, including supernumerary nipples, rudimentary polydactyly, preauricular tags, and palmoplantar keratoderma. For obvious reasons, such lesions do not represent nevi, which is why they are excluded by the new definition.
7.1 The Theory of Lethal Genes Surviving by Mosaicism
Most nevi reflect the action of a lethal gene that can only survive in proximity to a population of wild-type cells [75]. There are, however, many important exceptions from this rule such as the epidermolytic type of epidermal nevus being caused by nonlethal postzygotic KRT1 or KRT10 mutations (see Sect.7.3.1.7).
7.2 Pigmentary Nevi
This group comprises melanocytic nevi and other nevi reflecting pigmentary mosaicism.
7.2.1 Melanocytic Nevi
Melanocytic nevi represent the only type of nevi showing neoplastic proliferation [181]. They may be present at birth or develop throughout life.
7.2.1.1 Common Small Melanocytic Nevus
Both congenital and acquired small melanocytic nevi have a polygenic background, which means that they are not inherited as a Mendelian trait. In common acquired melanocytic nevi, events of LOH involving various gene loci including BRAF, NRAS, BRCA1, MC1R, and IRF4 were documented [174, 219], providing evidence that these skin lesions reflect mosaicism [157, 194, 217].
7.2.1.2 Common Atypical Melanocytic Nevus
In atypical nevi (“dysplastic nevi”), the number of LOH events appears to be even higher [127, 128, 219]. In the past, a so-called dysplastic nevus syndrome was described and said to be inherited as a Mendelian trait [48]. Other authors, however, argued that dysplastic nevi are a very common polygenic trait and do not constitute a distinct hereditary “syndrome” [78, 119, 218]. Recent molecular research has confirmed this alternative view.
Sometimes the atypical melanocytic nevi may occur in a segmental distribution, representing a particular form of mosaicism [206].
7.2.1.3 Large Congenital Melanocytic Nevus
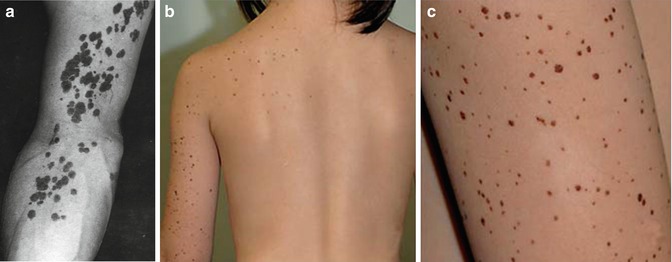
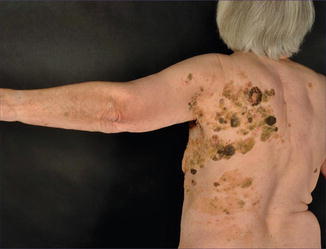
Large congenital melanocytic nevi including giant lesions usually occur sporadically. The assumption of a very early postzygotic mutation, surviving by mosaicism [77], was supported by several reports on nevus cell aggregates involving the placenta of mothers giving birth to a child with a giant melanocytic nevus [10]. In 2013, Kinsler et al. [136] found that most cases of multiple congenital nevi and neuromelanosis are caused by heterozygosity for a postzygotic NRAS codon 61 mutation originating from one single progenitor cell. If these results can be confirmed in larger series, the occurrence of extensive or giant congenital melanocytic nevi may be categorized as a monogenic disorder.
As a consequence, the hypothesis that large congenital melanocytic nevi may be explained as a superimposed mosaic manifestation of a polygenic trait [100] may today either be taken as refuted or it may only be valid for a rather small proportion of patients. In this minority of cases, a polygenic background would explain why the affected individuals often show, in addition, multiple small nevi scattered over their entire body (Fig. 7.2) [57, 192]. At an early developmental stage, LOH would occur at one of the predisposing gene loci, or a postzygotic new mutation would cause heterozygosity at an additional predisposing locus. Future research may show whether this etiological concept can be upheld at least for some cases of giant melanocytic nevi. It would be compatible with the description of several major genes causing large or giant melanocytic nevi, such as SOX10 [201] or MC1R [135].
