■ Punctate porokeratosis: onset in adolescence; 1- to 2-mm “seed-like” papules on palms/soles
■ Porokeratosis palmaris, plantaris, et disseminata (PPPD): onset in childhood/adolescence; occurs on palms/soles initially
■ Porokeratotic eccrine ostial and dermal duct nevus: clinically resembles a nevus comedonicus of palm or sole (Fig. 6-2), but histology shows abundant cornoid lamellae arising from acrosyringium
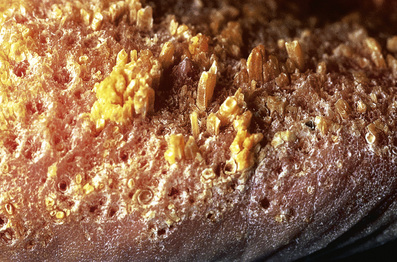
• Histology: cornoid lamella (angled column of parakeratosis w/ underlying hypogranulosis and dyskeratotic cells); centrally between two cornoid lamellae the epidermis may be atrophic, hyperplastic, normal, or BLK-like
• SCC can develop in any subtype except punctate form (0% risk); second lowest risk in DSAP; highest risk in linear form
Epidermal nevus
• Hamartoma of epidermis and papillary dermis; onset in first year of life
• Papillomatous, pigmented, linear plaques along Blaschko’s lines
■ Nevus unius lateris: extensive unilateral plaques on trunk
■ Ichthyosis hystrix: extensive bilateral lesions on trunk
■ Inflammatory linear verrucous epidermal nevus (ILVEN): along lines of Blaschko without associated neurologic defects
■ Epidermal nevus syndrome (Schimmelpenning syndrome): a/w developmental abnormalities (neurologic and musculoskeletal most commonly)
• Histology: epidermal papillomatosis; orthohyperkeratosis
■ May see epidermolytic hyperkeratosis as a result of genetic mosaicism (defects in keratins 1 and 10) → ↑risk bullous congenital ichthyosiform erythroderma in offspring
Flegel disease (hyperkeratosis lenticularis perstans)
• Rare disorder with AD inheritance; adult-onset
• Absent/altered lamellar granules (Odland bodies) on electron microscopy
• Disc-shaped keratotic papules in symmetric distribution; distal extremities including palms/soles
• Histology: discrete orthohyperkeratosis overlying atrophic epidermis; lichenoid dermal inflammation
Warty dyskeratoma
• Onset in fifth to seventh decade; M > F
• Solitary verrucous papulonodule w/ central keratotic plug usually on head/neck
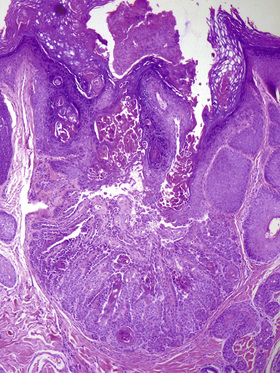
• Histology: cup-like epidermal invagination with acantholytic dyskeratosis and corp ronds/grains (Fig. 6-3)
■ “Cup-shape” and solitary nature distinguishes from Darier’s
Premalignant/malignant
Actinic keratosis
• Scaly, red plaques on sun-exposed areas; a/w chronic sun-exposure, male gender, older age, and fair skin phenotypes
• UVB responsible for AK development → induces thymidine dimers (C→T or CC→TT)
■ p53 mutations within keratinocytes → impaired apoptosis
• Histology: basal layer atypia (lower 1/3 epidermis) with budding/finger-like projections into dermis; “Flag sign”: overlying parakeratosis (pink) alternating with orthohyperkeratosis (blue); atypia and parakeratosis often spares follicles; solar elastosis in dermis
• Treatment: destructive measures (cryotherapy, ED&C, CO2 ablation, topical 5-FU, imiquimod, PDT, ingenol mebutate, topical diclofenac, and TCA peel)
Bowen’s disease (Squamous cell carcinoma in situ)
• Can progress from actinic keratosis or occur de novo
• Risk factors: elderly, chronic sun exposure, lightly pigmented skin, immunosuppression, arsenic exposure, ionizing radiation, HPV, and chronic irritation
• Hyperkeratotic erythematous patch or plaque; may affect any site
• Histology: acanthosis with full-thickness keratinocytic atypia, disorganized (“windblown”) architecture, ↑mitoses, dyskeratotic keratinocytes, and parakeratosis
• Variants: pigmented, pagetoid, verrucous, Bowenoid papulosis (multiple hyperpigmented penile papules, rarely progresses to invasive SCC), and erythroplasia of Queyrat (juicy red, erosive plaques on glans penis; more often progresses to invasive SCC)
Invasive squamous cell carcinoma
