(1)
Department of Dermatology, Freiburg University Medical Center, Freiburg, Germany
10.1.1 Trichoepithelioma
10.1.2 Trichodiscoma
10.1.3 Pilomatricoma
10.1.5 Fibrofolliculoma: A Hallmark of Hornstein-Knickenberg Syndrome (alias Birt-Hogg-Dubé Syndrome)
10.1.6 Syringoma
10.1.7 Spiradenoma
10.1.8 Eccrine Poroma
10.1.9 Cylindromatosis
10.1.10 Glomangiomatosis
10.1.10.2 Practical Aspects
10.1.11.1 Type 1 Segmental Involvement
10.1.11.2 Type 2 Segmental Involvement
10.1.11.3 Extracutaneous Type 2 Segmental Lesions
10.1.12 Lipomatosis
10.1.13 Neurofibromatosis 1
10.1.13.1 Type 1 Segmental NF1
10.1.13.2 Type 2 Segmental NF1
10.1.13.5 Other Practical Aspects
10.1.14 Neurofibromatosis 2
10.1.15 Schwannomatosis
10.1.16 Legius Syndrome
10.1.17 Leiomyomatosis
10.1.18 Gorlin Syndrome
10.1.18.1 Type 1 Segmental Involvement
10.1.18.2 Type 2 Segmental Involvement
10.1.20.4 Type 2 Segmental PTEN Hamartoma Syndrome
10.1.21 Cutaneous Mastocytosis
10.2.2 Darier Disease
10.2.3 Hailey-Hailey Disease
10.2.5 Acanthosis Nigricans
10.2.6 KID Syndrome
10.2.9.1 Practical Aspect
10.2.13 Costello Syndrome
10.2.14 Acrokeratoelastoidosis
10.3.1 Tuberous Sclerosis
10.3.1.1 Type 1 Segmental TS
10.3.1.2 Type 2 Segmental TS
10.3.1.3 Cases of Unclassifiable Mosaic TS
10.3.1.4 Genetic Counseling
10.3.2 Buschke-Ollendorff Syndrome
10.3.2.1 Type 2 Segmental Skin Lesions
10.3.3 Ehlers-Danlos Syndromes
10.3.4 Marfan Syndrome
10.3.7 Zimmermann-Laband Syndrome
10.4 Vascular Disorders
Abstract
A mosaic arrangement of skin lesions has been been noted in numerous autosomal dominant traits including hereditary multiple skin tumors, disorders of keratinization, diseases of the connective tissue or bones, and vascular disorders. Hereditary multiple skin tumors originate from allelc loss and thus represent mosaics. Multiple lesions may sometimes involve only a segmental area, in the form of type 1 mosaicism. Moreover, a pronounced segmental involvement may be superimposed on the ordinary, nonsegmental trait, in the form of type 2 mosaicsm. – Both type 1 and 2 manifestations have been documented in many autosomal dominant traits such as trichoepitheliomatosis, hereditary basaloid follicular hamartoma, multiple syringoma, multiple spiradenoma, glomangiomatosis, blue rubber bleb angiomatosis, neurofibromatosis 1, neurofibromatosis 2, leiomyomatosis, Gorlin syndrome, and hereditary non-syndromic basal cell carcinoma. – Examples of type 1 segmental involvement were reported in multiple trichodiscoma, cylindromatosis, schwannomatosis, and mastocytosis. – Cases suggesting a type 2 segmental manifestation were reported in Hornstein-Knickenberg syndrome (illegitimately named “Birt-Hogg-Dubé syndrome”), Legius syndrome, and PTEN hamartoma syndrome. Some hereditary tumor syndromes such as leiomyomatosis, neurofibromatosis 1, glomangiomatosis, and blue rubber bleb angiomatosis appear to be especially prone to develop a type 2 segmental involvement. Molecular proof of type 2 mosaicism has so far been provided in Gorlin syndrome, neurofibromatosis 1, Legius syndrome, and PTEN hamartoma syndrome. Disorders of keratinisation: Both type 1 and type 2 segmental involvement has been documented in patients with epidermolytic ichthyosis of Brocq, Darier disease, Hailey-Hailey disease, KID syndrome, disseminated superficial actinic porokeratosis, and plaque-type porokeratosis of Mibelli. Remarkably, a woman with porokeratosis of Mibelli gave birth to two sons who both had linear lesions in the form of a type 2 segmental involvement. Hence the underlying gene locus may be a hotspot for postzygotic recombination, as also assumed in disseminated superficial actinic porokeratosis. Molecular proof of type 1 mosaicism has so far been provided in epidermolytic ichthyosis of Brocq, Darier disease, and KID syndrome. Molecular evidence of type 2 mosaicism was presented in cases of Darier disease and Hailey-Hailey disease. – Cases of type 1 mosaicism with molecular proof were reported in pachyonychia congenita of the Jadassohn-Lewandowsky type and in the Galli-Galli variant of Dowling-Degos disease, and clinical examples without molecular data were reported in Costello syndrome and acrokeratoelastoidosis. – Moreover, cases suggesting type 2 mosaicism have been documented, without molecular data, in autosomal dominant acanthosis nigricans, autosomal dominant dyskeratosis congenita, and porokeratosis palmaris, plantaris et disseminata. Disorders of connective tissue or bones: Both type 1 and type 2 mosaicism has been documented in tuberous sclerosis and in autosomal dominant forms of Ehlers-Danlos syndromes. In tuberous sclerosis, molecular proof of type 1 segmental involvement was provided. Clinical features suggesting a type 2 segmental manifestation are frequently noted and described under various names such as cobblestone nevus, forehead patch, or fibrous hamartoma of infancy. – A type 1 segmental involvement in the form of pigmentary disturbances following Blaschko’s lines was documented in a girl with Brachmann-de Lange syndrome, a multisystem birth defect characterized by short stature, peculiar facial appearance, bone anomalies, and mental deficiency. – Cases suggesting a type 2 segmental involvement were reported, under the name “juvenile elastoma”, in