Introduction868
INTRODUCTION
NERVE SHEATH TUMORS
NEUROMAS
Traumatic neuroma
Histopathology13
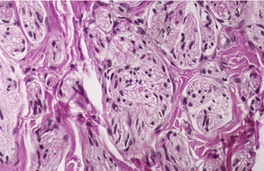
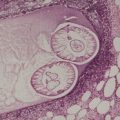
Fig. 37.1
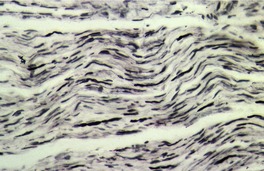
Fig. 37.2
Rudimentary polydactyly
Histopathology
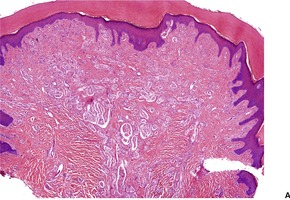
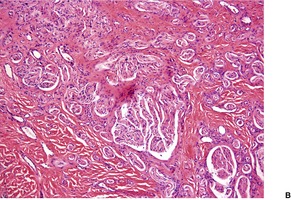
Fig. 37.3
Electron microscopy
Solitary circumscribed neuroma
Histopathology11
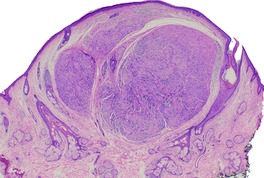
Fig. 37.4
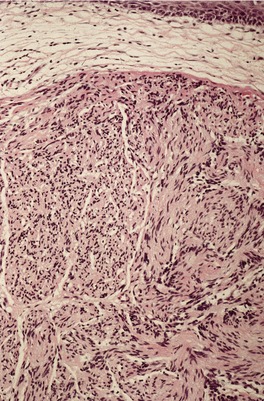
Fig. 37.5
Neuromas and the multiple endocrine neoplasia syndrome
Histopathology
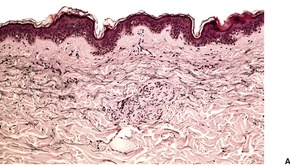
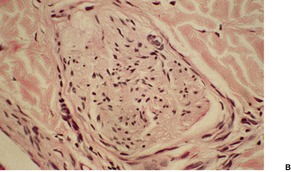
Fig. 37.6
Ganglioneuroma
Histopathology
EPITHELIAL SHEATH NEUROMA
Histopathology
RE-EXCISION PERINEURAL INVASION
PERINEURIOMA
Histopathology
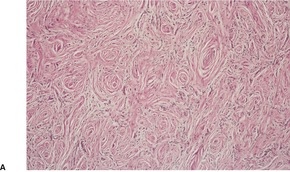
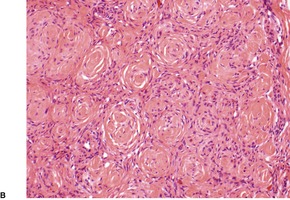
Fig. 37.7
Electron microscopy
SCHWANNOMA (NEURILEMMOMA)
Histopathology122
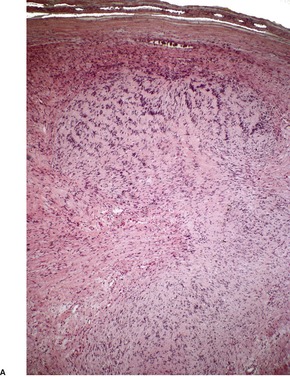
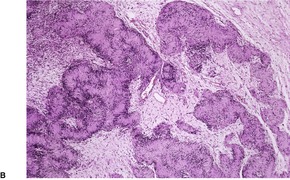
Fig. 37.8
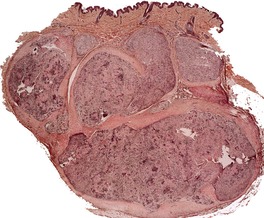
Fig. 37.9
PSAMMOMATOUS MELANOTIC SCHWANNOMA
Histopathology
NEUROFIBROMA AND NEUROFIBROMATOSIS
Classic neurofibromatosis (NF-1)
Segmental neurofibromatosis (NF-5)
Histopathology
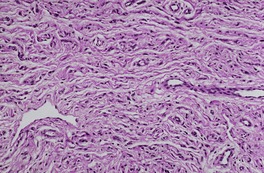
Fig. 37.10
Electron microscopy
PACINIOMA AND PACINIAN NEUROFIBROMA
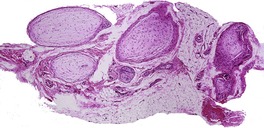
Fig. 37.11
NERVE SHEATH MYXOMA
Histopathology340.343.344. and 345.
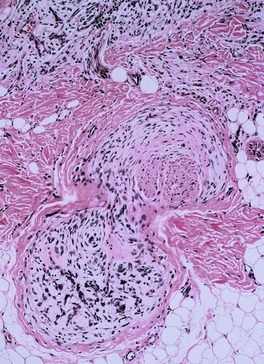
Fig. 37.12
NEUROTHEKEOMA
Histopathology
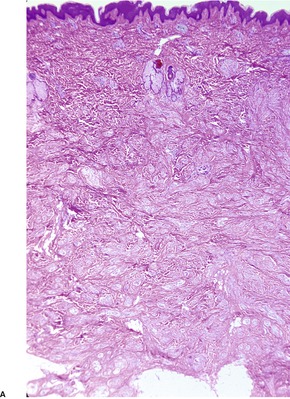
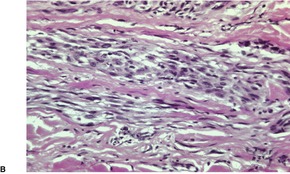
Fig. 37.13
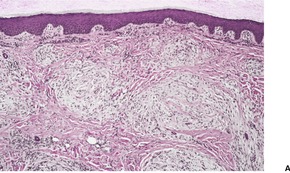
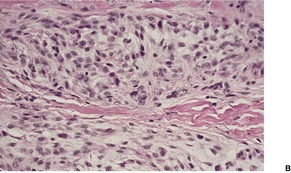
Fig. 37.14
PIGMENTED STORIFORM NEUROFIBROMA
GRANULAR CELL TUMOR
Histopathology379.380. and 426.
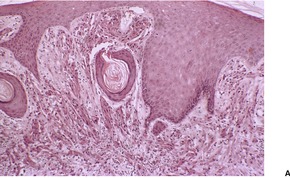
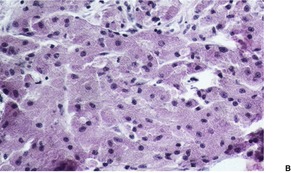
Fig. 37.15
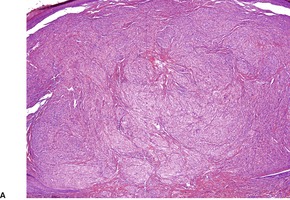
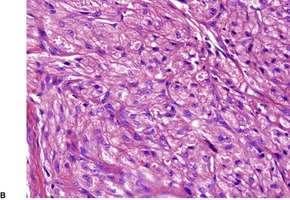
Fig. 37.16
Electron microscopy
MALIGNANT PERIPHERAL NERVE SHEATH TUMOR
Histopathology
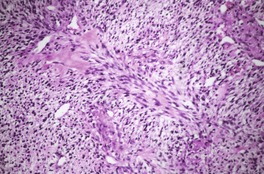
Fig. 37.17
HERNIATIONS AND ECTOPIAS
NASAL GLIOMA AND NEURAL HETEROTOPIAS
Histopathology508. and 517.
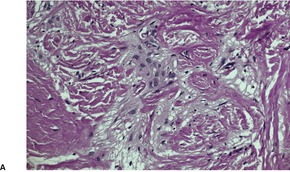
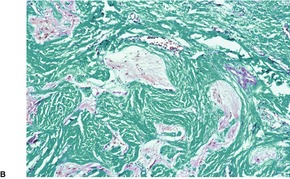
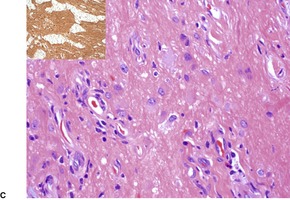
Fig. 37.18
CUTANEOUS MENINGIOMA
Histopathology526
NEUROENDOCRINE TUMORS
CUTANEOUS PARAGANGLIOMA
Histopathology
NEUROENDOCRINE ADENOMA
MERKEL CELL CARCINOMA
Treatment of Merkel cell carcinoma
Histopathology559. and 646.
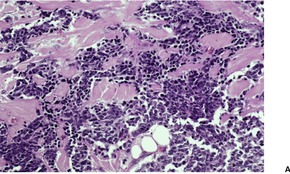
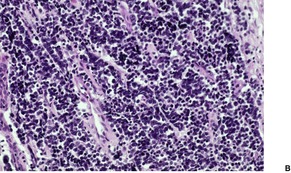
Fig. 37.19
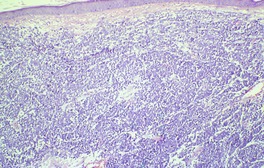
Fig. 37.20
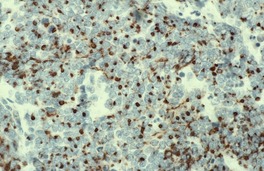
Fig. 37.21
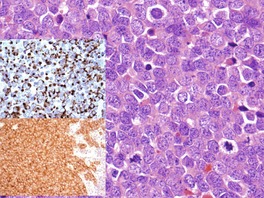
Fig. 37.22
Electron microscopy
MALIGNANT PRIMITIVE NEUROECTODERMAL TUMOR
Histopathology
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Neural and neuroendocrine tumors
Nerve sheath tumors868
Epithelial sheath neuroma871
Re-excision perineural invasion871
Perineurioma871
Schwannoma (neurilemmoma)872
Psammomatous melanotic schwannoma874
Pacinioma and pacinian neurofibroma876
Nerve sheath myxoma876
Neurothekeoma877
Pigmented storiform neurofibroma878
Granular cell tumor878
Malignant peripheral nerve sheath tumor880
Cutaneous neural tumors arise from, or differentiate toward, one or more elements of the nervous system. Most of the tumors found in the skin and subcutaneous tissue are derived from peripheral nerves or their neurocutaneous end organs. There are three principal cells comprising the sheath of peripheral nerves: the perineurial cell, the Schwann cell, and the fibroblast. Perineurial cells, which differ from Schwann cells by having no basement membrane, give rise to the perineurioma. Schwann cells give rise to the three main types of cutaneous neural tumors: neuromas, schwannomas (neurilemmomas), and neurofibromas. The tumors differ from one another by having a different proportion and arrangement of the various constituents of a peripheral nerve – Schwann cells, axons, fibroblasts, and supporting stroma. Although Schwann cells are generally viewed as neuroectodermal cells derived from the neural crest, it has been suggested that they may be of mesenchymal origin. 1 The perineurial cell may also be of neural crest derivation. 2
The other categories to be considered in this chapter are the herniations and heterotopias of glial and meningeal cells, giving rise to nasal gliomas, cutaneous meningiomas and related heterotopias, and the neuroendocrine carcinoma (Merkel cell tumor), which may be derived from another neural crest derivative, the Merkel cell. This cell subserves a neurosensory function in the skin. Merkel cell hyperplasia has been found in many settings, including sun- and radiation-damaged skin, as well as in some tumors of follicular origin. 3
Before considering the various tumors, it is worth noting a comparatively recent paper reviewing cutaneous innervation. 4 Itching and pain are symptoms of many of the conditions described in this book, and an understanding of the mechanisms involved is of some relevance.
The principal tumors in the nerve sheath category are neuromas, schwannomas (neurilemmomas), neurofibromas, and their variants. As malignant peripheral nerve sheath tumors are rare, and often deep-seated, only a brief account will be given. The granular cell tumor (myoblastoma) is possibly of Schwann cell origin and is also included. Nerve sheath tumors express both S100 protein and myelin basic protein. 5 The reaction for these substances is less intense in the malignant peripheral nerve sheath tumors. 5 Epithelial membrane antigen is present in the perineurial cell, the capsule of some nerve sheath tumors (see below), and in the nerve sheath myxoma. 6 Perineurial cells also express vascular endothelial cadherin. 7 The human progenitor cell antigen CD34 is present in a dendritic cell within the endoneurium of normal nerves, 8 as well as in neurofibromas, Antoni B areas of neurilemmomas, and dermatofibrosarcomas.9. and 10.
Neuromas are nerve sheath tumors in which the ratio of axons to Schwann cell fascicles approaches 1 : 1. 11 There are four distinct clinicopathological groups: traumatic neuromas, rudimentary polydactyly, solitary neuromas, and neuromas associated with the multiple endocrine neoplasia syndrome (type 2b). Ganglioneuromas will also be considered here.