• Erythematous scaly papulonodule/plaque; most commonly on head/neck and dorsal extremities
• Risk factors: chronic sun-exposure, male gender, older age, fair skin phenotypes, genetic syndromes, immunosuppression, HPV, radiation, chronic nonhealing wound (Marjolin’s ulcer), hypertrophic LE/LP, arsenic exposure, and chronic LS&A (genital)
• Histology: full-thickness keratinocytic atypia w/ dermal invasion; tumor often “paradoxically differentiated” (tumor cells are MORE eosinophilic/keratinizing than surrounding keratinocytes)
■ Variants: poorly differentiated, spindle cell, acantholytic, pseudoglandular, Bowenoid, and verrucous
• Treatment: WLE, Mohs, ED&C, and radiation
• ↑Risk of metastasis: immunosuppressed state, location on lip/ear, diameter >2 cm, Breslow depth >2 mm, arising in burn/scar (Marjolin’s ulcer), poorly differentiated, and acantholytic (debatable)
■ ↑risk of SCC: patients w/ CLL, tobacco users, vemurafenib, long-term voriconazole prophylaxis, RA patients on MTX and etanercept, and organ transplant (65 times ↑risk)
■ Genetic syndromes associated with SCC:
○ Dystrophic epidermolysis bullosa
○ Epidermodysplasia verruciformis
○ Keratitis, ichthyosis, deafness (KID) syndrome
Verrucous carcinoma
• Low-grade, locally destructive SCC a/w HPV-6 and HPV-11
• Large exo-endophytic nodule; three clinical variants:
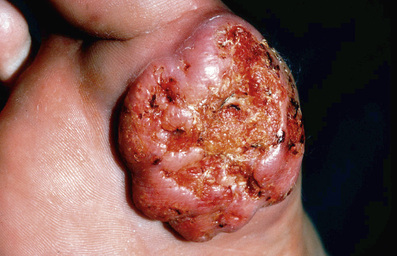
■ Epithelioma cuniculatum: slow-growing mass plantar foot (Fig. 6-4)
■ Buschke-Lowenstein tumor (giant condyloma): large cauliflower-like growth in anogenital region
■ Oral florid papillomatosis: widespread oral lesions
• Histology: very well-differentiated (minimal to no cytologic atypia); bulbous/pushing border, massive size and ↑depth of base = clues to malignancy
Keratoacanthoma
• Variant of SCC with unique features: initial rapid growth over weeks→ self-resolves/involutes over months
■ Subungual KAs are the exception (do NOT involute)
• Clinical variants: solitary, multiple, giant, intraoral, subungual, and keratoacanthoma centrifugum marginatum (can reach several centimeters)
■ Ferguson-Smith: AD inheritance, rapid onset of multiple KAs; onset third decade, sun-exposed areas, and resolves spontaneously
■ Grzybowski: sporadic; 1000s of milia-like KAs in later adulthood; can involve airway; a/w scarring, ectropion, and mask-like facies
○ Mnemonic: “Old (later onset) Grizzlies Growl (airway affected)”
• Other associations: Muir-Torre syndrome (classic KAs, or KAs w/ sebaceous differentiation), immunosuppression, and HPV
• Histology: crateriform, endophytic nodule w/ well-differentiated keratinocytes (lacks significant atypia), central keratin plug, and peripheral inflammation w/ eosinophils
• Treatment: excision or Mohs; may observe if certain involuting
Basal cell carcinoma
• Onset typically sixth to seventh decade, but can occur earlier; slow/indolent local growth; locally destructive (esp. morpheaform, infiltrative, and micronodular subtypes)
• Due to UV exposure (intermittent and intense > chronic and cumulative)
• PTCH (chromosome 9q) mutations (most common) > p53 point mutations (second most common)
• Sun-exposed skin, rare on palms, soles, and mucous membranes
• Numerous clinicopathologic variants (see Table 6-3)
Table 6-3
Basal Cell Carcinoma Variants
| Nodular | Favors head/neck; histology: large nests (centrally +/− necrosis, cystic spaces); centrally, cells lack organization, prominent peripheral palisading, and may be ulcerated |
| Superficial | Erythematous scaly patch, most common type in younger patients, trunk and extremities (>head/neck); histology: multiple buds from epidermis do not extend beyond papillary dermis |
| Morpheaform | Scar-like pink to white plaque; histology: small angulated nests and cords within a sclerotic stroma; retraction not prominent; may be more deeply invasive |