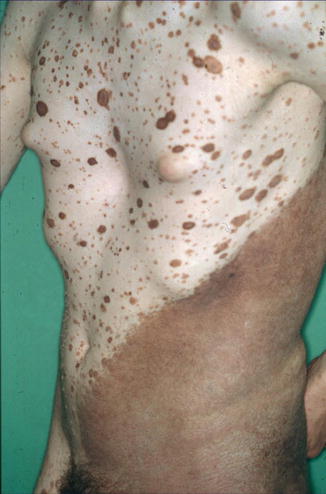
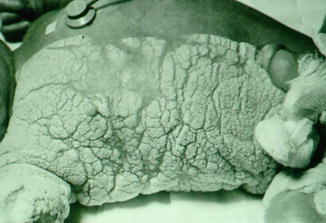
Fig. 7.2
Giant melanocytic nevus associated with multiple small melanocytic nevi, being partly congenital and partly acquired (Courtesy of Dr. Uwe Töllner, Fulda, Germany). Perhaps, the disseminated lesions are not “satellite nevi” but reflect a pronounced polygenic predisposition. According to this theory, the giant nevus would originate from an additional postzygotic mutation that occurred at an early developmental stage
An unusual linear arrangement of large melanocytic nevi, being superimposed on common acquired small nevi, has also been reported [43].
7.2.1.4 Spitz Nevus
Spitz nevi do apparently not develop from BRAF mutations, whereas HRAS mutations are often found in these melanocytic lesions [193, 220]. Such molecular findings may be related to the fact that Spitz nevi do usually not develop malignant growth.
Spitz nevi per se may be agminated but do not show a segmental arrangement. However, both macular and papular nevi spili are sometimes found to be covered with multiple Spitz nevi (see Sects. 7.2.1.6 and 7.2.1.7). Moreover, Menni et al. [167] have reported a case of flag-like hypomelanosis covered with Spitz nevi (Fig. 7.3).
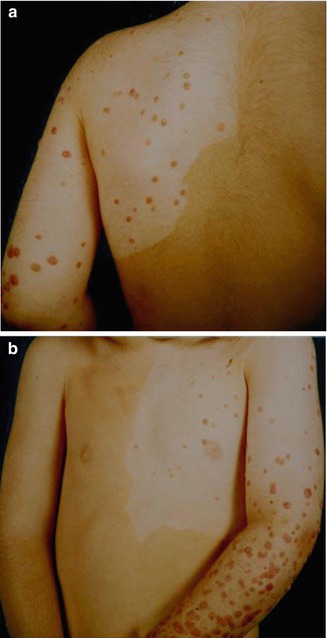
Fig. 7.3
(a, b) Unilateral flag-like nevus achromicus covered with multiple congenital Spitz nevi [167] (Reprinted with permission from John Wiley & Sons, USA)
7.2.1.5 Cellular Blue Nevus
7.2.1.6 Papular Nevus Spilus
Papular nevus spilus (speckled lentiginous nevus of the papular type) represents a light-brown macule containing multiple speckles in the form of dark brown papules, in addition to small macules (Fig. 7.4). These speckles are distributed in an uneven way, reminiscent of a star map. On histopathological examination, the papules are found to be melanocytic nevi of a dermal or compound type [97]. This nevus is caused by a postzygotic HRAS mutation [64].
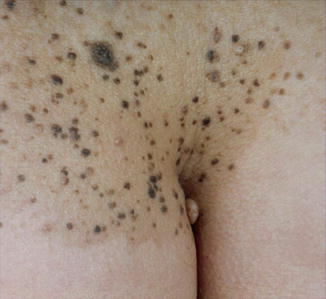
Fig. 7.4
Papular nevus spilus with speckles showing a characteristic star-map pattern
Papular nevus spilus syndrome: Large papular nevi spili are arranged in a flag-like pattern and may be associated with neurologic defects that are usually localized ipsilaterally and include hyperhidrosis, muscular weakness, dysesthesia, or thinning of a nerve [88, 184]. In the past this phenotype has been called “speckled lentiginous nevus syndrome,” but this term is ambiguous because two different types of SLN syndromes exist. The designation “papular nevus spilus syndrome” seems more appropriate. The disorder may also occur as a component of phacomatosis pigmentokeratotica that represents a variant Schimmelpenning syndrome (see Sect. 9.1).
7.2.1.7 Macular Nevus Spilus
This disorder is characterized by a light-brown macule containing multiple, small, completely flat, dark speckles that are rather evenly distributed, resembling a polka-dot pattern (Fig. 7.5) [97, 221]. Histopathologically, increased numbers of melanocytes are confined to the elongated rete ridges. At the tips of these structures, the melanocytes tend to form nests at the dermoepidermal junction, giving rise to a so-called “jentigo” pattern [158].
