Neurofibromatosis, type 1
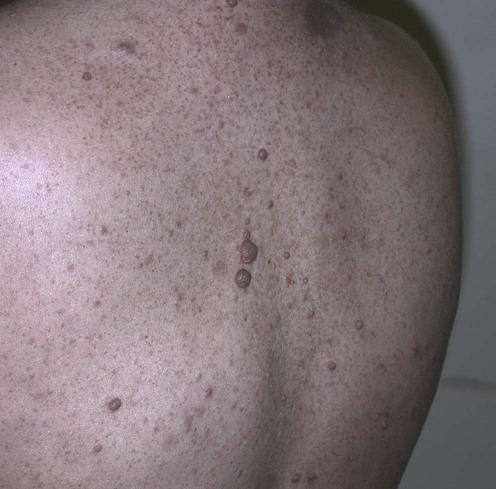
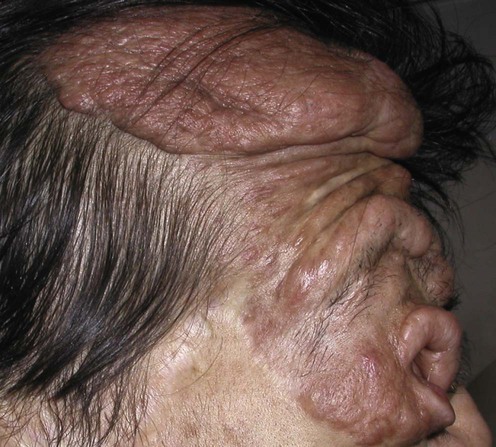
Specific investigations
 Annual complete cutaneous examinations, particularly in those patients with large plexiform lesions
Annual complete cutaneous examinations, particularly in those patients with large plexiform lesions
 Skin and eye exams for first-degree relatives
Skin and eye exams for first-degree relatives
 Regular blood pressure checks to screen for renal artery stenosis or pheochromocytoma
Regular blood pressure checks to screen for renal artery stenosis or pheochromocytoma
 MRI or PET scan, with and without contrast, for deep or changing plexiform neurofibromas
MRI or PET scan, with and without contrast, for deep or changing plexiform neurofibromas
 Biopsy or excision of changing or suspicious lesions
Biopsy or excision of changing or suspicious lesions
 Radiographs if osseous involvement is suspected
Radiographs if osseous involvement is suspected
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Neurofibromatosis, type 1