Traumatic neuromas result when a nerve is sectioned or traumatized in some way, and continuity cannot be re-established. 12 They are therefore found at sites of trauma, in scars, and in amputation stumps. 13 One case followed a human bite. 14 Traumatic neuromas are usually firm, oval, pea-sized nodules in the subcutis or deeper soft tissues. 12 They may be painful. Traumatic neuromas may represent a locus minoris resistentiae that may be used by neoplastic cells to spread outside of the main tumor mass. 15 This is an exceedingly rare phenomenon.
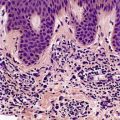
Traumatic neuromas are composed of an irregular arrangement of nerve fascicles embedded in fibrous scar tissue. There may be concentric condensations of fibrous tissue around individual fascicles, giving the appearance of multiple separate nerves (Fig. 37.1). Intraepithelial nerve fibers were present in one traumatic neuroma removed from the lower lip. 16 Perineurial cells, which contain epithelial membrane antigen, surround each fascicle, in contrast to solitary circumscribed neuromas, in which only the peripheral capsule contains these cells. 17 A Bodian stain will confirm the presence of numerous axons (Fig. 37.2). As in most neural tumors, mast cells are scattered throughout.
Traumatic neuroma composed of nerve fascicles separated by thin septa of fibrous tissue. (H & E)
Traumatic neuroma. Numerous axons are present. (Bodian preparation)
The term ‘rudimentary polydactyly’ is applied to a small nodular tumor which is found rarely near the base of the fifth finger, along its ulnar border.19.20. and 21. Most cases present in neonates or in childhood; presentation in adult life is uncommon. 22 The traditional view, that it is a variant of amputation neuroma at the site of a supernumerary sixth digit, presumed to have undergone spontaneous autoamputation, has been challenged. 23 It has been suggested that such lesions are neural malformations unrelated to a supernumerary digit. 23 A lesion with similar histology has been reported on the penis. 23 A family history of polydactyly is sometimes present. 22
Primary suture ligation of supernumerary digits in infancy may lead to the later development of a true amputation neuroma. Surgical excision of the lesion with high transection of the accessory digital nerve prevents the later development of amputation neuromas. 24
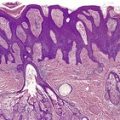
There are numerous bundles of nerve fibers embedded in connective tissue in the upper dermis, and in dermal papillae (Fig. 37.3). Many oval corpuscles, resembling small Meissner corpuscles, are also present. This latter feature, together with the indefinite outline and the position in the upper dermis, all differ from the usual traumatic neuroma.20. and 25. This supports the view that the traditional explanation for these lesions is incorrect. 23
(A) Rudimentary polydactyly. (B) There are many small nerves and some Meissner corpuscles in the upper dermis. (H & E)
There are normal Merkel cells along the basal layer of the epidermis, in addition to the nerve fibers and encapsulated corpuscles. 20
Solitary circumscribed neuromas, also described as ‘palisaded and encapsulated neuromas’, 11 are uncommon, slow-growing painless nodules found usually on the face of middle-aged individuals.26. and 27. An extremely rare site of involvement is the penis.28. and 29. They average 0.5 cm in diameter. A patient with multiple cutaneous neuromas, but no other abnormality, has been reported. 30 The patient reported as segmental neurofibromatosis of the face had lesions in which the published photomicrographs had some resemblance to a circumscribed neuroma. 31 The morphological resemblance of solitary neuromas to traumatic neuromas has led to the suggestion that they represent a regenerative lesion in response to minor trauma to the site.17. and 32.
Solitary circumscribed neuroma usually presents as a single nodule confined to the dermis (Fig. 37.4). A multinodular or plexiform growth pattern is uncommon. 33 The neuroma is composed of well-developed fascicles separated by a loose matrix resembling the endoneurium of normal nerve (Fig. 37.5). Rarely, cells with an epithelioid appearance are present. 34 A thin perineural connective tissue capsule often surrounds the tumor, but there is no fibrous tissue sheath around fascicles, as seen in a traumatic neuroma. 13 The perineurial cells in the capsule contain epithelial membrane antigen. 26 The fascicles contain axons, and many are also myelinated; 35 they stain for S100 protein.36. and 37. Kossard and colleagues have presented evidence challenging the concept that these tumors always contain axons. 38 They found that some tumors lacked a significant axonal content, thus merging with the features of a schwannoma. 38
Solitary neuroma. A circumscribed dermal tumor is present. (H & E)
Solitary neuroma composed of nerve fascicles with no intervening collagen. (H & E)
The superficial part of a solitary neuroma is sometimes more loosely arranged, with a myxomatoid stroma, resembling a neurofibroma. 11 In one case, most of the lesion was myxoid raising the possibility that the lesion was a nerve sheath myxoma with neuroma-like features. 39 Prominent stromal vascularity has been reported. 40 Occasionally a nerve can be traced into the lesion from below.
The overlying epidermis is usually normal or attenuated, but there is one report of overlying pseudoepitheliomatous hyperplasia. 41
Multiple mucosal neuromas may be the first manifestation42. and 43. of the multiple endocrine neoplasia syndrome (type 2b). This rare autosomal dominant condition (OMIM 162300), which also includes medullary carcinoma of the thyroid, pheochromocytoma, and somatic abnormalities,44.45.46.47. and 48. is due to a mutation in the RET gene on chromosome 10q11.2. Multiple cutaneous neuromas, macular amyloidosis, and medullary carcinoma of the thyroid have been reported in a patient who was a heterozygote carrier of a mutation in the RET proto-oncogene. 49 Multiple idiopathic mucosal and mucocutaneous neuromas have been reported in patients who had no features of MEN type 2b or other syndromes.50. and 51. Mucocutaneous neuromas have also been reported in association with germline mutations in the phosphatase and tensin homologue (PTEN) tumor suppressor gene on chromosome 10q23.31 (gene number 601728). 52 A spectrum of phenotypic expressions accompanies mutations in this gene, including Cowden’s syndrome, 53 Bannayan–Riley–Ruvalcaba syndrome (see p. 767), and some cases of Proteus-like syndrome. 52 They are all allelic.
Two different patterns may be seen. 11 The usual mucosal neuromas resemble solitary neuromas with haphazardly arranged bundles of Schwann cell fascicles. In the other pattern there are tortuous hyperplastic nerves with a thickened perineurium. 11
Hypertrophy of small nerves in the dermis (‘dermal hyperneury’) has also been noted in the clinically normal skin of patients with this syndrome. 54 Nerve hypertrophy has been reported in a patient with striated pigmentation and a marfanoid habitus, a possible forme fruste of this syndrome, but without the endocrine tumors. 55 Rarely, it may follow chronic irritation or rubbing of the skin (Fig. 37.6). 56
Dermal hyperneury. (A) A large nerve fiber is present just above the mid dermis. (B) Another large nerve in the deep dermis. (H & E)
Ganglioneuromas may be found in the skin in patients with neuroblastomas and mature cutaneous secondary deposits, and in von Recklinghausen’s disease where ganglion cells have been entrapped by a neurofibroma. 57 Primary cutaneous ganglioneuromas are exceedingly rare.58.59.60. and 61. Most cases develop after birth, but a congenital lesion has been reported. 62 They are clinically nondescript, but histologically distinctive. 63
Ganglioneuromas consist of mature ganglion cells, which are usually intermixed with fascicles of spindle cells.59. and 63. In one reported case the ganglion cells were separate from the neuromatous elements. 64 The term ‘ganglion cell choristoma’ has been used for rare cases with ganglion cells and no stromal component.65. and 66. At the other end of the spectrum is a case with abundant collagenous stroma containing scattered spindle and ganglion cells – a desmoplastic ganglioneuroma. 67 Overlying hyperkeratotic epidermal changes have been reported in two cases. 68
The fusiform cells are strongly positive for S100 protein, but not the ganglion cells. 69 In two recent reports both the spindle-cell component and the ganglion cells were positive for S100 protein and CD56.61. and 70. In one of these reports they were also positive for neuron-specific enolase (NSE) and glial fibrillary acid protein (GFAP), 61 and in the other report for neurofilament protein. 70
Epithelial sheath neuroma is a rare tumor that presents as papules and nodules on the upper back of elderly patients.71. and 72. Lesions may be solitary. 72
The tumor is characterized by a proliferation of enlarged nerve fibers in the upper dermis ensheathed by squamous epithelium. The nerve fibers are much larger than normal for the dermis, and the perineural squamous epithelium is mature with focal cornification. 73 These structures are surrounded by delicate fibroplasia and a mild inflammatory cell infiltrate of lymphocytes with a few plasma cells. 74 There is a superficial resemblance to perineural invasion by a squamous cell carcinoma.
Re-excision perineural invasion refers to the presence of mature squamous epithelium in the perineural spaces of normal cutaneous nerves in re-excision specimens.76. and 77. It was originally described in the re-excision specimens of melanocytic lesions, but it has also been noted in re-excision specimens of other cutaneous tumors, when its distinction from perineural invasion can be more difficult. The perineural epithelium, however, is mature. It expresses MNF116 and other keratin markers. The epithelium is negative for S100 protein, melan-A, and HMB-45. 76 The origin of the epithelium is uncertain as no connection can be demonstrated with surface or adnexal epithelium. 78 It may result from implantation during the initial procedure. 76 Other suggestions have been that this process may represent perineural squamous metaplasia, 79 or an unusual reactive eccrine duct regenerative response to surgical injury, with growth into a perineural space. 80
Perineurioma is a rare tumor, composed exclusively of perineurial cells, which develops in the dermis, subcutis, or deep soft tissue.69.81. and 82. There are two main forms of perineurioma: a rare intraneural perineurioma involving named peripheral nerves, 83 and a relatively more common extraneural variant which includes a conventional form, a sclerosing variant, a reticular (retiform) form,84. and 85. and a lipomatous variant, 86 which is a histological curiosity only. Another histological variant is the atypical, cellular form. 87 The malignant perineurioma is a variant of malignant peripheral nerve sheath tumor (MPNST). 88 Less than 5% of MPNSTs express perineurial differentiation; sometimes this change is only focal.
The conventional form is found on the trunk and extremities of middle-aged adults. An association with neurofibromatosis type 1 (NF-1) 89 and neurofibromatosis type 2 (NF-2) 81 has only been reported recently. Unfortunately, the term perineurioma was used in the past for localized hypertrophic neuropathy (‘dermal hyperneury’, see above), with which it has no relationship at all.90. and 91.
Rare cases of a granular perineurioma have been reported.92. and 93. The lesion presented as a polypoid mass 4.5 cm in diameter in one case, and as multiple lesions in another. 93
The sclerosing perineurioma is a distinctive variant with a predilection for the fingers and palms of young adults.94.95.96.97.98. and 99. This variant has been associated with aberrations of chromosome 10, 100 and with a cryptic deletion of the NF2 gene at chromosome 22q12.2.83. and 95. This supports other evidence linking other types of perineurioma to this chromosome.101. and 102. Despite the allelic loss or mutation of the NF2 gene in perineurioma, it is surprising that this tumor has only recently been described in association with NF-2. 103
Perineuriomas are firm, circumscribed nodules, usually 1.5–4 cm in diameter. Larger variants have been described, usually in the deep soft tissues. 81
The neoplasms are circumscribed non-encapsulated lesions composed of spindle cells with elongated, bipolar cytoplasmic processes.81. and 90. On low power they usually resemble neurofibromas, with fascicles or individual cells oriented either parallel to each other or forming small concentric collections (Fig. 37.7). 69 These concentric whorls (‘onion bulbs’) are an important clue to the diagnosis. 104 They sometimes have pacinian-like features. 102 Variants with granular cells and others with a plexiform or reticular pattern have been reported.84.105. and 106. The reticular (retiform) variant is composed of anastomosing cords of fusiform cells with bipolar cytoplasmic processes that wrap around islands of fibromyxoid stroma. 84 The stroma of the conventional type is also fibromyxoid; rarely, it contains calcospherites. 91 Myxoid features were prominent in one subungual perineurioma. 107 Uncommonly, there is a fibrous or sclerotic stroma. 108Lipomatous perineurioma refers to the presence of mature adipocytes admixed with spindle cells. 86
Perineurioma. (A) This is a paucicellular fibrous variant. Typical ‘onion bulb’ structures can be seen. (B) Another case. ‘Onion bulbs’ can be seen. (H & E)
The cases described by Mentzel and colleagues were more cellular lesions with a mixed lamellar and whorled pattern, sometimes with a marked storiform pattern. Focal stromal hyalinization and small aggregates of foam cells were present in some of their cases. 90 Such cases can be misdiagnosed as epithelioid histiocytomas. 109
The epithelioid perineurioma is composed of a syncytial-like proliferation of epithelioid cells with moderate amounts of eosinophilic cytoplasm. The cells in the reported case were positive for epithelial membrane antigen (EMA) and claudin-1. 110
The atypical cellular perineurioma is a well-circumscribed lesion composed of spindle cells arranged in a storiform pattern. 87 Focally, it may appear laminated. The cytoplasm of the cells is eosinophilic and well demarcated. The nuclei are elongated. Larger atypical cells are scattered through the tumor. A few cells have several nuclei. Mitoses are uncommon (1/20 high-power fields); atypical mitoses are not found. 87
Most of the tumor cells stain for EMA and vimentin. 97 Most cases show membranous positivity for claudin-1 protein, 88 a recently described tight junction-associated protein. 111 In recent reports, cells have also expressed GLUT-1, protein gene product 9.5 (PGP 9.5), CD57, 83 vascular endothelial (VE)-cadherin, 112 and CD99. 99 Focal positivity for factor XIIIa is often present. 109 The cells do not usually stain for S100 protein, CD34, chromogranin or NSE,90. and 91. but Hornick and Fletcher reported a significant number of cases (64%) that expressed CD34. 82 In the variant with granular cells (see above), these cells expressed S100 protein but not EMA, supporting their Schwann cell origin. 105
One of the reported granular perineuriomas expressed NK1/C3 but not S100 protein. 92
The sclerosing perineurioma is composed of abundant dense collagen and variable numbers of small, epithelioid, and spindled cells exhibiting corded, trabecular, and whorled (onion skin) patterns. 94 This tumor may be confused with the fibroma of tendon sheath, the sclerotic fibroma of Cowden’s disease (see p. 815), and the pacinian collagenoma (see p. 816). 113 A lipomatous variant of sclerosing perineurioma has been reported in the skin. 114 The cells stain for EMA and vimentin and often muscle-specific actin as well. 94 They also express GLUT-1, claudin 1, and collagen type IV. 114 The fibrous perineurioma is probably a less dense, more cellular, and better vascularized variant of the sclerosing perineurioma. 108
Intraneural perineuriomas cause fusiform expansion of a nerve, usually in the deep soft tissues. There are ‘pseudo-onion bulbs’ of concentrically arranged perineurial cells surrounding a central zone containing axons, Schwann cells, and fibrovascular stroma. 88
Multiple hybrid granular cell tumors with admixed perineurioma elements were present in one patient. 115 Schwannoma–perineurioma hybrids also occur.116. and 117.