patients with Buschke-Ollendorff syndrome. Remarkably, such pronounced segmental lesions are rather often noted in several members of a family, indicating that the underlying LEMD3 locus may be a hotspot for postzygotic recombination. Moreover, a particular bone disease called melorheostosis may occur as an extracutaneous type 2 manifestation of Buschke-Ollendorff syndrome. – Cases suggesting a type 2 segmental involvement have also been described in Marfan syndrome and Ehlers-Danlos syndrome type III. In Albright’s hereditary osteodystrophy, several persuasive examples of type 2 segmental involvement were found in the literature. – In hereditary osteomatosis cutis, cases of type 2 mosaic manifestation are so impressive that they have even got its own OMIM number 166350 under the name “progressive osseous heteroplasia”. – A girl with Zimmermann-Laband syndrome, a phenotype characterized by thick lips, bulbous nose, hypoplasia or absence of nails or terminal phalanges, increased joint mobility, hepatosplenomegaly, and mental deficiency, showed hemihyperplasia with enlargement of her external genitalia and ipsilateral segmental hypertrichosis and hyperpigmentation. A type 2 segmental manifestation is possible but not certain. Vascular disorders: In hereditary hemorrhagic telangiectasia (Osler-Rendu-Weber syndrome), one case of type 1 segmental lesions involving the face was reported. On the other hand, a case suggesting type 2 mosaicism in the form of a pronounced linear lesion involving the forehead has likewise been documented. Molecular studies are so far lacking. – Rhodoid nevus syndrome (“capillary malformation-arteriovenous malformation”) is characterized by a familial occurrence of multiple disseminated rhodoid nevi. These circular or oval lesions are often surrounded by an anemic halo. They are caused by RASA1 mutations. A type 2 segmental manifestation occurs rather frequently in the form of arteriovenous malformation with a concomitant large, segmentally arranged form of rhodoid nevus. For this reason, the phenotype had been described under the name “capillary malformation-arteriovenous malformation”, but it is now clear that the presence of an arteriovenous malformation is by no means a prerequisite for the diagnosis of the syndrome. Blistering skin disorders: Within the group of epidermolysis bullosa, revertant mosaicism is frequently noted, whereas reports on forward mosaicism are virtually absent. Two exceptional cases were found in the literature. – A self-limited form of dystrophic epidermolysis bullosa (“transient bullous dermolysis of the newborn”) is caused by COL7A1 mutations. It tends to heal spontaneously. A female neonate had linear lesions involving one of her legs, giving rise to scarring. Analysis of blood DNA revealed compound heterozygosity at COL7A1, without presence of mosaicism. Skin samples were not available for analysis, which is why the cause of this particular form of cutaneous mosaicism remains unknown. – Another peculiar mosaic phenotype in the form of superficial acantholysis arranged along Blaschko’s lines was reported by Jokiaho et al. (1989) under the term “miliaria rubra-like lesions” in a newborn girl with dysmorphism and a parietal meningocele. The linear acantholytic disorder resolved spontaneously within one week.
In autosomal dominant cutaneous traits, a type 1 mosaic involvement that originates from a postzygotic new mutation should be distinguished from a type 2 segmental manifestation being superimposed on the ordinary, nonsegmental phenotype.
10.1 Hereditary Multiple Skin Tumors
Three forms of mosaicism can be distinguished in hereditary benign cutaneous neoplasias. Firstly, all of them can be categorized as mosaic lesions that tend to originate from LOH [177]. Secondly, a type 1 segmental manifestation of these tumors is sometimes found, giving rise to a linear or otherwise mosaic pattern. Thirdly, a type 2 segmental involvement may be superimposed on the ordinary nonsegmental trait. In the following paragraphs we shall consider the two types of segmental arrangement.
10.1.1 Trichoepithelioma
Multiple trichoepitheliomas showing a bona fide type 1 segmental involvement could so far not be retrieved in the available literature. On the other hand, a convincing example of type 2 segmental manifestation has been documented by Geffner et al. [143] in a girl who later developed bilateral facial lesions. Reportedly, her mother and one of her brothers were likewise affected with trichoepitheliomas. Another case fulfilling all of the criteria of superimposed linear trichoepitheliomatosis was reported by Schirren et al. [389].
Some other cases of pronounced linear trichoepitheliomas that were present at birth (Fig. 10.1) or developed during infancy [185, 331] are likewise suggestive of a type 2 segmental manifestation.
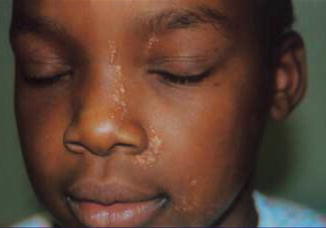
Fig. 10.1
This linear arrangement of trichoepitheliomas was present since birth [72], which is suggestive of a type 2 segmental involvement (Reproduced with permission from John Wiley & Sons, USA)
10.1.2 Trichodiscoma
10.1.3 Pilomatricoma
Multiple pilomatricomas are a feature of several hereditary syndromes, but a segmental arrangement of these tumors has not been reported so far.