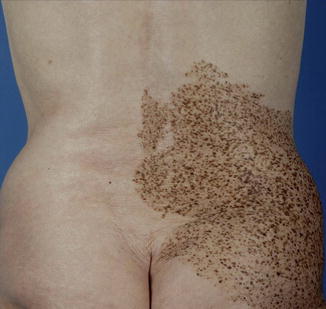
Fig. 7.5
Macular nevus spilus with speckles showing a characteristic polka-dot distribution (Courtesy of Dr. Alexander Stella, Vienna, Austria)
Macular nevus spilus sometimes occurs in combination with nevus roseus, giving rise to a peculiar disorder called “phacomatosis spilorosea” (see Sect. 9.5.2).
7.2.1.8 Linear Lentiginous Nevus
This unusual birthmark is characterized by multiple, tightly packed lentigines arranged along Blaschko’s lines. As a distinguishing feature, these speckles do not show any abnormal background in the form of hyper- or hypopigmentation (Fig. 7.6) [114].
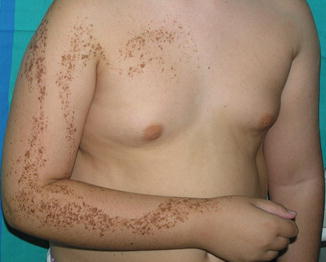
Fig. 7.6
Naevus lentiginosus linearis [114] (Reprinted with permission from the Society for Publication of Acta Dermato-Venereologica, Stockholm, Sweden)
7.2.1.9 Nevus Cesius (Segmental Dermal Melanocytosis)
This lesion (Fig. 7.7) is also called nevus fuscocoeruleus or aberrant Mongolian spot [137]. It should not be confused with the median lumbosacral blue spots being present in many neonates of indigenous populations of many parts of the world. The localization of nevus cesius in the ophthalmo-maxillary region has been called “nevus of Ota,” and in the shoulder region some authors use the name “nevus of Ito.”
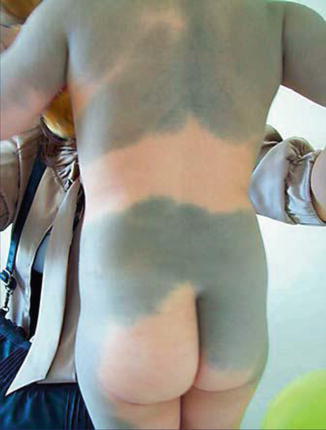
Fig. 7.7
Systematized nevus cesius (segmental dermal melanocytosis) [137] (Reprinted with permission from S. Karger AG, Basel, Switzerland)
Nevus cesius may be part of a syndrome called “phacomatosis cesioflammea” (see Sect. 9.5.1). At least some of these cases may constitute a binary genodermatosis in the form of nevus cesius coexisting with nevus flammeus. So far, however, it is not clear how many of these cases simply represent an arbitrary coincidence, especially in Latin American or Asian populations.
7.2.2 Other Nevi Reflecting Pigmentary Mosaicism
The following nevi are not characterized by abnormal numbers of melanocytes, but simply by increased or decreased amounts of melanin.
7.2.2.1 Linear Hypomelanosis in Narrow Bands (Pigmentary Mosaicism of the Ito Type)
These disorders are characterized by depigmented skin lesions following Blaschko’s lines (Fig. 7.8). If the lesions are not systematized, they are often described under the names nevus achromicus or nevus depigmentosus. The term “hypomelanosis if Ito” [38] should be avoided because it fosters the erroneous belief that there is a nosological entity with this name. In fact, linear hypomelanosis is an umbrella term that includes many different forms of cellular mosaicism that may or may not be discernible at the cytogenetic level [142].
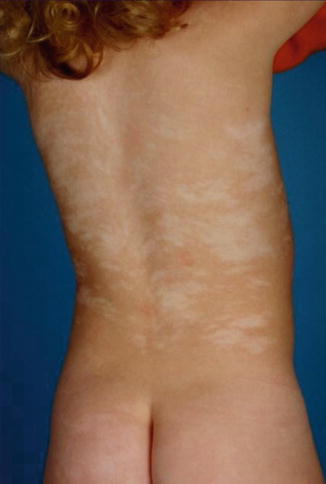
Fig. 7.8
Pigmentary mosaicism of the Ito type
Linear hypomelanosis is sometimes associated with neurologic or other extracutaneous defects, but again it should be borne in mind that such cases do not represent one distinct phenotype but a group of numerous different mosaic states. When percentages of the frequency of associated extracutaneous anomalies as noted in “hypomelanosis of Ito” are presented, this simply reflects absence of genetic thinking.