The spindle cells have lengthy, thin, bipolar cytoplasmic processes aligned in parallel and separated by small amounts of collagen. 88 Similar features are seen in the epithelioid cells of the sclerosing variant. 88 The cells contain few organelles, but pinocytotic vesicles are aligned along the plasmalemma of the cell processes.
Cutaneous schwannomas (neurilemmomas) are uncommon, slow-growing, usually solitary tumors with a propensity for the limbs of adults. 13 Pain, tenderness, and paresthesiae may be present in up to one-third of lesions.118.119.120. and 121. They measure 2–4 cm in diameter. 13 They are of Schwann cell origin. 122 Multiple tumors are uncommon and can occur in several clinical settings: in association with neurofibromas in von Recklinghausen’s disease; 123 or as multiple, localized tumors which may be a distinct disease;124. and 125. in neurofibromatosis type 2 (NF-2 – OMIM 101000), due to mutations in the NF2 gene, located at 22q12.2, and characterized by vestibular schwannomas and other tumors; and in (familial) schwannomatosis (OMIM 162091), characterized by the development of multiple spinal, cranial and peripheral schwannomas in the absence of a vestibular schwannoma. 126 Recently, mutations in the INI1 gene, also known as SMARCB1, a tumor suppressor gene located in the immediate vicinity of NF2, have been implicated as the gene responsible for schwannomatosis. 127 This same gene had previously been implicated in the pathogenesis of malignant rhabdoid tumors of infancy. 126 A further gene, CABIN1, also near NF2, is thought to contribute to the formation of schwannomatosis but further studies are needed to confirm whether it is co-located with either NF2 or INI1. 126 Hopefully, these new findings will end the controversy in this area.128.129.130.131.132.133. and 134.
In addition to vestibular schwannomas, patients with NF-2 may also develop meningiomas and ependymomas. The NF2 gene controls the synthesis of merlin, a protein found in the brain, nerves, eyes, and Schwann cells. Most gene mutations result in the production of a shortened version of merlin protein, which may then allow these cells to proliferate. 135Sporadic schwannomas may also have NF2 deletions. 135
Schwannomas also occur in the deep soft tissues, retroperitoneum, mediastinum, and tongue, 136 and on the vestibulocochlear nerve. Local recurrence and malignant transformation are exceedingly rare.137. and 138.
The tumor is gray-white in color, encapsulated, with a smooth, glistening appearance. 139 Cystic change is sometimes present, particularly in the larger, deeper tumors.
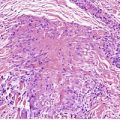
Schwannomas are circumscribed encapsulated tumors, usually confined to the subcutis. The nerve of origin may sometimes be seen along one border (Fig. 37.8). The agminate tumors and some of those in schwannomatosis are often situated in the dermis. 124 Multiple small tumor nodules may be present in these forms, as well as hypertrophied peripheral nerves.124. and 128. The term ‘plexiform schwannoma’ has been applied to this type (Fig. 37.9).140.141.142.143. and 144. It originates from nerve sheath (see below). 145
(A) Schwannoma. (B) There are Verocay bodies in the interlacing bundles of spindle-shaped cells (Antoni A tissue). (H & E)
Plexiform schwannoma. The tumor bulges into the subcutis. There are multiple nodules. (H & E)
Schwannomas are characteristically composed of two tissue types. 122 In the so-called ‘Antoni A’ areas there are spindle-shaped Schwann cells arranged in interlacing fascicles. The cells have indistinct cytoplasmic borders. The nuclei may be aligned in rows or palisades, between which the cell processes are fused into eosinophilic masses forming Verocay bodies. A variant in which the Verocay bodies form 75–100% of the tumor has been described. 146 Conspicuous Verocay bodies were present in a schwannoma of the penis. 147 Antoni B tissue consists of a loose meshwork of gelatinous and microcystic tissue with widely separated Schwann cells. Lipid-laden macrophages, dilated blood vessels with thick hyaline walls, old and recent hemorrhage, lipofuscin, and sometimes calcified hyaline areas may also be present in Antoni B areas.2.122. and 148. ‘Collagenous spherulosis’ (radiating eosinophilic collagen fibers) has been reported in one case. 149 Mast cells and non-specific cholinesterase may be demonstrated. 13 Merkel cells are increased in the epidermis overlying schwannomas. 150
Immunoperoxidase methods demonstrate S100 protein, 124 vimentin, 151 SOX 10, 152 and myelin basic protein153 in the tumor cells. Glial fibrillary acidic protein (GFAP) is present in a small number of cases.151. and 154. Epithelial membrane antigen is found in the perineurial cells in the capsule of schwannomas, as in that of solitary neuromas (see above).146. and 155. The conventional view has been that schwannomas, unlike neurofibromas, do not possess intratumoral axons. However, a recent study using an antibody to neurofilament protein found axons in 30–70% of schwannomas, the incidence varying with the histological subtype. 156 All types of Schwann-cell-derived tumors express the DNA-binding transcription factor, activating protein 2 (AP-2), mainly in their nucleus. 157
A cellular variant (cellular schwannoma), mostly found in the deeper tissues, has been described.158. and 159. There are compact spindle-shaped cells with mitoses and some storiform areas, and a near absence of Verocay bodies and Antoni B tissue. 158 Rare variants with multiple glandular elements, 160 with pseudoglandular structures, 161 and with sweat duct differentiation162 have been reported.
The plexiform schwannoma (see above) has a superficial resemblance to the plexiform neurofibroma with multiple interlacing and interconnecting fascicles and nodules, composed predominantly of Antoni A-type tissue. 145 Most tumors occur in the dermis and subcutis. A deep-seated variant has also been described. 163 In comparison with the superficial variety, the deep variant has a predilection for females, can occur in a congenital setting, and can show necrosis and myxoid change. 163 A further subset of plexiform schwannomas occurs in a congenital or childhood setting. It was previously considered a form of malignant peripheral nerve sheath tumor (MPNST). 164 Despite their cellular pleomorphism they are now known to have a benign behavior.
A neuroblastoma-like variant, with large rosette-like structures and fibrillary collagenous centers, has been described.165. and 166. The presence of intervening Antoni B areas and an absence of necrosis or mitoses assist in making a diagnosis. One case presented with anetoderma-like features. 167 The cells express S100 protein, but not neuron-specific enolase, as occurs in the smaller rosettes of neuroblastoma. 168 Only one of the several cases tested expressed GFAP. 166
The epithelioid schwannoma is an extremely rare variant composed largely of epithelioid cells, in which there is a lack of mitotic activity.169.170.171. and 172. In a study of 33 cases of benign epithelioid peripheral nerve sheath tumors (BEPNSTs), 18 were subclassified as epithelioid neurofibromas and 15 cases as epithelioid schwannoma, although in five of these cases the absence of nuclear palisading, the presence of uniform cellularity, and the presence of a significant CD34-positive spindle-cell population led to their classification as BEPNSTs of indeterminate histogenesis. 172 A collagen-rich variant of BEPNST has since been reported. 173 Features of classic schwannoma are usually present in some areas of the tumor. 170 There is a thin capsule containing EMA-positive cells although this was not present in a subsequent case. 174 Type IV collagen encircles individual cells within the tumor. 169 The cells stain for S100 protein but not HMB-45. An epithelioid schwannoma that was also plexiform has been reported. 175
The pseudoglandular schwannoma is an extremely rare variant composed of gland-like spaces containing mucinous material and lined by Schwann cells that are strongly positive for S100 protein and GFAP.176. and 177. In another case the glandular cells stained with CAM5.2 and EMA, but not for S100 protein. 178 This may be a true glandular variant. This variant is more common in schwannomas of the central nervous system. 177
Another type of schwannoma is the psammomatous melanotic schwannoma, which contains melanin and scattered psammoma bodies (see below).
The Wagner–Meissner schwannoma has a monomorphous morphology with sheets of corpuscles having a lamellar structure. 179 The nuclei are predominantly located at the periphery of the corpuscle. 179 They are separated by thin fibrous strands. There are some similarities to a perineurioma in the published photomicrographs.
The term ‘congenital neural hamartoma (fascicular schwannoma)’ has been used for an unencapsulated dermal tumor composed of fascicles of Schwann cells with frequent Verocay body-like structures. 180 Unlike cutaneous neuromas, there are no axons present.
The rare psammomatous melanotic schwannoma is an unusual component of Carney’s complex (myxomas, spotty pigmentation, and endocrinopathy).183. and 184. The most common locations are the posterior spinal nerve roots, gastrointestinal tract, bone, and soft tissue; 185 the skin is an uncommon site. 186 Multiple lesions may be present. 183
This tumor is usually well circumscribed but only partly encapsulated. There is a mixture of polygonal and fusiform cells, many of which are heavily pigmented melanocytes. 69 Psammoma bodies are present in varying numbers. Sometimes they coalesce to form larger, irregular masses. Adipocytes are often present.
The tumor cells stain positively for S100 protein, HMB-45, MART-1, synaptophysin, and vimentin. 187
Neurofibromas may occur as a solitary tumor or as multiple lesions in a segmental or widespread distribution, referred to as neurofibromatosis. The histopathology of the neurofibromas in these different clinical settings is similar and will be considered together.
Solitary neurofibromas are papular, nodular, or pedunculated tumors with a predilection for the upper trunk. 13 Rarely they have a diffuse pattern. 189 They are soft and tend to invaginate on pressure (the ‘buttonhole’ sign). Subungual lesions have been reported;190. and 191. in another case a toe was involved. 192
Neurofibromatosis, described by von Recklinghausen in 1882, is a clinically heterogeneous disorder with varied manifestations affecting the skin, soft tissues, blood vessels, and the peripheral and central nervous systems.193. and 194. It is said to affect 80 000 individuals in the United States. 195 It has a significant impact on quality of life. 196 Riccardi has proposed eight clinical subtypes of neurofibromatosis (NF-1 to NF-8). 197 NF-1 is classic von Recklinghausen’s disease, which accounts for 85–90% of all cases. NF-2 is associated with vestibular neuromas and sometimes other intracranial tumors. It has been discussed above. Cutaneous tumors, particularly schwannomas, are found in more than 50% of cases. 198 The inheritance of NF-2 is autosomal dominant, the gene being located on chromosome 22q12.2. NF-3 is a mixed form combining features of NF-1 and NF-2. NF-4 is a variant form with diffuse neurofibromas and café-au-lait pigmentation, but without many of the other clinical features that typify NF-1. NF-5 is the segmental form. NF-6 has prominent café-au-lait pigmentation as the sole manifestation. NF-7 is a late-onset type, and NF-8 is a miscellaneous group not categorized into the other subtypes.197. and 199.