| Micronodular | Smaller nests than nodular type; micronodules are separated by normal intervening collagen, and does not form a circumscribed contour at the deep aspect |
| Fibroepithelioma of Pinkus | Pedunculated, “soft/fleshy” lesion on lower back; histology: thin anastomosing strands form a network within pinker stroma; retraction and myxoid material are less prominent |
| Pigmented | Nodular pattern BCC with aggregates of melanin in the nests and dermal melanophages |
| Infundibulocystic (keratotic, follicular) | Well-circumscribed, comprised of basaloid and squamoid cells in anastomosing cords, w/ horn cysts → resembles benign follicular tumors (trichoepithelioma and basaloid follicular hamartoma) |
| Basosquamous | Ambiguous term w/ variable meanings; may refer to: 1) BCCs with “squamoid appearance” (pinker cells, more cytoplasm, and keratinization), 2) carcinomas with features indeterminate between BCC and SCC, or 3) collision lesions of BCC + SCC |
• General histologic features: nests of basaloid, uniform cells w/ high N : C ratio, peripheral palisading, epidermal connection (at least focally), myxoid stroma, stromal–epithelial retraction, and mitotic/apoptotic activity
• Treatment: WLE, Mohs, ED&C, radiation, imiquimod, topical 5-FU, and vismodegib (inoperable or metastatic BCC)
• Essentially no metastatic potential (dependent on stroma for growth)
■ Basosquamous subtype may behave more like SCC → ↑metastatic potential
6.2 Cysts
Epidermoid cyst
■ Firm dermal nodule with central punctum; any site, but most commonly head/neck/upper trunk
• Pathogenesis/histopathologic features
■ Derived from follicular infundibular epithelium; may arise primarily, or secondary to follicle disruption/traumatic implantation; lined by stratified squamous epithelium w/ intact granular layer and no adnexal structures in the wall (vs vellus hair cyst and dermoid cyst); laminated/flaky keratin centrally
■ Multiple epidermoid cysts may be a/w Gardner’s syndrome (often have pilomatricoma-like areas histologically)
Proliferating trichilemmal cyst/tumor
■ Slow-growing dermal nodule; scalp (90%); usually elderly women
• Pathogenesis/histopathologic features
■ Resembles trichilemmal cyst but more proliferative centrally w/ areas of multicystic architecture; well-circumscribed at periphery; variable cytologic atypia and mitotic activity
■ Mostly benign, but small percentage behave aggressively → complete removal recommended
Dermoid cyst
■ Infants; occur along embryonic fusion lines (most commonly lateral eyebrow)
• Pathogenesis/histopathologic features
■ Derived from entrapment of epidermis during embryogenesis; lined by stratified squamous epithelium with granular layer and adnexal structures (hair follicles and sebaceous glands) in cyst wall
Steatocystoma
■ Single or multiple (multiplex – AD inheritance) lesions; chest/axilla/groin; drain oily fluid if punctured
• Pathogenesis/histopathologic features
■ Lined by thin stratified squamous epithelium with no granular layer and thin bright pink corrugated (“shark-tooth”) cuticle; sebaceous glands in wall
■ Multiplex form with KRT17 mutations; a/w pachyonychia congenita type 2
Hidrocystoma
■ Translucent bluish cysts; face
• Pathogenesis/histopathologic features
■ Unilocular or multilocular cyst with low cuboidal lining +/− decapitation secretion (if apocrine); lumen appears empty
■ May be a/w Schöpf-Schulz-Passarge (multiple hidrocystomas, syringofibroadenomas, PPK, hypodontia, and hypotrichosis)
Bronchogenic cyst
■ Solitary; present at birth; suprasternal notch/anterior neck
• Pathogenesis/histopathologic features
■ Sequestration of respiratory epithelium during embryogenesis; pseudostratified, ciliated columnar cells with goblet cells; +/− smooth muscle/mucous glands/cartilage in wall