Sometimes it may be rather difficult or even impossible to determine whether the light or the dark component is the pathological one in a given case of linear pigmentary mosaicism. A conspicuous example was reported by Thapa et al. [213] (Fig. 7.9).
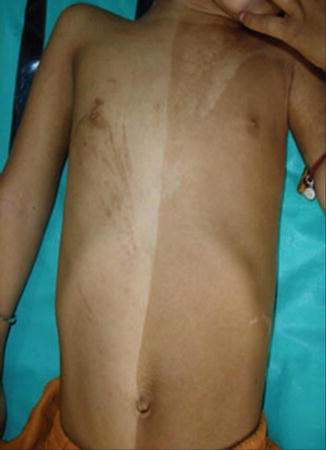
Fig. 7.9
Is this a linear hypo- or hypermelanosis? Sometimes it may be difficult to say which component is the abnormal one, but in this Indian boy it is certainly the light component [213] (Reprinted with permission from John Wiley & Sons, USA)
7.2.2.2 Linear Hypermelanosis in Narrow Bands
This is an umbrella term including many different states of pigmentary mosaicism that may or may not be associated with extracutaneous anomalies [5]. The disorder (Fig. 7.10) has often been called “linear and whorled nevoid hypermelanosis” [132, 162], but again it is important to realize that this term does not describe any distinct nosological entity.
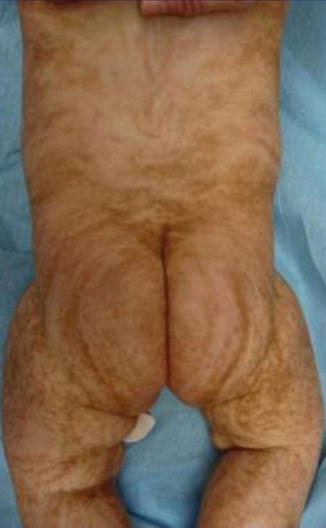
Fig. 7.10
Linear hypermelanosis in narrow bands [162] (Reprinted with permission from Elsevier Limited, UK)
7.2.2.3 Linear Hypermelanosis in Broad Bands
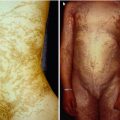
The McCune-Albright syndrome is characterized by very broad bands of hypermelanosis (Fig. 7.11), in combination with fibrous dysplasia of bones and endocrinological abnormalities [222]. When many cases are compared, it becomes obvious that these café-au-lait macules are arranged along Blaschko’s lines [81, 187]. These bands reflect the clonal outgrowth of cells carrying a postzygotic GNAS1 mutation [200, 222].
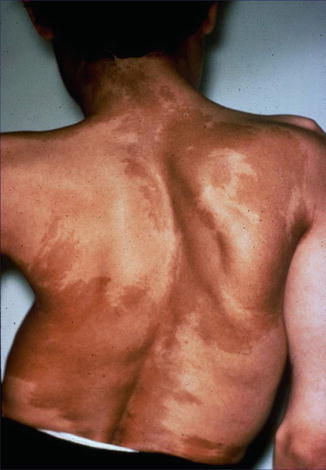
Fig. 7.11
McCune-Albright syndrome characterized by linear hypermelanosis in broad bands (Courtesy of the late Dr. Robert J. Gorlin, Minneapolis, Minnesota, USA)
In the past century it was a favorite point of controversy among pediatricians and endocrinologists whether the various hormonal disturbances as noted in patients with McCune-Albright syndrome were of “central” or “peripheral” origin. The concept of randomly distributed mosaicism has rendered this discussion pointless. For obvious reasons both mechanisms are possible [76].
7.2.2.4 Segmental Hypomelanosis Arranged in a Checkerboard Pattern
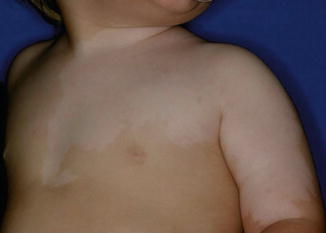
7.2.2.5 Segmental Hypermelanosis Arranged in a Checkerboard Pattern
Hyperpigmentation arranged in a checkerboard pattern likewise occurs rather rarely (Fig. 7.13). It was reported by Stoll et al. [207] in a boy with developmental delay, facial dysmorphism, and large, low-set ears. Cytogenetic analysis of lesional skin fibroblasts revealed a 12q;14q translocation present in 70 % of cells, whereas no chromosomal abnormality was found in peripheral blood lymphocytes.