10.1.4 Basaloid Follicular Hamartoma
Multiple basaloid follicular hamartoma (BFH) can be inherited as an autosomal dominant trait. Some authors prefer the name “infundibulocystic basal cell carcinoma” to categorize these benign skin tumors [78, 364]. I strongly recommend, however, to avoid this misleading term because otherwise an essentially benign tumor can easily be mistaken as a malignant neoplasia, resulting in overtreatment.
Two cases of unilateral distribution of multiple non-syndromic BFH [202, 229] can be categorized as a type 1 segmental manifestation. Two cases of superimposed arrangement suggesting a type 2 segmental involvement have also been reported (Fig. 10.2) [78, 422].
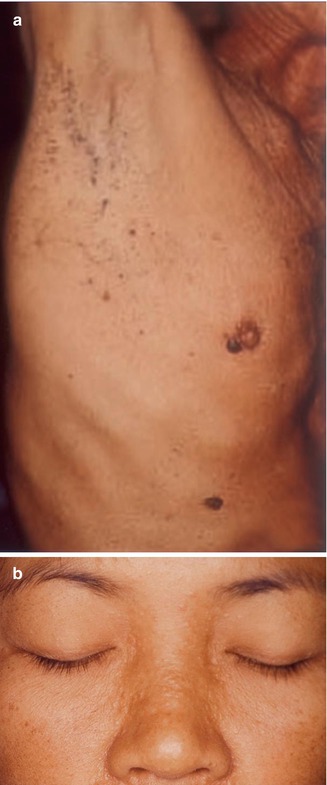
Fig. 10.2
(a) This 66-year-old man had multiple linear basaloid follicular hamartomas. Two brothers had nonsegmental lesions, and (b) his 39-year-old daughter showed likewise a diffuse involvement [78]. Because of such family constellation, a type 2 segmental manifestation is likely (Reprinted with permission from John Wiley & Sons, USA)
On the other hand, segmentally arranged basaloid follicular hamartomas are a hallmark of Happle-Tinschert syndrome (see Sect. 12.2.1.3).
10.1.5 Fibrofolliculoma: A Hallmark of Hornstein-Knickenberg Syndrome (alias Birt-Hogg-Dubé Syndrome)
Multiple fibrofolliculomas (Fig. 10.3a) are a hereditary cutaneous trait that was previously described under the name “multiple perifollicular fibromas” [81]. They are a cutaneous hallmark of a syndrome that is caused by folliculin mutations and includes lung cysts, pneumothorax, renal cysts, proneness to renal cell carcinoma, and colorectal polyps showing proclivity to malignant degeneration [301, 318]. The term “Birt-Hogg-Dubé syndrome” is an improper eponymic designation because these authors have not added any new data to what had completely and scholarly been described by Hornstein and coworkers [208–210] as a distinct autosomal dominant trait being associated with proneness to extracutaneous cancer. In fact, Birt et al. [34] referred to the pioneering work of Hornstein and Knickenberg [209]. They reiterated Hornstein’s meticulous description of perifollicular fibromatosis cutis and, by giving it another name, incorrectly maintained that they had found “a previously unrecognized hereditary pilar hamartoma.” The renaming “Hornstein-Knickenberg syndrome” gives credit to the original authors [193, 334].
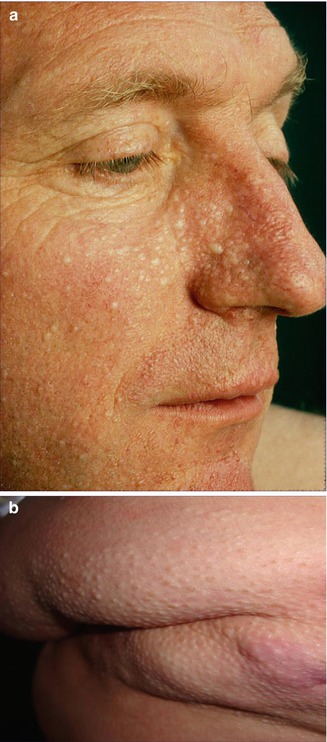
Fig. 10.3
Hornstein-Knickenberg syndrome. (a) Multiple fibrofolliculomas diffusely involving the face in 56-year-old man who died from colon carcinoma; (b) type 2 segmental involvement of the left chest wall in a 75-year-old woman. Note characteristic gooseskin-like surface of the huge lipomatous lump (b: Courtesy of Dr. Derek Lim and Dr. Eamonn R. Maher, Birmingham, UK)
10.1.5.1 Type 1 Segmental Manifestation of Hornstein-Knickenberg Syndrome
An unusual case of multiple unilateral facial fibrofolliculomas was published by Casalá et al. [66]. Whether this report can be taken as an example of type 1 segmental manifestation of Hornstein-Knickenberg syndrome cannot be said with certainty as long as molecular data are lacking.