Prenatal diagnosis of neurofibromatosis has not been routinely available because of the large size of the gene. Furthermore, the high rate of new mutations (approximately 50% of all cases) means that the index of suspicion that a fetus may be affected is consequently lowered. 200 In the last decade, prenatal screening has become more effective. 201 Both prenatal and preimplantation genetic diagnosis can be used for individuals with NF-1. 202
Neurofibromatosis type 1 (NF-1 – OMIM 162200) is inherited as an autosomal dominant trait, although spontaneous (new) mutations account for up to 50% of all NF-1 probands.200. and 203. Its prevalence is approximately 1 in 3000–4000 individuals worldwide. 201 It is linked to the NF1 gene located on the long arm of chromosome 17 (17q11.2). 204 Specific mutations of the NF1 gene may be related to the expression of a specific phenotype. 205 Neurofibromin is the protein encoded by the gene, which appears to have a tumor suppressor function. It is capable of down-regulation of the p21-ras oncogene and it negatively regulates MAPK (mitogen-activated protein kinase) signaling. 206 Recently a germline loss-of-function in the SPRED1 gene on chromosome 15 has been reported that resulted in an NF-1-like phenotype. 206 Several phenotypically overlapping syndromes (Noonan syndrome, LEOPARD syndrome, Costello syndrome, and cardio-facio-cutaneous syndrome) all result from mutations in genes encoding key components of the RAS-MAPK pathway. 206 Other signaling pathways, such as hedgehog one, may be involved in tumorigenesis in NF-1. 207
Café-au-lait pigmentation, which varies from small macular areas to large patches of pigmentation, is present in 99% of cases. 195 The pigmentation is present at birth or appears early in childhood before other stigmata of the disease. 208 It may be found in other conditions, 209 but the presence of six or more patches is said to be diagnostic of neurofibromatosis. 210 A giant garment-like café-au-lait macule, involving the lower half of the trunk, has been reported in a patient with NF-1. It was regarded as a possible type 2 segmental manifestation of the disease. 211 Axillary freckling is present in about 20% of patients. Small lentiginous melanocytic nevi are increased in number. 212
The neurofibromas are multiple and sometimes disfiguring. They appear before puberty, although late onset has been recorded.213. and 214. Congenital, disseminated disease is extremely rare. 215 Sex incidence has been equal in many studies. The location of the neurofibromas has been variable, with the extremities the predominant site in one African series. 214Neurofibromas may develop for the first time in pregnancy; women who already have tumors may experience an increase in the size and number of the lesions during pregnancy. 216 The screening of school-aged children for neurofibromatosis is controversial.217. and 218. Plexiform neurofibromas, 13 which were thought to be pathognomonic of neurofibromatosis, may rarely present as solitary lesions without any features of neurofibromatosis. 219 They may present as large deep tumors or localized areas of deformity.220. and 221. They are found in about 25% of cases.210. and 222. In about 10% of cases plexiform neurofibromas are multiple. 223 Schwannomas are sometimes present as well.
Other clinical features of the classic form are pigmented hamartomas of the iris (Lisch nodules),224.225. and 226. poliosis circumscripta, 227 macrocephaly, learning disability, kyphoscoliosis, bone hypertrophy, pseudoarthrosis, and vascular lesions.224.228.229.230. and 231. Somatic mutations may explain the variable expressivity of the NF1 phenotype. 232 The classic form has been reported in a patient with tuberous sclerosis, 233 and in one with the McCune–Albright syndrome. 234 The association with mycosis fungoides was probably fortuitous. 235 Elephantiasis neuromatosa, with localized gigantism or thick redundant folds of skin, is rare.228. and 236. Neurofibromas, each with a halo of depigmentation, have been reported in one patient with presumptive mild neurofibromatosis. 237 Generalized vitiligo was present in another case in which the neurofibromas showed the halo phenomenon. 238 Another rare association is hypertrophy of pacinian corpuscles. 239
Malignant degeneration of neurofibromas occurs in 2–3% of patients, 210 although higher figures have been reported in some series. 222 Pain, neurological deficit, and tumor enlargement are features suspicious for malignant transformation. 240 In addition, various other sarcomas, 241 melanomas,222.242. and 243. and visceral carcinomas222 have been reported, although the latter may not be increased in incidence. 244 Clinical follow-up seems to be as effective as imaging studies in detecting these complications. 245 Subcutaneous neurofibromas and the absence of cutaneous lesions are two independent risk factors for mortality in patients with NF-1. 246
The segmental variant (NF-5) is a heterogeneous group.247.248.249.250.251.252.253.254. and 255. Café-au-lait pigmentation is present in some cases;220.256.257.258. and 259. there are usually no other stigmata of neurofibromatosis (other than neurofibromas) and no family history.260.261.262. and 263. This localized form may be due to mosaicism resulting from a postzygotic NF1 gene mutation.264. and 265. There is a tendency now to disregard the Riccardi classification and to refer to ‘mosaic NF-1’ for all cases resulting from a postzygotic NF1 lesion. 266 Such inactivations may result in generalized mild disease, segmental disease, or gonadal mosaicism. 266 Determining when, and in what cell types, inactivation of the NF1 gene occurs is critical for understanding the various clinical manifestations of segmental NF1. 266 Variants of the segmental form include a localized, 267 a unilateral,247. and 261. and a bilateral247.265.268.269.270. and 271. segmental form, a form associated with deep neural tumors, 272 and a rare hereditary form. 273 Most cases involve the trunk; the face is uncommonly involved. 274 Over 100 cases of NF-5 have been reported. An association with an internal malignancy is very rare. 265 The simultaneous occurrence of NF-5 and clustered incomplete tuberous sclerosis has been reported. 275
Neurofibromatosis localized to the vulva and perianal area can be classified with this group.276. and 277.
Cutaneous neurofibromas are non-encapsulated, loosely textured tumors centered on the dermis.13. and 278. There is often extension into the subcutis, sometimes in a diffuse infiltrative pattern. 278 An overlying grenz zone separates the lesion from the undersurface of the epidermis. The lesion is composed of delicate fascicles, usually only a single cell thick. 11 The cells have an oval or spindle-shaped nucleus and scant, indefinite cytoplasm (Fig. 37.10). Vacuolated cells are found focally in a small number of tumors. 279 Floret-like multinucleated giant cells were present in a neurofibroma arising in a patient with NF-1. 280 There is sometimes nuclear pleomorphism, but mitoses are rare. Neurofibromas with high cellularity and atypia have been called dysplastic neurofibromas. Patients with such lesions require increased surveillance because of a potential risk for malignant transformation. 281 The matrix is pale staining with delicate wavy collagen; rarely, the matrix is rich in mucin or is sclerosing or hyalinized.191.278. and 282. Solitary neurofibromas are sometimes more compact than those in neurofibromatosis. 122 Blood vessels are increased in number in the stroma. 283 A Bodian stain will demonstrate some axonal material, but not in the 1 : 1 ratio with Schwann cells, as occurs in neuromas. 11 Mast cells, 284 non-specific cholinesterase, 263 S100 protein, myelin basic protein, 153 and factor XIIIa285. and 286. can be demonstrated by the appropriate techniques. Podoplanin expression, using the D2-40 antibody, is found in a small number of neurofibromas. It is found more often in neurilemmomas. 287 The mast cells may not always be quiescent as evidenced by the report of abundant eosinophils in a solitary neurofibroma. 288
Neurofibroma composed of thin fascicles of cells, each with a ‘wavy’, spindle-shaped nucleus. (H & E)
Various growth factors and their receptors are also present. 289 Neural cell adhesion molecule (NCAM – CD56) was present in one recent study in 100% of schwannomas and MPNSTs, in 86% of neurofibromas, 76% of neurotized nevi, but only 50% of desmoplastic/spindle-cell melanomas. 290 The staining was less intense in the melanomas than in the neural tumors. 290
Plexiform neurofibromas are a distinct variant in which there is irregular cylindrical or fusiform enlargement of a subcutaneous or deep nerve; 13 rarely, they arise in the dermis. 291 Numerous large nerve fascicles are embedded in a cellular matrix containing abundant mucin as well as collagen, fibroblasts, and Schwann cells. 11 Initially, this proliferation of nerve fibers is confined within the epineurium of the involved nerve. 11 The massive soft tissue neurofibroma is highly specific for NF-1. It is worrisome because its size may mask the development of a malignant peripheral nerve sheath tumor. 292
The pigmented plexiform neurofibroma is a subset of plexiform neurofibromas with hyperpigmentation and hypertrichosis of the overlying skin. 293 There is melanocytic hyperplasia at the dermoepidermal junction and single pigmented melanocytes with occasional small nests in the papillary dermis, and scattered within the underlying neurofibromatous tissue. 293 Immunohistochemical staining with melan-A (MART-1) confirms the melanocytic differentiation of these scattered nests and individual cells, whereas GFAP and Leu-7 are detected only within plexiform areas and neuroid spindle cells. 293 The case reported as a cutaneous melanocytoneuroma had an intraneural proliferation of large epithelioid cells with eosinophilic cytoplasm with prominent cell borders, imparting a plant-like pattern. 294 Involvement of smaller nerve twigs gave the lesion a plexiform appearance. The cells expressed S100 protein, melan-A, HMB-45, microphthalmia-associated transcription factor (MITF), and PGP9.5. 294 The lesion was surrounded by an EMA-positive perineurial layer. 294
Other findings in neurofibromatosis are schwannomas (neurilemmomas), tumors with features of both neurofibroma and schwannoma (hybrid lesions),295. and 296. and neurofibromas containing scattered, possibly entrapped, ganglion cells. Pilar dysplasia and folliculosebaceous proliferations have been reported overlying neurofibromas.297. and 298. Vascular changes are rarely found. 299 They include smooth muscle islands in the intima of vessels. 229 Fibromuscular dysplasia of larger cutaneous vessels was so extensive in one case that it resulted in cutaneous ulceration. 300 If random biopsies are taken from apparently normal skin in patients with NF-1, microscopic (occult) neurofibromas are sometimes found. Abundant S100-positive cells may also be found within the perifollicular fibrous tissue.301. and 302. In elephantiasis neuromatosa there is a diffuse proliferation of Schwann cells and axons in the subcutis. 122 Islands of cartilage are present, rarely, in this tissue. 122 Glandular epithelial structures (epithelioid neurofibroma) have been reported in the stroma.172.251. and 303.
In a review of the histopathological variants of neurofibroma, Megahed listed 10 variants: 278 classic, cellular, myxoid, hyalinized, epithelioid, plexiform, diffuse, pigmented, granular cell, and pacinian. Some of these variants are regarded by others as discrete entities, whereas others represent morphological curiosities.
An additional variant is the recently described dendritic cell neurofibroma with pseudorosettes.304. and 305. Its development within the confines of the perineurium in the skin in one case supports its origin as a peripheral nerve sheath tumor and not a melanocytic nevus as was thought for a brief period. 306 It does not appear to be associated with NF-1 or NF-2. 307 The dermis is filled with tumor nodules that are oval shaped and oriented more or less vertically. Two types of cells are present: type I cells are small, dark, lymphocyte-like cells with inconspicuous cytoplasm, and the type II cells are larger, with pale-staining vesicular nuclei and abundant pale eosinophilic cytoplasm. 306 Type I cells are arranged concentrically around type II cells, forming pseudorosettes. Both cell types express S100 protein and CD57, and varying proportions of both cell types express CD56 and PGP9.5. 306
The sclerotic neurofibroma is an extremely rare variant in which there are scant cells and thick collagen bundles arranged chiefly in an interweaving pattern with prominent clefts.308. and 309. The tumor cells have uniform fusiform nuclei. There are some similarities to sclerotic fibroma. 310
The rare pigmented neurofibroma, six cases of which were described by Bird and Willis in 1969, 311 had received scant attention until a report of 19 cases. 312 It is composed of whorled fibrillar ovoid structures resembling Wagner–Meissner bodies.312. and 313. Melanin is present in macrophages and in some of the tumor cells, which stain for S100 protein, HMB-45, and melan-A. In the few cases tested, the cells also expressed CD34. 312 There is also co-expression of MET proto-oncogene and MITF. 314
The cellular neurofibroma can be difficult to distinguish from a malignant peripheral nerve sheath tumor (MPNST), particularly if atypia and low-grade mitotic activity are present. It has been suggested that ancillary studies (p53 expression, Ki-67 immunostaining, and flow cytometry to assess DNA content) may be useful in confirming a benign diagnosis in these cases. 315 Further studies are needed to confirm these findings. 316
The lipomatous neurofibroma is a recently described variant of neurofibroma characterized by the presence of intratumoral fat.317.318. and 319. In one study of 320 cases of neurofibroma, fat was observed in 22 (6.9%). 320 It was focal in 18 of these cases, while in the other four it was diffuse with mature adipocytes regularly interspersed with the spindle cells of the neurofibroma. 320
Giant pigment granules (macromelanosomes) are often present in melanocytes and basal keratinocytes in the café-au-lait spots of neurofibromatosis. 321 The granules can just be seen with the light microscope. They are probably increased in older individuals. 322 Macromelanosomes are not present in all cases of neurofibromatosis; their presence is not pathognomonic, as they can also be found in several other conditions.321. and 323.