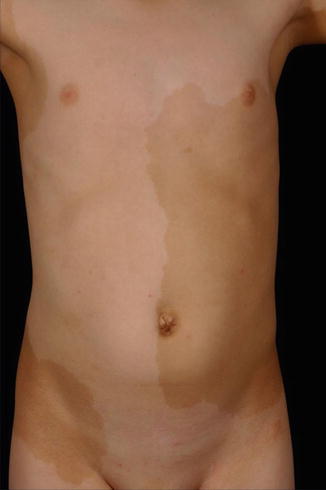
Fig. 7.13
Segmental hypermelanosis arranged in a checkerboard pattern (Courtesy of Dr. Regina Fölster-Holst, Kiel, Germany)
7.2.2.6 Phylloid Hypomelanosis
The pattern of this pigmentary disorder (Fig. 7.14) reflects mosaicism but cannot be described as “segmental.” Most cases of phylloid hypomelanosis are caused by mosaic trisomy 13q [39, 61, 85] (see Sect. 5.3). Hence, this phenotype cannot simply be taken as a mosaic manifestation of complete trisomy 13 (Patau syndrome) [131], although it is closely related to this severe form of numeric chromosomal aberration.
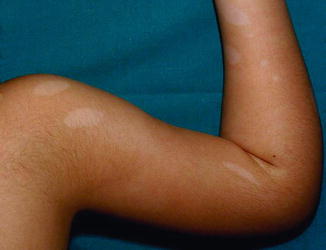
Fig. 7.14
Phylloid hypomelanosis (Courtesy of Dr. Antonia González-Enseñat, Barcelona, Spain)
7.2.2.7 Phylloid Hypermelanosis
In 1993, the term “phylloid pattern” was proposed in an article describing an arrangement of leaf-like hyperpigmented macules reminiscent of floral ornaments in a 12-year-old girl who had an arteria subclavia originating from the aorta descendens [81]. Phylloid hypermelanosis (Fig. 7.15) can be taken as a hyperpigmented counterpart of phylloid hypomelanosis. Contrasting with the hypopigmented phenotype, however, it represents a class of heterogeneous disorders [101]. Oiso et al. [175] reported a case of phylloid hypermelanosis and mental deficiency. In this patient, analysis of blood lymphocytes showed three different cell types containing either a dicentric chromosome 13, or monosomy 13, or a ring chromosome (13)(p11.2q34). Remarkably, no normal karyotype was found in 30 cells examined. Already in 1956, Dockx et al. [41] had documented a similar pattern of hypermelanotic macules in a girl with mental deficiency and abnormalities of the bones and joints (Fig. 7.16). Additional cases have recently been reported [23, 110]. Another example of phylloid hypermelanosis has been mistaken for “linear and whorled nevoid hypermelanosis” [150].
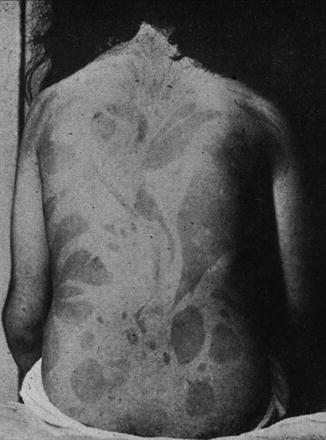
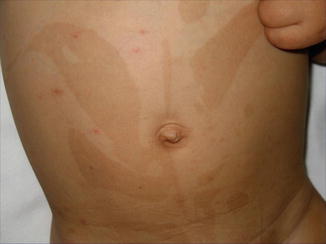
Fig. 7.16
A historical case of phylloid hypermelanosis described by Dockx et al. in 1956 [41] (Reprinted with permission from Elsevier Masson SAS, France)
7.3 Epidermal Nevi
There are two large categories of epidermal nevi. The keratinocytic nevi are “true” epidermal nevi because they are exclusively characterized by epidermal changes, whereas in the group of organoid nevi the essential changes involve the adnexal structures of the skin. In principle, the designation “epithelial nevi” would be a better generic term to denote both categories, but the name “epidermal nevi” has firmly been entre-nched in our literature and will be maintained in the following paragraphs.
7.3.1 Keratinocytic Nevi
Most keratinocytic nevi follow the lines of Blaschko. An exception from this rule is the CHILD nevus that is characterized by a peculiar lateralization pattern but may also show an arrangement along Blaschko’s lines.