10.1.5.2 Type 2 Segmental Manifestation of Hornstein-Knickenberg Syndrome
Possible examples of a type 2 segmental manifestation of the syndrome (Fig. 10.3b) have been documented by several authors. In a 49-year-old man with this disorder, Weintraub and Pinkus [472] described “a large connective tissue nevus” involving a unilateral segment of the thorax. The firm plaque was present “as long as the patient could remember.” Its surface showed a “pigskin-like graining,” being studded with umbilicated papules that contained central keratin plugs or hairs. Histopathologically these papules corresponded to fibrofolliculomas. Similar plaques have been noted by Toro et al. [439] in 3 out of 125 patients. Kluger et al. [246] found such plaques in 2 out of 22 patients and assumed that they may reflect a type 2 segmental manifestation of the trait.
10.1.6 Syringoma
Multiple syringomas represent an autosomal dominant trait. A type 1 segmental involvement has been documented in many reports [70, 89, 175, 382, 489]. Cases of type 2 involvement superimposed on the ordinary nonsegmental phenotype have likewise been described [241, 288, 478]. A case of a 4-year-old girl with pronounced lesions of a congenital “linear syringomatous hamartoma” [475] can likewise be categorized as an example of type 2 segmental involvement (Fig. 10.4). In an additional case, type 2 mosaicism is likely but less certain [371].
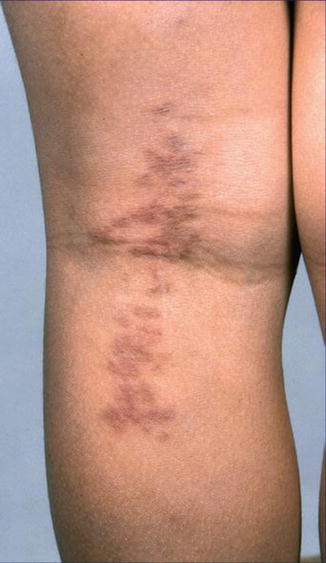
Fig. 10.4
Linear arrangement of syringomas in a 4-year-old girl [475]. Because the lesions were present at birth, a type 2 segmental involvement is very likely (Reprinted with permission from John Wiley & Sons, USA)
10.1.7 Spiradenoma
Multiple spiradenomas are inherited as an autosomal dominant trait [431]. Several cases suggesting a type 1 involvement have been reported [8, 48, 321, 487].
An unquestionable case of type 2 segmental spiradenomatosis was described in a family with multiple eccrine spiradenoma and cylindroma present in three consecutive generations [479]. In three additional cases a type 2 segmental involvement is almost certain because a pronounced segmental involvement was already present at birth (Fig. 10.5) [117, 328, 370]. Some other cases of segmentally arranged eccrine spiradenomas [28, 162, 163, 267, 405, 450, 494] are difficult to categorize according to the dichotomous types of mosaicism.
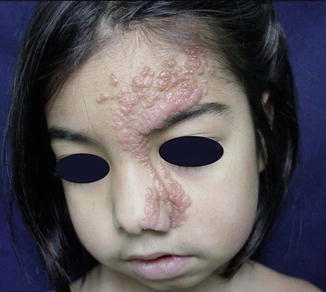
Fig. 10.5
An 8-year-old girl with congenital linear eccrine spiradenomas suggesting a type 2 segmental manifestation [117] (Reprinted with permission from John Libbey Eurotext, Montrouge, France)
10.1.8 Eccrine Poroma
A case of multiple eccrine poroma arranged in a linear pattern was described by Ogino [329].
10.1.9 Cylindromatosis
Cutaneous cylindromatosis is caused by CYLD1 mutations. A case of multiple linear cylindromas as reported by Martinez et al. [291] probably represents a type 1 segmental manifestion.
10.1.10 Glomangiomatosis
The vascular lesions of this autosomal dominant trait are true angiomas, which is why the presently prevailing term “glomuvenous malformation” [45, 52] appears to be inappropriate. Glomangiomatosis is caused by mutations in the glomulin gene [52]. It is a rather rarely occurring phenotype, but in affected families a type 2 segmental involvement is very common. Apparently, the glomulin locus represents a hotspot for somatic recombination [189]. The lesions of segmental glomangiomatosis do not follow Blaschko’s lines but tend to be arranged in a flag-like or checkerboard pattern [248, 286].
Cases suggesting a type 1 segmental involvement have rarely been reported [263, 438, 485], whereas a type 2 involvement (Fig. 10.6) appears to occur far more frequently [11, 24, 43, 53, 102, 107, 195, 205, 223, 286, 287, 303, 312, 315]. This may in part reflect a bias of ascertainment because a type 2 involvement is more conspicuous. Notwithstanding, the prevalence of such pronounced mosaic manifestation is extremely high as compared to most other autosomal dominant skin disorders. The superimposed segmental lesions tend to be much more painful than the nonsegmental glomangiomas [11, 77].
Cases of “congenital plaque-like” glomangioma [65, 232, 486] or “giant glomangioma” [411] can today be categorized as examples of type 2 mosaicism even when nonsegmental lesions are absent in the patient and his family members.
10.1.10.1 Type 2 Segmental Involvement of Internal Organs
The lesions of glomangiomatosis are usually limited to the skin. Remarkably, however, a type 2 segmental manifestation may also involve internal organs. Goujon et al. [154] described two newborn children with extensive mosaic plaque-type glomangiomatosis. They had been diagnosed in utero with pleural effusion and ascites, suggesting “a pathogenic link” between the conditions. In one of these cases, a glomulin mutation was found in the boy and his mother who had nonsegmental glomagiomatosis. No molecular analysis was perfomed in the other newborn. Similarly, Tejedor et al. [430] reported that in a newborn girl with plaque-type glomagiomatosis, isolated fetal ascites had been documented, 1 week prior to birth, by sonographic examination. After birth, the ascites was found to be of chylous origin. The patient’s father had multiple glomangiomas being grouped on his left thigh. According to present knowledge, these cases cannot be understood without accepting the theory that at the underlying gene locus, the corresponding wild-type allele has been lost at an early developmental stage.