Neurofibromas are composed of fusiform or stellate cells which are widely separated by individual collagen bundles and matrix.18. and 181. They have usually been interpreted as Schwann cells, 18 but one report has claimed that the principal cell is the perineurial cell, although scattered Schwann cell–axon complexes are also present. 2
The term ‘pacinioma’ has been applied to the rare finding of a hamartomatous overgrowth of mature Vater–Pacini corpuscles (Fig. 37.11).324. and 325. Another term used is ‘hyperplasia and hypertrophy of pacinian corpuscles’. 326 Bale reported two such lesions in the sacral region associated with spina bifida occulta. 324 Pain and local tenderness are often present. 327 Sometimes a history of prior trauma to the site can be elicited. 326 An association with neurofibromatosis (NF-1) is extremely rare. 239
Pacinioma composed of enlarged Vater–Pacini corpuscles. (H & E)
Pacinian neurofibroma (pacinian neuroma) is a rare tumor of the digits, hands, and feet. It is composed of round or ovoid corpuscles with multiple concentric lamellae.328.329. and 330. They do not have the perfect structure of a Vater–Pacini corpuscle, but the resemblance is close. The report of a case with multiple hairy lesions on the buttock composed of rudimentary Vater–Pacini corpuscles suggests that the distinction between the hamartomatous pacinioma and the pacinian neurofibroma is artificial. 331 In another case, multiple tiny lesions were present on the ring finger in one patient, associated with marked vascular changes of the glomus type of arteriovenous anastomoses. 332 Some of the cases reported in the literature as pacinian neurofibromas would now be reclassified as nerve sheath myxomas. 333 These latter tumors have less resemblance to Vater–Pacini corpuscles and they contain more stromal mucin than true pacinian neurofibromas.
The nerve sheath myxoma has been a controversial entity for many years, 334 particularly since the description in 1980 of the neurothekeoma by Gallagher and Helwig. 335 For many years nerve sheath myxoma and neurothekeoma were regarded as neural tumors at either end of a histological spectrum.336.337.338. and 339. This approach was adopted in the last edition of this book. A study, in 2005, of a large series of tumors coded at the Armed Forces Institute of Pathology (AFIP) as a nerve sheath myxoma or neurothekeoma resulted in 57 cases of nerve sheath myxoma being reviewed. 340 The authors concluded that this tumor was unrelated to so-called cellular and mixed-type neurothekeoma. 340
Nerve sheath myxomas are rare tumors that occur at all ages, but the peak incidence is in the fourth decade of life. There is a predilection for the extremities, particularly the hands, 341 and the knee/pretibial region. 340 The tumors are solitary, slow-growing multinodular masses. They are usually painless. Tumor size varies from 0.5 to 2.5 cm in diameter. Recurrences are common after simple excision, particularly if the lesion is incompletely removed. 340
This lesion is a peripheral nerve sheath tumor of Schwann cell origin as shown by their highly reproducible immunohistochemical findings (see below).340. and 342.
The nerve sheath myxoma is multilobulated and non-encapsulated (Fig. 37.12). It is centered on the reticular dermis but often extends into the superficial subcutis. 345 The lobules are composed of spindle-shaped, stellate, and sometimes epithelioid cells arranged in a swirling, lamellar, and often concentric pattern. Syncytial-like aggregates are also present. Vacuolated cells are often seen. The cellular elements are embedded in a myxoid stroma, which is usually abundant in those tumors in which stellate and bipolar cells predominate. Chondroitin-4 or chondroitin-6 sulfate is the principal heteroglycan present.334. and 344. There is a peripheral fibrous border. There is variable nuclear hyperchromatism and sometimes nuclear atypia. Mitoses are uncommon. In one case, melanin was present in some tumor cells. 342
Nerve sheath myxoma. Spindle-shaped cells are arranged in a swirling and concentric fashion. The stroma is myxoid. (H & E)
The cells are immunoreactive for S100 protein, GFAP, NSE, and CD57.340.346.347. and 348. They are bordered by collagen IV. Perineurial cells expressing EMA are present in small numbers, primarily in the fibrous tissue directly adjacent to the myxoid nodules. 340 Both nerve sheath myxoma and neurothekeoma express S100A6. 349
Neurothekeomas (cellular neurothekeomas) are distinctive cutaneous tumors of uncertain histogenesis.350. and 351. Because some lesions have a myxoid stroma, they were regarded for many years as part of a histological spectrum that included nerve sheath myxoma. However, there is no good evidence that they even show nerve sheath differentiation. 350 Until this is clarified, this benign tumor will be considered in this section.
Neurothekeomas are found mostly on the face and upper extremities.350.351.352. and 353. They are not associated with neurofibromatosis. There is a predilection for females, 354 and for young adults; cases in children are well documented.354. and 355. They present as asymptomatic, dome-shaped nodules that measure 0.3–6 cm or more in diameter.356. and 357. The mean tumor size in one series was 1.1 cm. 350 Neurothekeomas are usually solitary, but multiple lesions were present in two cases.351. and 358. Uncommonly, they recur if inadequately or marginally excised.
The histogenesis of these tumors is controversial. 359 The finding of PG-M1, a reliable marker for histiocytic differentiation, in one case led to speculation that the tumor was of fibrohistiocytic lineage. 360 Hornick and Fletcher concluded that there is no compelling evidence for a neuroectodermal derivation. 350 Another study postulated an origin from fibroblastic cells with the ability to differentiate into myofibroblasts, and a tendency to recruit histiocytic cells. 351
Although neurothekeoma is usually a dermal tumor, subcutaneous extension and/or skeletal muscle involvement may occur.350. and 361. It is composed of nests and fascicles, giving a multilobular appearance (Fig. 37.13). The intervening stroma may be hyalinized; often there are myxoid foci. 362 The nests of spindle cells sometimes have a concentric or whorling arrangement. A plexiform pattern may sometimes be present. 363 There may be a poorly differentiated interface between the fascicles and the dermal collagen. 364 Some of the cells may have large hyperchromatic nuclei and some mitoses are common (Fig. 37.14). 365 Dermal melanocytosis, in which the cells expressed NKI/C3 but not S100 protein, has been reported in the stroma of neurothekeomas. 366 Extensive dystrophic calcification367 and ossification have been reported. 368 Osteoclast-like giant cells are present in nearly 40% of cases. 351 Several cases of a tumor with extensive ossification and features similar to ossifying neurothekeoma have been called ossifying plexiform tumor, as the authors believed it was probably a lesion sui generis. 369 A variant of neurothekeoma, with epithelioid as well as spindle-shaped cells and a cellular morphology, has been reported as a ‘cellular neurothekeoma’, a term often used now to include all neurothekeomas.350.370. and 371. The authors speculated that it was an epithelioid variant of pilar leiomyoma, although only three were positive for smooth muscle actin. 370
(A) Neurothekeoma. (B) Tumor fascicles merge with the intervening dermal collagen. (H & E)
(A) Neurothekeoma. (B) This is a further case with both epithelioid and spindle cells and some nuclear atypia. (H & E)
In 1998, Busam and colleagues described an atypical cellular neurothekeoma, 356 characterized by at least one of the following features: large size, deep extension of the lesion, infiltrating borders, high mitotic activity (>3 mitoses/10 high-power fields), marked cytological pleomorphism, and signs of vascular invasion. 356 Based on a limited number of cases, it appears that surgical excision may be curative. 372
The immunohistochemical findings are different from those found in nerve sheath myxomas. Neurothekeomas do not express S100 protein, but all express NKI/C3.346.350. and 353. This latter marker is frequently positive in histiocytic tumors, limiting its usefulness. 373 The marker PGP9.5 (protein gene product 9.5) is often expressed but some experts do not use it because of its profound lack of specificity. 350 Interestingly, anti-PGP9.5 targets ubiquitin carboxyl terminal esterase L1, an enzyme said to be found exclusively in neurons. 4 In one study, 89% of cases expressed NSE, and half showed at least focal positivity for smooth muscle actin. 350 S100A6 is frequently positive. 349 Focal positivity for factor XIIIa has also been noted.374. and 375. One case expressed PG-M1, a reliable histiocytic marker. 360
The pigmented storiform neurofibroma, also known as the Bednar tumor, is now regarded as a variant of dermatofibrosarcoma protuberans that shows some neural differentiation. It is discussed further in Chapter 34, page 833.
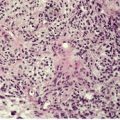
The granular cell tumor, previously known as granular cell myoblastoma, is an uncommon, benign tumor of disputed histogenesis. It may develop in many anatomical sites:379.380.381. and 382. most are found in the oral cavity, especially the tongue, and in the skin and subcutaneous tissue.379.383. and 384. Rare sites include the clitoris385 and the penis. 386 There is a female predominance and a predilection for black races. Familial cases are rare.387. and 388. The average age of presentation is 40–50 years, but the tumors may arise in children.389.390. and 391. Most lesions are asymptomatic solitary skin-colored nodules, less than 2 cm in diameter. They are multiple in about 10% of cases.389.392.393.394.395.396.397.398. and 399. Sometimes there are associated visceral granular cell tumors400 or defects in other organs.396.397. and 401. Multiple granular cell tumors have been associated with neurofibromatosis,402. and 403. with growth retardation, 391 and with Hodgkin’s disease. 404
Morphologically similar tumors are found on the anterior alveolar ridge of neonates, almost exclusively in females. 405 Occasionally they are multiple, 405 or they may involve other areas of the oral cavity. 406 These gingival giant cell tumors (congenital epulis) may regress spontaneously, or following inadequate removal. They are about one-tenth as common as the acquired variant.
Malignant granular cell tumors are exceedingly rare, accounting for only 1–3% of all acquired granular cell tumors.379.407. and 408. Very few have been reported in the skin.409.410.411.412.413.414.415. and 416. They may metastasize to regional lymph nodes, or more widely.387. and 411. Some pathologists require metastasis to have occurred before accepting a case as malignant. 417 The atypical granular cell tumor is benign. 418 Of interest is the report of a patient with neurofibromatosis in whom a malignant granular cell tumor developed. 419
Some immunohistochemical studies have been interpreted as indicating an origin from a ‘neural crest-derived peripheral nerve-related cell’, 420 but most investigators still favor a Schwann cell origin.382. and 421. Ultrastructural studies of gingival giant cell tumors of the newborn have suggested an origin from undifferentiated mesenchymal cells;406. and 422. by analogy, a similar origin has been proposed for the acquired variant. 406 These theories are not mutually exclusive.420. and 423.
The polypoid granular cell tumors of the skin described by LeBoit et al are thought to be from a different cell lineage. 424 Lazar and Fletcher have since reported a further 13 cases of this entity, which they called ‘primitive nonneural granular cell tumors’. 425 Their cases ranged in age from 5 to 83 years (median, 16 years). They involved mainly the trunk. They ranged in size from 0.2 to 2.8 cm in diameter and were present for months to years prior to removal. One case metastasized to a lymph node but the patient was disease-free 70 months after lymphadenectomy. 425 They do not appear to be neural or Schwannian in nature. 425
The tumors are non-encapsulated and composed of irregularly arranged sheets of large polyhedral cells with a small central hyperchromatic nucleus and abundant fine to coarsely granular eosinophilic cytoplasm (Fig. 37.15). Large cytoplasmic granules, surrounded by a clear halo (pustulo-ovoid bodies of Milian) are present in varying numbers in all tumors. 427 Dermal tumors often extend into the upper subcutis. Cells infiltrate between collagen bundles and may displace them. They surround appendages, but may extend into the arrector pili muscle. Rarely, a plexiform or dermatofibroma-like pattern is present.428.429.430. and 431. Cytoplasmic borders are not always distinct. The clear cell variant is also very rare, but focal clear cell change is not uncommon in conventional cases. 432 Prominent lymphoid nodules were present at the periphery in the clear cell variant. 432 The cytoplasmic granules are PAS positive and diastase resistant. They are also well seen with Movat’s pentachrome technique. 426 The nuclei contain one or two nucleoli. Elastosis is common in the stroma of granular cell tumors. 433
Granular cell tumor. (A) The epidermal hyperplasia may overshadow the dermal granular cells. (B) The eosinophilic granular cytoplasm is characteristic. (H & E)
The overlying epithelium often shows prominent pseudoepitheliomatous hyperplasia, which may be misdiagnosed as squamous cell carcinoma if only a superficial biopsy is available for examination. 379 Attempts have been made to quantify the morphometric differences between these two processes. 434 This epithelial response is usually not present in congenital gingival tumors, 406 and is absent in some cutaneous tumors, especially if multiple. 393 The rare granular cell tumors that extend into the epidermis are potential mimics of melanocytic neoplasia. An immunohistochemical panel will distinguish these two processes, if there is any doubt, on light microscopy. 435
Congenital gingival tumors have a more prominent vascular stroma, often with perivascular collections of lymphocytes and histiocytes. 422 The amount of stromal collagen increases as the lesion ages. Small nerve fibers are sometimes found in and around acquired granular cell tumors. 393
The polypoid granular cell tumors of the skin are mostly in the upper dermis and associated with an epithelial collarette. 425 Some tumors are situated more deeply in the reticular dermis with limited extension into the subcutis. They are composed of spindled to ovoid cells with abundant granular, eosinophilic cytoplasm and vesicular nuclei with small prominent nucleoli (Fig. 37.16). 425 Cytological atypia and scattered mitoses are often features of this tumor. 424 A majority of polypoid granular cell tumors express NKI/C3, vimentin, PGP9.5, and CD68, and some also express NSE. They are negative for S100 protein, as well as for epithelial and myoid markers.425. and 436.