The different epidermal nevus syndromes characterized by particular keratinocytic nevi are listed in Table 7.1.
Table 7.1
Epidermal nevus syndromes characterized by keratinocytic nevi
Syndrome | Type of epidermal nevus | Pattern of epidermal nevus | Discriminating clinical criteria | Formal genetic classification | Molecular basis |
|---|---|---|---|---|---|
Proteus syndrome | Linear Proteus nevus (nonepidermolytic keratinocytic nevus of a soft, flat type) | Blaschko lines | Cerebriform connective tissue nevi of palms or soles; asymmetric macrodactyly | Not heritable | Mosaic activating |
AKT1 mutation | |||||
CLOVES syndrome | Nonepidermolytic keratinocytic nevus | Blaschko lines | Disproportionate overgrowth of fatty tissue, truncal vascular malformations, nonprogressive overgrowth of toes (“ballooning”), “wrinkling” of palms or soles, cerebral defects | Not heritable | Mosaic PIK3CA mutations |
Type 2 segmental PTEN hamartoma syndrome (including type 2 segmental Cowden disease) | Linear PTEN nevus (nonepidermolytic keratinocytic nevus of a soft and rather thick, papillomatous type) | Blaschko lines | Macrocephaly, overgrowth of a limb, colon polyps, focal segmental glomerulosclerosis; family members with nonsegmental PTEN hamartoma syndrome | Type 2 segmental manifestation of PTEN hamartoma syndrome | PTEN germline mutation, with loss of the wild-type allele in the involved segment |
FGFR3 epidermal nevus syndrome (García-Hafner-Happle syndrome) | Nonepidermolytic keratinocytic nevus of a soft type | Blaschko lines | Presence of keratinocytic nevus of the common type; absence of disproportionate overgrowth | Not heritable | Mosaic FGFR3 mutation |
CHILD syndrome | CHILD nevus | Two patterns in the form of lateralization and Blaschko lines | Lateralized, inflammatory skin lesions; ptychotropism; ipsilateral limb defects | X-linked dominant inheritance with lethality in male embryos | NSDHL mutations |
NEVADA syndrome (nevus epidermicus with angiodysplasia and aneurysms) | Nonepidermolytic, hystrix-like epidermal nevus | Blaschko lines or lateralization pattern | Dysplasia of large vessels, arteriovenous shunts; dysplasia of retinal vessels | Not heritable | Unknown |
7.3.1.1 Common Keratinocytic Nevi of the Soft Type, Including Seborrheic Keratoses
These nevi have a soft, velvety surface and are usually of gray or brown color. The earlier the postzygotic mutation has occurred during embryonic life, the more widespread the involvement will be. Some lesions are so small that a linear arrangement is barely discernible. Some form one single band, whereas others show a systematized pattern involving many parts of the body (Fig. 7.17). When the postzygotic mutation occurs later in life, the resulting keratinocytic nevus is small and nonlinear and called, by tradition, “seborrheic keratosis.”
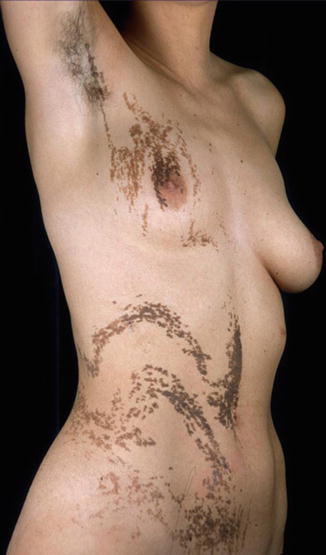
Fig. 7.17
Systematized keratinocytic nevus of the common, soft type
In about one third of such cases, mosaicism has been proven at the cellular level by documenting a heterozygous R248C hotspot mutation in the FGFR3 gene within the nevus [71]. A different FGFR3 mutation has likewise been documented [177]. In other cases such nevi are caused by postzygotic activating PIK3CA mutations [72]. Both FGFR3 and PIK3CA mutations are likewise found in seborrheic keratoses [66, 68, 69]. Moreover, many keratinocytic nevi are associated with postzygotic HRAS or, more rarely, KRAS mutations [21, 69, 70].