10.1.10.2 Practical Aspects
The lesions of type 2 segmental glomangiomatosis are far more difficult to treat than the nonsegmental glomangiomas [23].
10.1.11 Blue Rubber Bleb Angiomatosis (“Blue Rubber Bleb Nevus Syndrome”)
In 1958, William Bean [27] coined the misnomer “blue rubber bleb nevus” to describe the peculiar color and consistence of cutaneous vascular tumors that also involve extracutaneous organs in the form of an autosomal dominant “blue rubber bleb nevus syndrome.” These vascular lesions, however, are true angiomas and can, therefore, not be categorized as nevi [133, 190]. The molecular cause of blue rubber bleb angiomatosis is not known as yet.
10.1.11.1 Type 1 Segmental Involvement
10.1.11.2 Type 2 Segmental Involvement
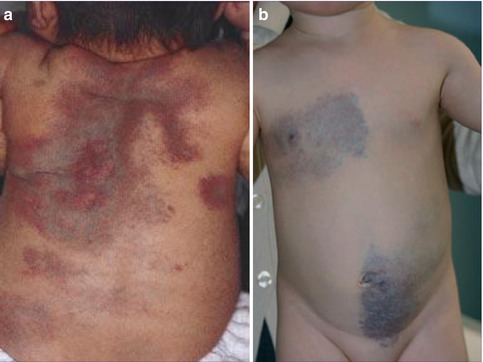
In 2010, five cases suggesting a type 2 segmental manifestation have been reviewed (Fig. 10.7) [190]. In one of these patients, the right forearm was amputated in childhood because a giant congenital blue rubber bleb angioma had rendered the limb functionless [133]. A type 2 segmental involvement can render the disorder detectable even before birth (Fig. 10.8). In such cases, prenatal sonographic surveillance revealed very large cutaneous vascular tumors that were complicated by life threatening consumptive coagulopathy soon after birth [307].
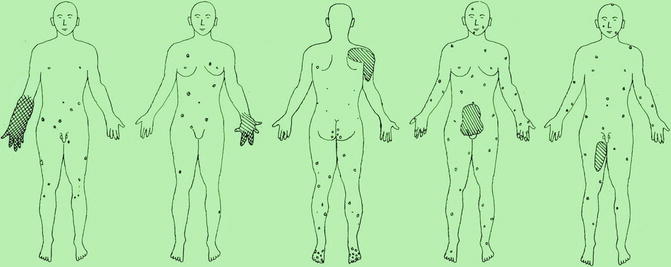
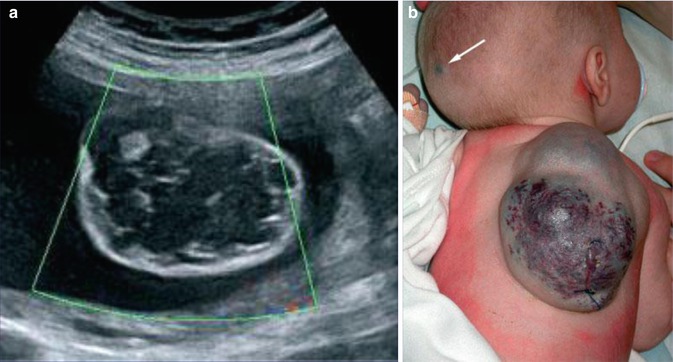
Fig. 10.7
Schematic representation of case reports suggesting a type 2 segmental manifestation of blue rubber bleb angiomatosis. Crossed hachures indicate areas of both cutaneous and subcutaneous involvement, whereas simple hachures symbolize either subcutaneous or extracutaneous lesions [190] (Reprinted with permission from John Libbey Eurotext, Montrouge, France)
Fig. 10.8
Type 2 segmental blue rubber bleb angiomatosis [307]. (a) Prenatal ultrasound image at 25 weeks showing a “cystic” dorsal lesion with many septa and thromboses; (b) the same angiomatous tumor in the newborn. Note the additional small lesion (arrow) heralding nonsegmental involvement (Reprinted with permission from John Wiley and Sons, USA)
10.1.11.3 Extracutaneous Type 2 Segmental Lesions
Type 2 segmental blue rubber bleb angiomatosis may also give rise to extensive extracutaneous lesions. For example, Atten et al. [19] described a woman with numerous cutaneous vascular lesions and episodes of rectal bleeding. During childhood she had an exploratory laparotomy and was told she had a large pelvic vascular malformation for which no treatment was proposed. At 20 years of age, magnetic resonance imaging showed a large pelvic hemangioma, measuring 60 cm and extending into the abdomen. It had eroded the rectal wall to produce rectal bleeding. Because of the enormous size of the pelvic tumor, bleeding was controlled by conservative measures and an expectant attitude was chosen. In retrospect, this appears to be a classic example of extracutaneous type 2 segmental manifestation of the disorder (see case 4 in Fig. 10.7). An excessive intraperitoneal and retroperitoneal involvement was also reported by Patel et al. [345]. In another case of blue rubber bleb angiomatosis, a giant mass of angiomatous lesions had to be resected in the tranverse colon [319].