(A) Primitive non-neural granular cell tumor (polypoid granular cell tumor). (B) There is some cytological atypia. (H & E)
Immunohistological studies of granular cell tumors have given conflicting results. Granular cell tumors usually express S100 protein,416.437.438. and 439. CD68, 440 low-affinity nerve growth factor receptor (NGFR-5), 386 microphthalmia transcription factor (MITF), 441 inhibin-α, 440 NSE, 420 PGP9.5, 442 and the melanoma-associated antigen NKI/C3. 420 Myelin basic protein (MBP) and calretinin have been present in some cases;386. and 443. MBP was absent in all 25 cases in one series. 420 The cells are negative for myoglobin,439. and 444. HMB-45, 441 and GFAP. 154 Melan-A is focally positive in rare cases only. 441 Some reports have suggested the presence of carcinoembryonic antigen (CEA), 444 but this appears to be a false positive result, due to a related antigen in the tumor cells.445. and 446. The cells are also positive for esterase and acid phosphatase. 426
There are no well-defined criteria for malignancy. Tumor size greater than 5 cm, vascular invasion, necrosis, and rapid growth are important indicators of malignant behavior.400. and 414. Mitoses, apoptotic cells, and pleomorphic nuclei may also be present, but not necessarily so; they are invariably absent in benign variants. 415 Cell spindling and angiogenesis have been reported in malignant lesions. So-called atypical variants have one or two of the following features: spindling of the tumor cells, necrosis, diffuse pleomorphism, prominent nucleoli, a high nuclear–cytoplasmic ratio, and a mitotic rate >2 per 10 high-power fields. 417 Three or more of these findings are present in malignant tumors. 417 The number of cells positive for Ki-67 and phosphohistone-H3 (PHH3) is higher in atypical forms.417. and 440.
The exact nosological position of the congenital lesion reported as CD34-positive granular cell dendrocytosis is currently unknown. 447 The patient had numerous papules and plaques on the face and extremities. The lesions were composed of S100-negative, CD34-positive dermal dendrocytes, with a granular morphology due to an abundance of phagolysosomes and an appearance resembling a granular cell tumor. 447 A similar case with CD34-positive cells has been reported as a congenital granular cell tumor. 388
The tumor cells are surrounded by a basal lamina. 400 The cytoplasm contains numerous granules of various sizes and shapes; the majority of these are phagolysosomes.382.444. and 448. Microfilaments and microtubules have been reported. 448 Angulate bodies may also be found in acquired tumors, 444 and sometimes these are also found in satellite fibroblasts. 449 Congenital tumors have immature mesenchymal cells as well as forms transitional between these and the granular cells. 406
Malignant peripheral nerve sheath tumor (MPNST) 2 is the preferred designation for a tumor which has in the past been called neurosarcoma, neurogenic sarcoma, neurofibrosarcoma,450. and 451. and malignant schwannoma.452.453. and 454. This latter tumor is not always included under this ‘umbrella’ term. 455 MPNST is a rare tumor, accounting for approximately 2% of all nerve sheath tumors. 456 Most cases are thought to arise by malignant transformation of a neurofibroma. 292 Only two forms of neurofibroma – the plexiform, and massive soft tissue variant – are significant precursors of MPNST. 292 However, Schwann cells are not the only component of MPNSTs. 457 Other cells contribute to the heterogeneous components present in this tumor. 457 Although tumor is sometimes present in the dermis,458. and 459. this usually represents extension of a growth that originated in the deeper soft tissues. 460 Superficial tumors have had a better prognosis than deeper ones, in most series. 461
A recently described variant is the perineurial malignant peripheral nerve sheath tumor derived from perineurial rather than Schwann cells. Only 4% of MPNSTs show perineurial cell differentiation. 462 The biological potential of the malignant perineurioma is not known, as few pure cases have been reported to date. 463
The t(X;18) translocation found in synovial sarcomas has now been reported in MPNST. 464
Neurofibromatosis is present in more than 50% of cases of MPNST.465. and 466. It was present in only 22% of patients with MPNST reported from Milan. 467 There is a predilection for the deep soft tissues of the proximal extremities of young and middle-aged adults. The mean survival for patients with these tumors is 2–3 years.468. and 469. Soft tissue sarcomas, particularly rhabdomyosarcomas, developing in children and adolescents with NF-1 have an aggressive course. 470 In the series of 205 patients from Milan, the disease-specific mortality was 43% at 10 years. 467 This series included tumors occurring in all body sites.
Treatment is usually surgical resection. The role of radiotherapy and/or chemotherapy has not been studied in a controlled fashion. 467
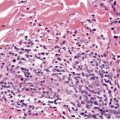
There is usually a spindle-cell growth pattern with cells arranged in tight wavy or interlacing bundles (Fig. 37.17). 471 There are sometimes densely cellular areas alternating with more loosely textured areas. 472 A rare plexiform variant has been reported. 473 The cellularity and number of mitoses determine the grading of the tumor.
Malignant nerve sheath tumor. This cellular tumor is composed of interlacing bundles of spindle-shaped cells. Several mitoses are present. (H & E)
Purely epithelioid variants have been reported (malignant epithelioid schwannoma).474.475.476.477.478. and 479. There appears to be a tendency for MPNSTs arising in a benign schwannoma to show epithelioid morphology. 480 Cartilaginous differentiation was present in one case. 481 Metastatic melanoma needs consideration in the diagnosis. 482
Focal divergent differentiation, with the formation of foci of osteogenic sarcoma, chondrosarcoma, angiosarcoma, 483 rhabdomyosarcoma, or an epithelial element, is present in about 15% of tumors.484. and 485. The epithelial element is usually glandular in type. 303 The presence of rhabdomyosarcomatous elements can be confirmed by immunoperoxidase stains for myoglobin, 486 desmin, muscle-specific actin, and myogenin. 487 This variant is known as a ‘triton tumor’.479.486.487. and 488. More than half of the patients have NF-1. 487 It has a worse prognosis than the classic MPNST. This may be due to its expression of c-myc. 487 A breakpoint involving 11p15 (the region of the myogenic differentiation 1 gene) has been identified in one case. 489 It should not be confused with the rare neuromuscular hamartoma of infancy composed of nerve fibers admixed with well-differentiated skeletal muscle. 490
In the past, the diagnosis of a malignant nerve sheath tumor was difficult, particularly in the absence of a clinical history of neurofibromatosis, and in those tumors in which no anatomical relationship to a nerve trunk could be demonstrated. The development of immunoperoxidase techniques has assisted considerably in making a specific diagnosis. 491 These tumors contain S100 protein, neuron-specific enolase, neurofilament protein, and myelin basic protein, although sometimes this staining is weak.5.451. and 454. Some cases do not express S100 protein. 457 Tumor cells do not express melan-A. 492 Vimentin, AE1/AE3, SOX 10, and EMA may also be present.152.154. and 493. Expression of CD117 is extremely uncommon.494. and 495. Whereas the majority of monophasic synovial sarcomas stain for one or both of cytokeratins 7 and 19, most MPNSTs do not express these cytokeratin subsets. 493 A high Ki-67 labeling index (>25%) is associated with a reduced survival rate. 496
The perineurial variant is composed of spindle cells with long processes disposed in whorls or storiform patterns. Necrosis is sometimes present. Their characteristic immunohistochemical profile is EMA positive, S100 negative.462. and 463.
The unsatisfactory term ‘nasal glioma’ refers to the presence of heterotopic neural tissue, predominantly glial in nature, at or near the root of the nose. Approximately 60% of these lesions are confined to the subcutaneous tissue, whereas 30% are intranasal in location.498.499. and 500. The remainder have both external and intranasal components. In approximately 20% of cases intracranial connections are present, sometimes with an associated bony defect in the nasofrontal region,501. and 502. but there is no fluid-filled space connecting with the ventricular system, as in an encephalocele. Nasal gliomas present at birth, or in early infancy, as a red to blue, firm smooth tumor near the bridge of the nose. Those confined to the intranasal region may present later with nasal obstruction or epistaxis, or as a nasal polyp.
Rarely, similar tissue has been reported in the scalp, 503 either as a midline parietal nodule504 or as multiple subcutaneous nodules in the scalp. 505 A ring of dark long hair encircling a congenital scalp lesion (the ‘hair collar sign’) is sometimes present as a marker of an associated encephalocele or heterotopic brain tissue. Hair anomalies may also overlie sequestrated meningoceles. 506 Heterotopic glial tissue has been reported in the subcutaneous tissues overlying T12. There was no associated spinal defect. 507 The term ‘heterotopic neural tissue’ is the preferred collective term for these lesions, and for nasal gliomas. 504 They are hamartomas, resulting from sequestration of neural tissue early in embryogenesis. 508
Encephaloceles are abnormal herniations of brain tissue through a bony defect. 509 They are broadly divided into anterior (facial) and posterior (scalp) lesions. In general, an anterior lesion portends a more favorable outcome. 510 Anterior lesions may rarely present as nodules on the cheek. 510
Spinal dysraphism refers to a spectrum of congenital anomalies in which there is incomplete fusion of the midline mesenchymal, bony, or neural elements of the spine.511. and 512. Cutaneous defects may also occur. Occult spinal dysraphism is the term used for skin-covered lesions without exposed neural tissue. This defect occurs predominantly in the lumbosacral region. Markers of occult spinal dysraphism include lipomas, dermal sinuses, midline dimples, hemangiomas, and hypertrichosis (faun tail).511. and 513.
Ependymal rests are relatively common lesions arising in the skin and subcutaneous tissues of the sacrococcygeal region. 514 There may be overlying dimples or pores. They represent remnants of the embryonic filum terminale externum. 514 In one case the ependymal rest presented as a skin tag; this may have been related to the concomitant eccrine hamartoma. 514 The subcutaneous sacrococcygeal myxopapillary ependymoma arises from these ependymal rests.
The phakomatous choristoma is a rare congenital hamartoma of lens tissue. 515 It presents in newborns or young infants as a subcutaneous mass in the medial lower eyelid. About 20 cases have been reported. They consist of large cuboidal cells with faintly eosinophilic cytoplasm arranged in nests, tubules, and cords set in a fibrotic stroma. Some nests contain proteinaceous pink material, with secondary dystrophic calcification. Some cells show vacuolization and degenerative changes. The epithelial cells express S100 protein but not cytokeratin or epithelial membrane antigen. 515
The meningeal–cutaneous relationships in anencephaly have been reviewed. 516 This study is mentioned for completeness.
In nasal gliomas there are islands of neural and fibrovascular tissue in the subcutis. The neural tissue is composed of astrocytes enmeshed in a neurofibrillary stroma (Fig. 37.18). Multinucleate astrocytes are not uncommon, but neurons are usually inconspicuous and few in number. They were the predominant element in one case. 518 The nodules are interlaced with vascular fibrous septa. A markedly sclerosed stroma may be seen in older lesions. 519 Calcification and sweat duct hyperplasia are two other rare associations. 520
Heterotopic glial tissue admixed with collagen bundles. (A) This lesion was removed from the occipital region. (H & E) (B) The pale pink glial fibers contrast with the green stromal collagen. (Masson trichrome) (C) Another case with an inset stained for glial fibrillary acid protein. (H & E with immunoperoxidase inset)
Heterotopic neural islands occurring on the scalp may be surrounded by a capsule that structurally resembles the leptomeninges.504. and 521. Clusters of ependymal cells with central vascular channels and myxoid stroma (myxopapillary ependymal rests) are found rarely in the sacrococcygeal region. 522
Immunoperoxidase staining demonstrates glial fibrillary acid protein (GFAP) and S100 protein in the glial tissue. 519
Meningiomas (cutaneous meningothelial tumors, cutaneous heterotopic meningeal nodules) are found only rarely in extracranial sites, including the skin.523.524.525.526.527. and 528. The scalp, forehead, and paravertebral areas are most commonly involved. Three distinct clinicopathological groups have been recognized. 526Type I lesions arise in the subcutaneous tissue of the scalp, forehead, and paravertebral region. They are usually present at birth, and are thought to be derived from ectopic arachnoid cells misplaced during embryogenesis. A familial occurrence has been reported. 529 This group includes lesions with only scattered meningothelial cells in a collagenous nodule (acelic meningeal hamartoma), lesions with a rudimentary cystic channel (rudimentary meningocele variant), and those with well-circumscribed nodules resembling intracranial meningiomas.506.509.530.531.532.533.534.535.536. and 537. Type II lesions are found in adults around sensory organs of the head (periorbital, aural, and paranasal) and along the course of cranial and spinal nerves. They are thought to be derived from nests of arachnoid cells which are found along the course of these nerves after they penetrate the dura. Type III lesions represent the direct cutaneous extension or distant metastasis of an intracranial tumor.526.538. and 539.