Seborrheic Keratoses are Acquired Keratinocytic Nevi
Seborrheic keratoses (Fig. 7.18) have recently turned out to be acquired epidermal nevi. Histopathologically, they very closely resemble linear epidermal nevi of the common type. The spectrum of postzygotic mutations as found in seborrheic keratoses corresponds to that observed in linear keratinocytic nevi of the nonepidermolytic type [66, 68, 72]. In particular, R248C mutations of the FGFR3 gene as well as activating PIK3CA mutations are frequently found. The R248C FGFR3 mutation, when present in the germline, causes thanatophoric dysplasia [71]. This allele can, therefore, be categorized as a lethal mutation surviving by mosaicism.
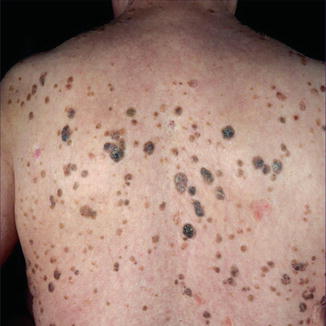
Fig. 7.18
Seborrheic keratoses can today be categorized as acquired keratinocytic nevi of the common type
Activating PIK3CA mutations are present in a broad spectrum of malignant tumors [68]. Paradoxically, however, epidermal nevi harboring such mutations do usually not show any proclivity to develop malignant growth.
An Early Postzygotic FGFR3 Mutation May Cause a Distinct Neurocutaneous Syndrome
In a 5-year-old girl with severe structural CNS defects causing mental deficiency and epileptic seizures, García et al. [54] described a systematized, soft, velvety epidermal nevus of the common, nonorganoid, nonepidermolytic type (Fig. 7.19). A mosaic R248C mutation of the FGFR3 gene was found in the blood and within the nevus, whereas the normal skin contained the wild-type allele only. This disorder, for which the name “FGFR3 epidermal nevus syndrome” was proposed [102], can be categorized as a mosaic manifestation of thanatophoric dysplasia. A similar patient was found to be affected with a different FGFR3 mutation [177]. A case of FGFR3 epidermal nevus associated with mild facial dysmorphism [30] can be taken as an oligosymptomatic example of the same syndrome.
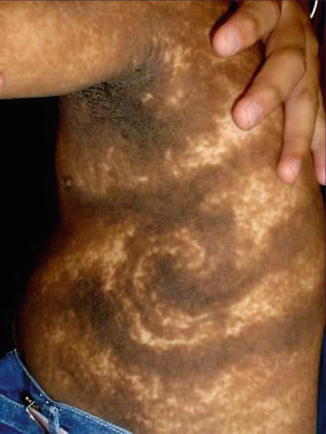
Fig. 7.19
Systematized keratinocytic nevus in a girl with severe cerebral defects (FGFR3 epidermal nevus syndrome) [54] (Courtesy of Dr. Alejandro García Vargas, Guadalajara, Mexico)
Early Postzygotic PIK3CA Mutations Cause CLOVES Syndrome
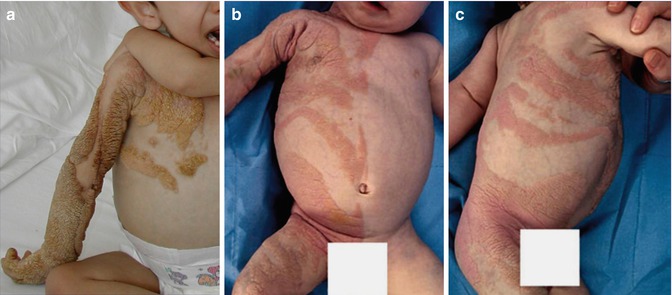
The term CLOVE was proposed as an acronym for congenital lipomatous overgrowth with vascular and epidermal anomalies [198]. In 2009 the name was expanded to CLOVES syndrome to denote the additional presence of skeletal anomalies [7]. The associated keratinocytic nevus is of a soft, velvety type and may sometimes show a rather broad segmental involvement (Fig. 7.20). In the past the phenotype was confused with Proteus syndrome, but it represents a distinct nosological entity being caused by postzygotic activating mutations in the PIK3CA gene [140].