10.1.12 Lipomatosis
Lipoma is a very common tumor. Its molecular basis is unknown. Lipomatosis is inherited as an autosomal dominant trait (OMIM 151900). A mosaic arrangement has so far not been reported.
10.1.13 Neurofibromatosis 1
Neurofibromatosis 1 (NF1) is caused by mutations in the neurofibromin gene. Today the expression “segmental NF1” should be regarded as an umbrella term. The type 1 segmental NF1 is a well known phenomenon that has so far been reported in more than 150 cases. In my view, however, the type 2 segmental manifestation is even more common but widely neglected until today, although the existence of this superimposed form of mosaic NF1 has been proven at the molecular level.
10.1.13.1 Type 1 Segmental NF1
In the past century this type was simply called “segmental NF1” because it was generally believed that this was the only form of mosaic NF1. Around 1990 many experts erroneously considered segmental neurofibromatosis to represent a distinct entity in the form of “NF5,” to be separated from NF1 [360, 367, 368]. Similarly, a classification of segmental neurofibromatosis into four subtypes as proposed by Roth et al. [373, 443] should today be taken as a historical error. This outdated concept implied that in patients with bilateral or otherwise systematized segmental neurofibromatosis, one had to assume multiple postzygotic mutations [233, 373, 425, 443], although one single postzygotic mutational event is sufficient to plausibly explain such cases.
Cases that can today be categorized as legitimate examples of type 1 segmental NF1 have been described by many authors [60, 61, 90, 97, 233, 306, 335, 375, 385]. In the involved area, either neurofibromas alone (Fig. 10.9) or pigmentary changes alone (Fig. 10.10) or both lesions are noted. Ipsilateral Lisch nodules may be an additional feature of this mosaic disorder [40, 376, 473].
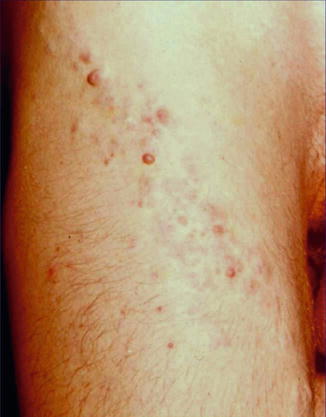
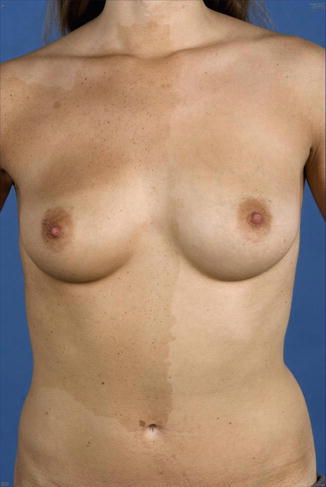
Fig. 10.9
Type 1 segmental neurofibromatosis 1 showing neurofibromas alone
Fig. 10.10
Type 1 segmental neurofibromatosis 1 showing hyperpigmentation alone
Molecular proof that postzygotic mosaicism causes type 1 segmental NF1 has been provided by Tinschert et al. [434] and Vandenbroucke et al. [456]. Interestingly, in one of these cases [456] almost the entire surface of the body was affected by NF1 manifestations, leaving a few segments unaffected (Fig. 3.1). The authors were able to exclude revertant mosaicism, which clearly demonstrates that type 1 segmental NF1 may affect, by way of exception, far more than half of the body tissue.
10.1.13.2 Type 2 Segmental NF1
The lesions of type 2 segmental NF1 tend to be rather pronounced, and they are usually noted at birth or during early infancy. They may adopt various clinical appearances. In part this superimposed form of mosaicism may consist of a large band-like café-au-lait hyperpigmentation with intralesional cutaneous or subcutaneous neurofibromas [14, 242] (Fig. 10.11), or of multiple segmentally arranged neurofibromas [221], or of a giant café-au-lait macule without any hint of underlying neurofibromas [482]. The most frequently noted manifestation, however, is a large plexiform neurofibroma. Indeed, all plexiform neurofibromas of appreciable size represent a type 2 segmental NF1 [179]. It is in line with this view that virtually all of these tumors are present at birth [368]. In particular, all cases of “giant neurofibroma” [359] appear to result from LOH occurring at an early developmental stage.