■ Main clues for boards: cartilage, smooth muscle, and ↑↑goblet cells
Thyroglossal duct cyst
■ Children/young adults; midline anterior neck; moves w/ swallowing
• Pathogenesis/histopathologic features
■ Columnar, cuboidal or stratified squamous lining with thyroid follicles in the wall (low cuboidal epithelium with bright pink contents)
■ Main clue for boards: pink thyroid follicles (pathognomonic)
Branchial cleft cyst
■ Second or third decades; lateral neck (anterior SCM, preauricular, and mandibular)
• Pathogenesis/histopathologic features
■ Pseudostratified columnar or stratified squamous epithelium with surrounding dense lymphoid tissue including lymphoid follicles w/ germinal centers
■ Main clue for boards: very prominent lymphoid aggregates/follicles
6.3 Melanocytic neoplasms
Ephelides (freckles)
• 1- to 3-mm areas of ↑pigmentation; darken w/ sun-exposure; sun-exposed areas of body, mainly face, dorsal upper extremities, and upper trunk
• More common in blonde or red haired individuals; absent at birth, but appear in first 3 years of life
• ↑melanogenesis and ↑melanin transfer to keratinocytes
• Histology: ↑basilar keratinocyte pigmentation +/− enlarged melanocytes without increased melanocyte density
• No propensity for malignant transformation, however are a marker of UV damage
Lentigo simplex
• Well-demarcated, evenly pigmented brown to black macule; any age and any anatomic site
• Histology: basal layer hyperpigmentation; elongated rete ridges with mild ↑melanocyte density
• Conditions a/w multiple lentigines:
■ Peutz-Jeghers (especially oral/perioral)
■ Bannayan-Riley-Ruvalcaba (penile)
Mucosal melanotic macule
• Compared with lentigo simplex can be more irregular and mottled
• Oral lesions usually occur in adults > 40 years old on vermillion border > gingiva, buccal mucosa, or palate; genital lesions most common on labia minora
• Histology: acanthosis; mild basilar hyperpigmentation +/− subtle increase in melanocyte density
Dermal melanocytosis
• Congenital (Mongolian spot): present at birth in most Asians and blacks; lumbosacral region; presents with (p/w) gray-blue patch (as a result of the Tyndall effect where shorter light wavelengths are reflected by melanocytes); often resolves during childhood
■ Histology: sparsely distributed elongated dendritic melanocytes in lower 2/3 dermis, lying parallel to epidermis
• Nevus of Ota: presents in first year of life or around puberty; ↑incidence in pigmented individuals (Asians and blacks); p/w coalescing gray/blue macules in V1/V2 distribution and frequent scleral involvement (60%); unilateral (90%) > bilateral; persists for life; may enlarge under hormonal influences; 10% develop glaucoma; rare malignant degeneration to uveal melanoma (perhaps higher risk in Nevus of Ota lesions with activating mutations in GNAQ)
■ Histology: elongated dendritic melanocytes more numerous than in congenital dermal melanocytosis; involves upper dermis
■ Nevus of Ito: located on the shoulder, supraclavicular, and scapular regions; essentially no risk of progression to melanoma
■ Hori’s nevus: acquired nevus of Ota-like macules bilateral zygomatic region; East Asian females
■ Sun’s nevus: acquired, unilateral variant of Hori’s nevus
○ Mnemonic: “There is only 1 Sun, but the (w)HOle face is affected in HOri’s”
• Histologically, dermal melanocytoses are distinguished from blue nevi by their ↓cellularity, poor circumscription, and lack of dermal sclerosis
Blue nevus
• Onset in childhood/adolescence, but can also occur in older patients, 25% of cellular blue nevi are congenital
• Most common sites: scalp, sacral area, and distal extensor extremities
• Derived from dermal melanocytes (persist during embryogenesis rather than populating epidermis)
• Activating mutations in GNAQ and GNA11 seen in 83%; results in downstream MAPK pathway activation