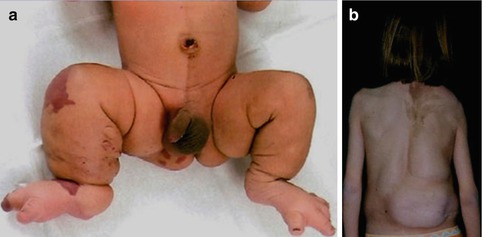
Fig. 7.20
CLOVES syndrome. (a) Overgrowth of right leg, vascular nevi, contralateral epidermal nevus, and wide feet with broad first interdigital space [120] (Reprinted with permission from John Wiley & Sons, USA); (b) 14-year-old girl with large subcutaneous lipomas and keratinocytic nevus (Courtesy of Dr. Sigrid Tinschert, Berlin, Germany)
7.3.1.2 Common Keratinocytic Nevi of the Hard, Verrucous Type
7.3.1.3 SASKIA Nevus (Segmentally Arranged Seborrheic Keratoses with Impending Atypia): A New Skin Disorder?
Multiple seborrheic keratoses arranged along Blaschko’s lines have been suggested to reflect genetic mosaicism [155]. Livingstone et al. [149] described a 76-year-old woman who had since birth multiple seborrheic keratosis-like lesions arranged in a systematized linear pattern on the left side of her body (Fig. 7.21). Some years ago, a squamous cell carcinoma had developed within one of these lesions. Subsequently, carcinomata in situ in association with seborrheic keratosis-like structures were found in four out of seven biopsies obtained from rather hyperkeratotic and erythematous lesions. An FGFR3 mutation was present in three of the lesions, and an additional PIK3CA mutation was found in one of them. No mutation could be documented in specimens from the squamous cell carcinoma. The authors raised the question whether this unusual nevoid disorder may represent a new nosological entity.
7.3.1.4 Linear PTEN Nevus (Linear Cowden Nevus Included)
7.3.1.5 Epidermal Nevus of the Proteus Type
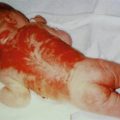
Proteus syndrome is characterized by disproportionate and progressive overgrowth of various tissues including asymmetrical macrodactyly, cerebriform connective tissue nevi of the soles, lipomas, cystic lymphangiomas, and vascular nevi (Fig. 7.23a, b) [15, 102]. The frequently associated epidermal nevus is a soft and rather flat linear lesion (Fig. 7.23c) [94, 102]. The underlying mutation involves the AKT1 gene [148]. Remarkably, the mutant gene cannot be found in blood DNA because its lethal action eliminates all lymphocytes carrying the mutation.
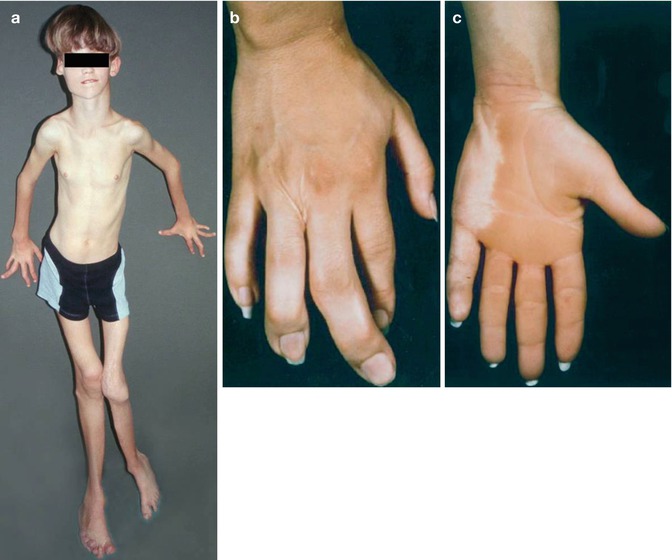
Fig. 7.23
Proteus syndrome (a) A 9-year-old boy who had reached a height of 186 cm (Courtesy of Dr. Sigrid Tinschert, Berlin, Germany) (b) Macrodactyly of the right hand and (c) flat, soft epidermal nevus of the left forearm and hand in a 24-year-old woman [28] (Reprinted with permission from John Wiley & Sons, USA)
7.3.1.6 Hystrix-Like Epidermal Nevus of NEVADA Syndrome
A peculiar keratinocytic nevus characterized by a thick, hystrix-like white or brownish hyperkeratosis has been described in patients with multiple vascular malformations (Fig. 7.24) [160]. The acronym NEVADA stands for “nevus epidermicus verrucosus with angiodysplasia and aneurysms” [103]. It is likely, albeit unproven, that this nevus represents a distinct entity.
7.3.1.7 Epidermolytic Epidermal Nevus
This nevus (Fig. 7.25) represents a mosaic manifestation of epidermolytic ichthyosis of Brocq [178]. Affected individuals run an increased risk to give birth to a child with the diffuse form of this disorder (see Sect. 3.1.1.3).