Primary cutaneous meningioma has been reported in association with von Recklinghausen’s disease, 540 and in association with a sinus pericranii. 541 Another rare presentation is as an aural polyp. 542
Whereas type I lesions are usually confined to the subcutis, type II and III tumors may also involve the dermis. Some type I lesions consist merely of irregular strands of meningothelial cells set in a collagenous stroma, 543 whereas others resemble type II and III tumors in being circumscribed and more cellular, akin to intracranial meningiomas and including spindle-cell areas and meningothelial whorls. Psammoma bodies are variable in number. Inflammatory cells may be present in type II and III lesions.
When meningoceles and related malformations are studied, a range of tissue types can be identified, including dura-like tissue, blood vessels, lipoma formation, hypertrophy of the arrector pili muscle, nerve fibers, meningothelial cells, and neural tissue. 509 The infant who presented with several nodules on the face composed of meningeal tissue, smooth muscle, and connective tissue was regarded as a hamartoma of ‘cephalic neural-crest derived tissues’. 544
Immunoperoxidase studies for S100, vimentin, and epithelial membrane antigen have been positive.523.524. and 545. A recent case was S100 negative but positive for epithelial membrane antigen and vimentin. 537 Ultrastructural studies have shown a similar appearance to intracranial meningiomas.525. and 526.
Barr and colleagues have described three cases of an unusual cutaneous tumor with a superficial resemblance to a meningioma or the canine hemangiopericytoma. 546 The tumors had a whorled configuration of spindle cells, some concentrically arranged around blood vessels. Only vimentin positivity was present.
Cutaneous neoplasms of neuroendocrine origin are almost invariably malignant. Malignant tumors may be primary – neuroendocrine carcinoma, Merkel cell carcinoma – or secondary, usually from the lung. As the earlier literature used the terms neuroendocrine carcinoma and Merkel cell carcinoma synonymously, it is difficult, in retrospect, to comment on these early cases. Furthermore, immunoperoxidase markers were not at the sophisticated level they are today.
Primary neuroendocrine carcinoma of the skin is exceedingly rare. There is a recent case involving the male nipple that was initially misdiagnosed as Merkel cell carcinoma because of their histological similarities. 547 As the tumor expressed neuroendocrine markers (synaptophysin, NSE, and CD56), but not CD20, it was reclassified as neuroendocrine carcinoma. 547 The other neuroendocrine marker, chromogranin A, was not performed.
Before discussing Merkel cell carcinoma, a brief description of the rare cutaneous paraganglioma and neuroendocrine adenoma will be given.
Paragangliomas are neuroendocrine tumors derived from the autonomic nervous system. 548 Approximately 10% are extra-adrenal in location. Carotid body paragangliomas are one such group. The first case of a primary cutaneous paraganglioma was reported in 2006. 548 It was a dermal nodule on the scalp of a 10-year-old boy. There was no evidence of the multiple endocrine neoplasia syndrome.
Paragangliomas are characterized by nests of large polygonal cells with central oval nuclei and abundant eosinophilic and granular cytoplasm. 548 The nests are separated by thin fibrous septa, giving a ‘zellballen’ (cell-ball) appearance.
The cells express vimentin, neuron-specific enolase, chromogranin, and synaptophysin. 548
Neuroendocrine adenomas are rare tumors of the middle ear of uncertain nature. A case has been reported in the external ear that presented as a granular mass composed of closely packed glands, in a somewhat trabecular pattern. The glands were lined by cuboidal cells with abundant granular cytoplasm. 549 An amorphous pink substance was present in scattered glandular lumina. Pagetoid spread of tumor cells was present, but the tumor behaved in a benign fashion.
The cells expressed neuron-specific enolase and synaptophysin but not S100 protein, cytokeratin 20 (CK20), or chromogranin. 549
In 1972, Toker reported five cases of this tumor as trabecular carcinoma. 550 Some years later, on the basis of the electron microscopic findings of neurosecretory granules, Tang and Toker suggested that the tumor had a Merkel cell origin. 551 Because a Merkel cell origin is not established beyond doubt, 552 and because a trabecular pattern is not usually a dominant feature, the designation ‘neuroendocrine carcinoma’ has been favored by some.553. and 554. Cases continue to be reported that indicate that neuroendocrine carcinoma is not synonymous with Merkel cell carcinoma.547. and 555. Another term used is ‘primary small-cell carcinoma of the skin’. 556 Most reports in the literature use the term ‘Merkel cell carcinoma’.
Merkel cells, the presumptive cell of origin of Merkel cell carcinoma, are primarily localized in the basal layer of the epidermis, and they are concentrated in touch-sensitive areas in glabrous and hairy skin. 557 They cannot be identified by standard histological staining (H & E). Immunohistochemistry or electron microscopy analysis is required. 557 They take part in mechanoreception, but little is known about their interactions with other epidermal cells. They synthesize numerous peptides. 557 Interestingly, in a study using rats, the electromagnetic radiation from a cellular telephone led to significantly higher exocytotic activity in Merkel cells compared with a control group. 558
Merkel cell carcinoma usually arises on the sun-exposed skin of elderly patients, particularly on the head and neck, and extremities.559.560. and 561. Perianal and eyelid involvement have been recorded.562. and 563. The synchronous onset of multiple cutaneous lesions, localized to the scalp, has been reported. 564 It occurs, rarely, in children. 565 Its incidence in the United States in 2001 was estimated to be 0.44 cases per 100 000. 566 Rare associations include chronic lymphocytic leukemia,567.568. and 569. chronic arsenicism,570. and 571. HIV infection,572.573. and 574. postmastectomy lymphedema, 575 sarcoidosis, 576 a paraneoplastic syndrome, 577 therapeutic immunosuppression,578.579. and 580. and ectodermal dysplasia.581. and 582. It is uncommon in organ-transplant recipients. 583 There is a relatively high incidence of second neoplasms in patients with Merkel cell carcinoma.569.584. and 585. A collision tumor composed of Merkel cell carcinoma and lentigo maligna melanoma has been reported. 586 Two cases of Merkel cell carcinoma of the parotid, each associated with a Warthin tumor, have been reported. 587 There has been a female preponderance in some series and a predilection for males in others.588.589.590.591. and 592. The tumors are often clinically indistinguishable from other skin cancers. They may have a reddish, nodular appearance which sometimes resembles an angiosarcoma or granulation tissue.593. and 594. They average 2 cm in diameter, although ‘giant’ and small variants have been recorded.595. and 596. The acronym AEIOU (asymptomatic, expanding rapidly, immune suppression, older than 50 years, and ultraviolet-exposed site) has been proposed as a useful reminder of the clinical features. 597
Local recurrences occur in about one-third of cases. The tumor spreads to the regional lymph nodes in up to 75% of cases,559.598. and 599. and distant metastasis with eventual death occurs in one-third or more. 600 Sentinel lymph node biopsy detects tumor spread in one-third of patients whose tumors would have otherwise been understaged.601. and 602. There was significant prognostic benefit of adjuvant nodal therapy, but only when the sentinel lymph node biopsy was positive. 602 Common metastatic sites include regional lymph nodes, skin, liver, lungs, bones, and brain. 603 Leukemic dissemination of tumor cells has been reported in a patient with systemic lupus erythematosus. 604 Spontaneous regression of the tumor has been reported.605.606.607.608. and 609. In a large series reported from the Memorial Sloan-Kettering Cancer Center, the overall 5-year disease-specific survival rate was 75%. 610 In another series, the 5-year relative survival was 75%, 59%, and 25% for localized, regional, and distant Merkel cell carcinoma, respectively. 590 Female sex, limb presentation, localized disease, and younger age were positive predictors of survival. 590 In another study, adverse prognostic features included tumor size, invasion into subcutaneous fat, and a heavy lymphocytic infiltrate. 592 Advanced clinical stage is another adverse prognostic feature. 611 No significant difference in outcome has been found in patients who have Merkel cell carcinoma in combination with another cutaneous tumor. 612 Merkel cell carcinoma involving lymph nodes and lacking an identifiable primary site may result from regression of a cutaneous primary, although an origin in lymph nodes may be the explanation in other cases.613. and 614. Cases in other ectopic sites have also been reported. 614
Ectopic peptide production is not uncommon (see below), 615 but the levels are not high enough to produce a clinical endocrinopathy. 616 Paraneoplastic syndromes are rare. 617
Nearly 50% of Merkel cell carcinomas exhibit trisomy 6. 618 A distal deletion involving chromosome 1p35–p36 is also quite common, a feature shared with other neoplasms of neural crest derivation. 619 In one-third of cases there is loss of the whole of chromosome 10 or part of its long arm. 620 Other chromosomes implicated include 18q and 20. 621 The T1796A BRAF mutation is absent. 622 The chemoresistance gene ABCG2 is expressed only very focally, and in a small number of cases of Merkel cell carcinoma. 623
Now that tumor cell lines have been established in the laboratory, it is hoped that further studies may give some insight into the origin of this enigmatic tumor.624. and 625. There is no evidence of an association with Epstein–Barr virus. 626 In 2008, a report appeared linking Merkel cell carcinoma to a new polyomavirus which the authors called Merkel cell polyomavirus (MCV). 627 They detected MCV sequences in 80% of the tumors (10) studied. 627 They concluded that the virus ‘may be a contributing factor in the pathogenesis’ of this tumor. 627 The kinetics of this tumor have been studied. 628 There is superficial aneuploid DNA content. Apoptosis follows proliferation in superficial compartments, being less variable and proliferation independent in the deeper compartments. 628 The WNT-signaling pathway is not implicated in tumorigenesis. 629
It is generally agreed that the primary tumor should be treated aggressively.630. and 631. The treatment of choice for Merkel cell carcinoma is wide surgical excision combined with radiotherapy.591. and 632. Adjuvant nodal therapy appears to have a benefit when sentinel lymph node biopsy is positive. More specific recommendations were made in a 2007 report that recommended margins of 1 cm when the tumor was <2 cm diameter and 1–2 cm margins for larger tumors. 633 Radiotherapy to the primary site was recommended for tumors >2 cm in diameter. This contrasts to other studies that have found that adjuvant local or locoregional radiotherapy has resulted in a better disease-free survival compared with surgery alone.634.635. and 636. Tumor size was not specifically mentioned in these reports. Radiotherapy alone for inoperable tumors is the treatment of choice.637.638. and 639. Earlier studies had suggested that the role of postoperative radiotherapy was unclear, 640 and that surgery was the primary treatment. 566 In support of this is a study using Mohs micrographic surgery, in which radiotherapy seemed to confer no additional benefit. 641
There is an anecdotal report of the complete remission of Merkel cell carcinoma of the scalp with local and regional metastases after topical treatment with dinitrochlorbenzol. 642 It has been said that systemic chemotherapy should be restricted to clinical trials, 636 although in the past it was used for patients with metastatic or locally advanced disease. 643 A high incidence of toxic death due to chemotherapy has been reported in the literature. 644 However, the recent finding of mutations in the platelet-derived growth factor receptor alpha (PDGFRA) gene in one-third of the cases tested raises the possibility that the kinase inhibitor imatinib mesylate may be effective in these cases. 645
The tumor is composed of small, round to oval cells of uniform size with a vesicular nucleus and multiple small nucleoli (Fig. 37.19). Mitoses and apoptotic bodies are usually numerous. 647 The cytoplasm is scanty and amphophilic and the cell borders are vaguely defined. Tumor cells may be larger in recurrences after radiotherapy. 648 An initial large cell variant also occurs. 649 A few tumors have scattered areas with spindle-shaped nuclei. The cells are present as sheets and solid nests, infiltrating the entire dermis and sometimes extending into the subcutis (Fig. 37.20). Some authors have attempted to define three distinct patterns of histological differentiation: trabecular, small cell, and intermediate. Overlap features are common and the trabecular pattern may be limited to the peripheral areas of the tumor. 650 Pseudorosettes are quite uncommon. 651 There is a dissociation of tumor cells in poorly fixed areas. Other features include focal necrosis, particularly in large tumors, frequent involvement of dermal lymphatics, 650 and a scattered infiltrate of lymphocytes and sometimes plasma cells. Vascular proliferation is present in approximately 20% of cases. 652 Increased vascular density is associated with a worse prognosis. 653 Strongly hematoxyphilic blood vessels, due to nuclear DNA deposition from tumor necrosis (the Azzopardi phenomenon), are most uncommon. 654 Stromal desmoplasia is a rare finding. 655 Amyloid has been demonstrated in a few tumors.656. and 657. Focal argyrophilia is sometimes present, and this is enhanced by fixation in Bouin’s solution. 647