■ Same mutations are the most common mutations in uveal melanoma (46%; concomitant BAP-1 loss in uveal melanoma leads to increased risk of metastasis and death)
• Multiple blue nevi and epithelioid blue nevi (latter is much more specific) a/w Carney complex
○ Blue/gray macules or papules usually less than 1 cm
○ Elongated, dendritic melanocytes containing melanin pigment usually in the upper 2/3 dermis with associated sclerotic collagen; no junctional component
○ Blue/gray/black plaques or nodules; often larger (1–3 cm); favor buttocks or scalp
○ Dense proliferation of plump/fusiform pale gray melanocytes containing little pigment + admixed dendritic melanocytes resembling common blue nevus cells; characteristically bulges into subcutis (“dumbbell configuration”)
○ Heavily pigmented; usually seen and a/w Carney complex; histologically resembles “animal type melanoma,” but lacks mitoses and atypia
■ Malignant blue nevus (= melanoma): often arises within cellular blue nevus; scalp (#1); commonly see benign precursor within specimen; frequently have concomitant GNAQ/GNA11 mutations and BAP-1 loss (a/w more aggressive behavior, similar to uveal melanoma)
Recurrent melanocytic nevus
Balloon cell nevus
• Clinically indistinguishable from ordinary nevi
• Histology: >50% dermal melanocytes are “balloon cells” (large, pale, and polygonal melanocytes with foamy/vacuolated cytoplasm and variable pigmentation); balloon cell change is as a result of melanosome degeneration
■ Boards tip: can always identify conventional nevus somewhere within lesion
Halo nevus (Sutton nevus)
• Pigmented nevus with surrounding hypopigmented zone; most commonly second decade; most commonly on the back; most commonly benign
■ May be a/w vitiligo or melanoma (rarely) at another site
• Multiple lesions can occur idiopathically or w/ infliximab
• Histology: bland nevus w/ lymphocytes intertwined (“mingling”) with melanocytes
■ In contrast, lymphocytes form a lichenoid band (“riot police barrier”) under and around melanoma
Spitz nevus
• Acquired, usually solitary lesions in first two decades (use caution in diagnosing a patient in the fourth decade and older); most common on head/neck > extremities
■ Recently described subset of atypical epithelioid Spitz nevi with loss of BAP-1 tumor suppressor gene (“BAPomas”; have unique histology, and unlike most Spitz nevi, have BRAF mutations)
• Rapidly growing pink-red papulonodule; usually <1 cm
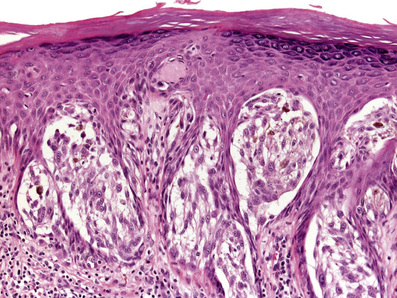
• Histology (Fig. 6-5):
■ Symmetric and circumscribed; most often compound
■ Epidermal hyperplasia (vs consumption of epidermis in melanoma)
■ Large junctional nests with clefting around entire nest (vs discohesion of nests in melanoma, where the nest itself becomes fragmented)
■ Parallel “raining-down” orientation of nests and cells
■ Kamino bodies: pink clumps of BMZ material (collagen IV) within epidermis
■ “Spitzoid” cytology: large epithelioid and spindled cells w/ abundant pink-purple (amphophilic) cytoplasm and prominent lilac-colored nucleoli (vs cherry red nucleoli in melanoma); usually not pigmented
■ Dermal component “matures” with depth (reduction in density and cell size)
■ Superficial mitoses allowable, especially in young patients → if numerous (>2–3); deep or atypical mitoses are present, raises concern for melanoma
• Immunostains: S100A6+, S100+, Melan-A+, and p16+
■ p16 is frequently lost/diminished in atypical Spitz tumors and spitzoid melanoma
• Treatment: controversial, but often complete excision is recommended
• Boards fodder: FISH analysis very helpful in risk stratification of atypical Spitzoid lesions
■ Homozygous loss of 9p21 (most predictive gene locus; corresponds to p16/CDKN2a gene) → ↑risk of metastasis and death
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