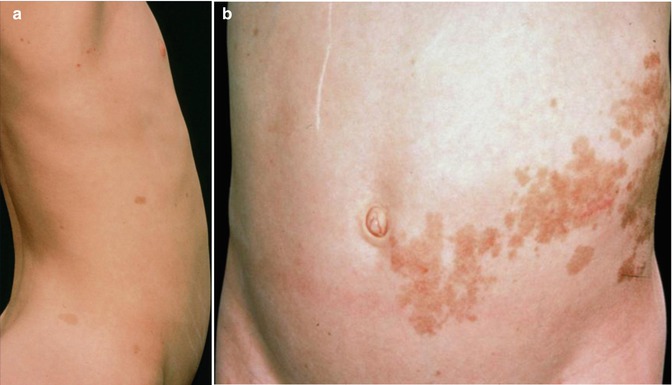
Fig. 10.11
Type 2 segmental neurofibromatosis 1 in an 11-year-old girl. (a) Right side showing nonsegmental café-au-lait macules; (b) left side showing pronounced segmental involvement [14] (a: Courtesy of Dr. David Atherton, London, UK; b: Reprinted with permission from John Wiley & Sons, USA)
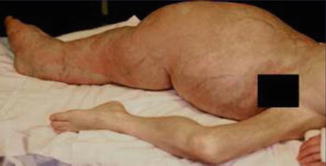
According to Pivnick and Riccardi [352], 5 % of NF1 patients have large plexiform neurofibromas. In a population-based study, Huson et al. [215] found that even 26.7 % of individuals with NF1 had a plexiform neurofibroma evident by physical examination. These tumors may be inconspicuous during infancy but later tend to show aggressive growth. Involvement of the face may result in severe compromise and death [13], whereas overgrowth of a limb likewise constitutes a ponderous handicap [253]. The overlying skin is usually thickened and hyperpigmented. The acanthotic epidermis has sometimes been mistaken as an epidermal nevus [88]. Hypertrichosis in the form of dark, coarse terminal hair is usually noted (Fig. 10.12). On the other hand, plexiform neurofibroma may involve all of the underlying structures such as nerves, muscles, and bones, resulting in giant overgrowth of a limb (Fig. 10.13) or half of the face [253]. Large plexiform neurofibromas may involve the brain [41] or the liver [74], or show intrathoracic growth [449], or even originate from the colon [483]. By way of exception, such segmental overgrowth may also be caused by diffuse proliferation of the fusiform cells of classical neurofibromatous tissue [292]. In short, it is the type 2 segmental involvement that, to a large degree, contributes to morbidity in patients with NF1.
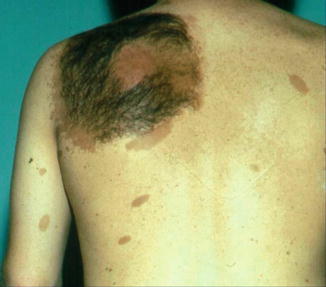
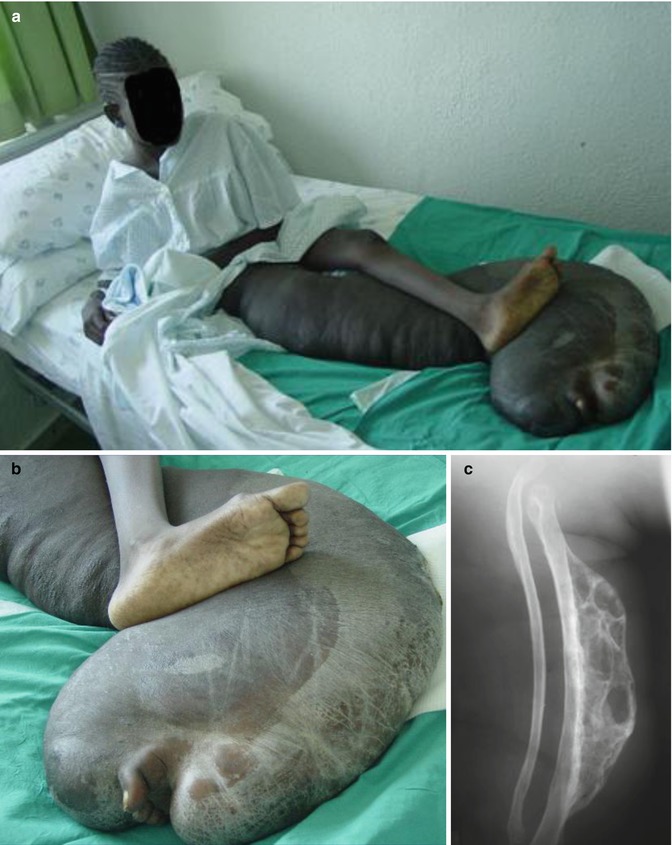
Fig. 10.12
Plexiform neurofibroma showing pronounced hypertrichosis (Courtesy of Dr. Howard Pride, Danville, Pennsylvania, USA)
Fig. 10.13
Type 2 segmental neurofibromatosis 1 giving rise to huge overgrowth of a leg. (a) General view; (b) the right foot being hidden within the tumor mass; (c) cystic periosteous overgrowth of the tibia [292] ( a and c: Reprinted with permission from John Wiley and Sons, USA; b: Courtesy of Dr. Silvestre Martínez-García, Málaga, Spain)
It should be noted that until today the concept of type 2 segmental NF1 is far from being generally accepted by the scientific community. For example, in 2008 Pascual-Castroviejo et al. [343] presented a series of 43 cases of segmental NF1 occurring in children or adolescents. The least frequently noted form was an exclusively cutaneous manifestation, whereas in 81 % of cases underlying structures such as the bones were additionally affected. In most patients the segmental involvement was characterized by a plexiform neurofibroma. Moreover, a family history of nonsegmental NF1 was noted in 15 cases (35 %). From this well-documented study we can conclude that most of the patients had in fact a type 2 segmental involvement in the form of plexiform neurofibroma. Similarly, numerous photographs as published in books or chapters on NF1 [271, 368, 377] inadvertently depict classical cases of type 2 segmental NF1. Many other examples of type 2 segmental NF1 can be found in the literature (Table 10.1)
Table 10.1
Some case reports that can today be reclassified as examples of type 2 segmental NF1
Reference | Age at onset of segmental lesions | Coexistence of nonsegmental lesions | Family members with nonsegmental lesions |
|---|---|---|---|
Butterworth [58] | Birth | + | |
Diekmann et al. [106] | Infancy | + | + |
Bingham and Burrows [33] | Birth | + | + |
Archer et al. [14] | Childhood | + | |
Boltshauser et al. [41] | [17 patients] | + | |
Chen et al. [74] | Early childhood | + | |
Ettl et al. [122] | Birth | + | |
Pivnick et al. [351] | Birth | + | |
Kim et al. [242] | 20 years | + | |
Oguzkan et al. [330] | Childhood | + | + |
Yang et al. [482] | Birth | + | |
Pascual-Castroviejo [343] | [35 children] | + | |
Rallis [359] | Infancy | + | |
Menon and Kumar [302] | Birth | + | |
Bano et al. [22] | Childhood | + | |
Ji et al. [228] | Birth | + | |
Thammaiah et al. [432] | <18 months | + | + |
Kantaputra et al. [236] | Birth | + | |
Sharma et al. [402] | Birth | + | |
Nasir et al. [320] | <7 years | + | + |
Salvitti et al. [383] | <3 months | + | + |
Molecular evidence for the concept of type 2 segmental NF1 has been provided by several groups who documented absence of the corresponding wild-type allele in plexiform neurofibromas [96, 230, 247]. Moreover, Steinmann et al. [421] found postzygotic recombination to be a frequent cause of LOH in plexiform neurofibromas.