(A) Merkel cell carcinoma. (B) The cells are small, with indistinct cytoplasmic borders and a hyperchromatic nucleus. (H & E)
Merkel cell carcinoma. Sheets of small cells with hyperchromatic nuclei extend throughout the dermis. (H & E)
The overlying epidermis is ulcerated in about 20% of cases. Sometimes there is epidermal hyperplasia. Epidermotropism of tumor cells is uncommon.552.583.658.659. and 660. Epidermal involvement was present in 11 of 132 cases in one series. 661 Several cases have been reported in which the tumor cells were confined exclusively to the epidermis.662. and 663. Cases with prominent epidermal involvement have been used as evidence of a Merkel cell origin of these tumors. 659 Overlying and contiguous Bowen’s disease has also been reported,559.664. and 665. as has the admixture of a Merkel and squamous cell carcinoma.666.667. and 668. No transition was seen between the two cell types. 667 Bowen’s disease and concurrent intraepidermal Merkel cell carcinoma have been reported. There was no dermal component of either tumor. 669 Another rarity is the finding of a tumor composed of Merkel, squamous, and basal cell carcinoma. 670 Areas that resemble basal cell carcinoma are not uncommon. 671 This can lead to misdiagnosis on small biopsy specimens, and on frozen sections.671. and 672. Patients with Merkel cell carcinoma have often had numerous skin cancers in the past. 667 Small swirls of squamoid differentiation, 673 and even sweat duct differentiation,661.664.674. and 675. are occasionally seen within the tumor itself. Merkel cell carcinoma with squamous and sarcomatous differentiation is another rare finding. 676 Tripartite differentiation (squamous, glandular, and melanocytic) has been recorded. 677 This has led to the suggestion that the tumor arises from a primitive cell that can differentiate in either a neuroendocrine or a sudoriferous direction. 664 Prominent microcystic features mimicking eccrine carcinoma have been reported. 678 Other exceedingly rare patterns of differentiation include skeletal muscle, leiomyosarcomatous, fibrosarcomatous, lymphoepithelioma-like, and atypical fibroxanthoma-like.679.680.681.682.683.684. and 685. Well-differentiated follicular structures were present in one of these cases. 681 They have since been reported in the more usual type of Merkel cell carcinoma. 686 Cases of Merkel cell carcinoma arising in an epidermal cyst,687. and 688. and others associated with a trichilemmal cyst have been reported.689. and 690. In-situ Merkel cell carcinoma was present in a trichilemmal cyst in one case. 691
Examination of the sites of a regressed lesion shows perivascular lymphocytes, some lymphoid nodules, variable numbers of foamy histiocytes, and some fibrosis. No tumor cells remain. 609 In partial regression, there is usually a heavy lymphocytic infiltrate with prominent apoptosis of tumor cells and some fibrosis. 692
Histological features associated with a worse survival include lymphovascular invasion, high tumor mast cell numbers, 693 increased vascular density, 653 small cell size, and a high mitotic rate. 611 The expression of bcl-2, bax, and p53 did not correlate with survival in one study, 694 although in another small series, the presence of p53 did correlate with increased recurrence/death. 695 This tumor expresses Cox-2 in low levels and its expression is not a prognostic factor. 696 The expression of p63, survivin, and Ki-67 are associated with a worse prognosis.697.698.699. and 700. High expression of certain matrix metalloproteinases (MMP-1 and MMP-3) may influence the invasive and metastatic potential of this tumor. 701
Merkel cell carcinomas need to be differentiated histologically from anaplastic primary and secondary tumors; therefore there have been numerous immunoperoxidase studies assessing the specificity of various markers.702. and 703. The variability in the reported results for the same marker probably reflects the different sensitivity of the techniques used and the level of specificity of the monoclonal antibodies. Most tumors are positive for neuron-specific enolase673. and 704. and epithelial membrane antigen. 702 Cytokeratin is usually present as paranuclear globules (Fig. 37.21),705.706.707. and 708. which are present in mitotic as well as interphase cells. 709 Both the CAM5.2 and CK20 stains will show this paranuclear dot-like immunoreactivity.710. and 711. Cytokeratin 20 (CK20) was initially regarded as a sensitive and specific marker for Merkel cell carcinoma, which allowed distinction from other small cell carcinomas.712.713.714. and 715. However, one study has shown that one-third of small cell carcinomas of the lung are positive for CK20. 716 In this same study, 28 of 33 of the lung tumors stained for thyroid transcription factor-1 but none of the 21 Merkel cell carcinomas stained. 716 Other studies have confirmed the value of a negative TTF-1 in distinguishing Merkel cell carcinoma from other small cell carcinomas.717. and 718. Positive staining for CK20 and neurofilaments (NF), performed in association with negative TTF-1, completes this distinction between the two tumors. 718 MOC-31, an antibody that recognizes epithelial cell adhesion molecule (Ep-CAM), can be positive in both tumors. 719 A series of seven cases of CK20−/CK7+ Merkel cell carcinomas has been described. 720 All tumors expressed synaptophysin. 720 CK20 can also be used for the detection of micrometastases in lymph nodes. 721 It improves the detection of these deposits.722. and 723. Tumor cells may also stain with Ber-EP4, an antibody directed against an epithelium-specific membrane antigen. 661 CD117 is expressed in 55–95% of cases, as granular cytoplasmic staining.710.724.725.726. and 727. Nuclear HUR is expressed in Merkel cell carcinoma and normal skin, while cytoplasmic staining is found only in a subset of tumors, but not normal skin. 728 Neurofilaments, 729 chromogranin,730.731. and 732. synaptophysin, guanine nucleotide-binding proteins, 733 CD171, 734 CD24, 734 CD56, 735 and polypeptides such as calcitonin, 736 gastrin, 737 somatostatin, and corticotropin are sometimes present (Fig. 37.22). 553 Chromogranin was present in all 20 cases in one series. 738 CD99 was detected in 11 of these cases. 738 Expression of CD44 may indicate a metastatic potential. 739 Pax5, a marker of B-cell lineage, is expressed in Merkel cell carcinomas.740. and 741. So too is terminal deoxynucleotidyl transferase (TdT), which is also expressed in some lymphomas/leukemias.742. and 743. Acetylcholine receptors are also expressed. 744 The tumor cells are negative for S100 protein, laminin, CD45 (leukocyte common antigen), 702 and metenkephalin,616. and 706. the latter being a marker for normal Merkel cells. Tumor cells often contain high levels of bcl-2 protein.745. and 746. The cells of secondary ‘oat-cell’ carcinomas and carcinoids, some of which are spindle shaped, usually have more cytoplasm; they may be immunoperoxidase positive for bombesin, leucine, enkephalin, and β-endorphin, markers which are absent in Merkel cell carcinomas. 703
Merkel cell carcinoma. Paranuclear filaments are seen as small globules (‘dots’). (Immunoperoxidase preparation for cytokeratin)
Merkel cell carcinoma. Apoptotic tumor cells can be seen. (H & E) The insets show dot positivity with CAM5.2 and cytoplasmic positivity for synaptophysin. (Immunoperoxidase stains)
In one case, the nodal metastasis of a Merkel cell carcinoma of the skin showed rhabdomyosarcomatous differentiation. 747
The tumor cells contain dense core neurosecretory granules in the cytoplasm, 748 but these tend to be lost in formalin-fixed material. 749 A characteristic feature is the presence of paranuclear aggregates of intermediate-sized filaments.750. and 751. Complex intercellular junctions and cytoplasmic spinous processes are also present.559.752. and 753.
The term ‘malignant primitive (peripheral) neuroectodermal tumor’ (MPNET) is the preferred designation for a rare small cell malignancy of the skin that differs from Merkel cell carcinoma on immunohistochemical and ultrastructural features.754.755.756. and 757. Other terms used for this tumor include ‘malignant neuroepithelioma’758 and ‘peripheral neuroepithelioma’.
Extraskeletal Ewing’s sarcoma (ES) and MPNET are generally regarded as two ends of a morphological spectrum of the same biological entity.759.760. and 761. They both contain the cell surface product of the MIC2 gene, CD99, and both have a consistent chromosomal translocation t(11;22)(q24;q12), found in up to 90% of all cases.762. and 763. Variant translocations, all of which involve the EWS gene on chromosome 22q12, have been reported. 762 Rearrangement of the EWS gene can be detected using new fluorescent in-situ hydridization probes for EWSR1 on paraffin-embedded tissue. 764 This gene rearrangement can also be found in desmoplastic small round cell tumor, and clear cell sarcoma. 764
Most MPNETs/EWs are situated in the deep soft tissues. 758 The subcutis and dermis are involved quite uncommonly.765. and 766. The peak incidence is in the 20s, with a slight male predilection. 767 Most involve the trunk and lower extremities. The vulva and vagina are uncommonly involved.767. and 768. They are highly aggressive tumors that eventually metastasize widely. The overall 5-year survival rate for lesions designated as extraskeletal Ewing’s sarcoma was 61% in one series. 769 A younger age and wide initial surgical excision correlated with improved overall survival. 769 Superficial Ewing’s sarcomas have a reasonably good prognosis. 770
Malignant primitive neuroectodermal tumors are composed of sheets of hyperchromatic cells with small amounts of cytoplasm. Homer–Wright rosettes are frequently present, and central neurofibrillary material may be seen in them. 766 Glycogen may be present, a feature once thought to be restricted to Ewing’s sarcoma (see below). 771 The tumor cells contain neurosecretory granules and are usually positive for neuron-specific enolase, CD99, and PGP9.5. 771 They show restricted expression of S100 protein and the various neuropeptides.
Extraskeletal Ewing’s sarcoma has cells that often contain glycogen, but there are no neurosecretory granules or well-developed desmosomes. 772 Several histological variants have been reported in skeletal tumors. 773 The cells express CD99 in a characteristic membranous pattern,762. and 774. but are negative for S100 protein and desmin. 775 CD117 and FLI-1 are sometimes expressed.495. and 767. The presence of p16INK4a alteration is associated with a poorer prognosis. 776 Merkel cell carcinomas are frequently positive for epithelial membrane antigen and cytokeratin 20 (CK20), features not seen usually in MPNETs. Neuroblastomas are usually negative for CD99. 777
A final diagnosis can usually be made only after the fluorescent in-situ hybridization studies and immunohistochemistry results have been correlated. 773 Electron microscopy is now used infrequently.
References
Introduction
1. Feigin, I, Skin tumors of neural origin, Am J Dermatopathol 5 (1983) 397–399.
2. Erlandson, RA; Woodruff, JM, Peripheral nerve sheath tumors: an electron microscopic study of 43 cases, Cancer 49 (1982) 273–287.
3. Hartschuh, W; Schulz, T, Merkel cell hyperplasia in chronic radiation-damaged skin: its possible relationship to fibroepithelioma of Pinkus, J Cutan Pathol 24 (1997) 477–483.
4. Oaklander, AL; Siegel, SM, Cutaneous innervation: Form and function, J Am Acad Dermatol 53 (2005) 1027–1037.
Nerve sheath tumors
5. Nadji, M, Immunoperoxidase techniques II. Application to cutaneous neoplasms, Am J Dermatopathol 8 (1986) 124–129.
6. Theaker, JM; Fletcher, CDM, Epithelial membrane antigen expression by the perineurial cell: further studies of peripheral nerve lesions, Histopathology 14 (1989) 581–592.
7. Smith, MEF; Jones, TA; Hilton, D, Vascular endothelial cadherin is expressed by perineurial cells of peripheral nerves, Histopathology 32 (1998) 411–413.
8. Jimenez, FJ; Grichnik, JM; Clark, RE, CD34-positive spindle cells in nerves, J Cutan Pathol 21 (1994) 189–190.
9. Weiss, SW; Nickoloff, BJ, CD-34 is expressed by a distinctive cell population in peripheral nerve, nerve sheath tumors, and related lesions, Am J Surg Pathol 17 (1993) 1039–1045.
10. Khalifa, MA; Montgomery, EA; Ismiil, N; Azumi, N, What are the CD34+ cells in benign peripheral nerve sheath tumors? Am J Clin Pathol 114 (2000) 123–126.
11. Reed, RJ; Fine, RM; Meltzer, HD, Palisaded, encapsulated neuromas of the skin, Arch Dermatol 106 (1972) 865–870.
12. Mathews, GJ; Osterholm, JL, Painful traumatic neuromas, Surg Clin North Am 51 (1972) 1313–1324.
13. Reed, ML; Jacoby, RA, Cutaneous neuroanatomy and neuropathology, Am J Dermatopathol 5 (1983) 335–362.
14. Thami, GP; Kaur, S; Kanwar, AJ, Traumatic neuroma following a human bite, Clin Exp Dermatol 27 (2002) 76–77.
15. Diaz-Cascajo, C, Multiple recurrences of cutaneous carcinomas as a consequence of local tumor spread along nerves of traumatic neuroma, Am J Surg Pathol 26 (2002) 814–816.
16. Tashiro, A; Imafuku, S; Furue, M, Traumatic neuroma of the lower lip with intraepithelial nerve fibers, J Cutan Pathol 35 (2008) 320–323.
17. Argenyi, ZB; Santa Cruz, D; Bromley, C, Comparative light-microscopic and immunohistochemical study of traumatic and palisaded encapsulated neuromas of the skin, Am J Dermatopathol 14 (1992) 504–510.