10.1.13.3 The Issue of “Genetic Transmission of Segmental NF1”
Because the concept of dichotomous types of segmental NF1 was unknown, some authors have developed, by conflating the two forms, the erroneous idea of “genetic transmission of segmental NF” [330, 373, 374, 395, 452]. In fact, authentic cases of familial occurrence of type 1 segmental NF1 have not been reported until today. Two asserted cases observed by Huson and Ruggieri [216] have not been documented in detail and can, therefore, not be accepted as legitimate examples of vertical transmission of type 1 segmental NF1. Most cases of alleged transmission of segmental NF1 can be explained in the following way. The parent had type 1 segmental NF1 heralding gonadal mosaicism. The child had nonsegmental NF1 being superimposed by a type 2 segmental involvement that may constitute, during childhood, the only recognizable feature of the disorder. On the other hand, the possibility that both parent and child had type 2 segmental lesions should also be considered.
10.1.13.4 Genetic Counseling in Cases of Segmental NF1
Because a type 1 segmental NF1 implies the possibility of gonadal mosaicism, patients run an increased, albeit rather small, risk to give birth to a child affected with nonsegmental NF1 [40, 87, 174, 216]. The risk cannot be expressed in terms of a given percentage, but apparently the danger of gonadal mosaicism increases with the size of the body area involved [377]. On the other hand, it should be borne in mind that germline mosaicism may even be present in individuals showing no clinical feature of NF1 [264].
Patients with type 2 segmental NF1 have a 50 % risk to give birth to a child with nonsegmental NF1. The possibility that an affected child will show, in addition, a type 2 segmental involvement is likewise present because all children born with nonsegmental NF1 run this risk. Hence, it is of utmost practical importance that clinicians are able to discriminate between a type 1 and type 2 segmental manifestation of NF1.
10.1.13.5 Other Practical Aspects
In type 2 segmental NF1, the involvement of extracutaneous structures can be recognized by application of the presently available imaging techniques [343].
Dermatologists in practice should be warned that attempts to remove plexiform neurofibromas by operative procedures may cause diffuse bleeding. Such therapeutic interventions should only be performed by a fully equipped and well-trained surgical team, including anesthesiological support.
Appropriate knowledge of type 2 segmental NF1 is important because the vast majority of malignant peripheral nerve sheath tumors develop from large plexiform neurofibromas [297, 368]. Apparently, the inherent early loss of the corresponding wild-type allele represents a first step in the multistage process of carcinogenesis giving rise to such malignant tumors.
10.1.14 Neurofibromatosis 2
In this disorder, a type 1 mosaicism appears to occur rather frequently [123], although cutaneous manifestations have so far not been reported. In such cases, a unilateral or otherwise segmental involvement of the brain has been confirmed by MRI examination [376]. A type 2 segmental involvement of the skin has been documented by Susan Huson from Manchester (Fig. 10.14).
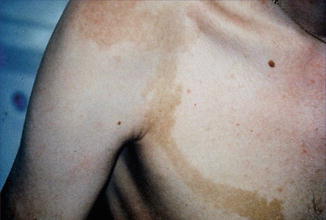
Fig. 10.14
Type 2 segmental manifestation of neurofibromatosis 2 (Courtesy of Dr. Susan M. Huson, Manchester, UK)
10.1.15 Schwannomatosis
The disorder is characterized by multiple cutaneous and spinal schwannomas. The tumors that are also called neurilemmomas originate from mutations in SMARCB1 [213]. Remarkably, however, evidence has been provided that these schwannomas may be caused by a four-hit mechanism [397] (see Sect. 3.1.1.1).
Several patients with mosaic subcutaneous lesions have been reported [266, 355, 427, 442]. Most, if not all, of these cases appear to represent a type 1 segmental involvement (Fig. 10.15). Moreover, germline mosaicism in a mother of two affected children has been documented [212].
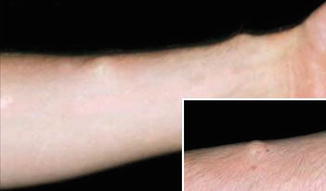
Fig. 10.15
Type 1 segmental schwannomatosis [266] (Reprinted with permission from John Wiley & Sons, USA)
10.1.16 Legius Syndrome
The features of Legius syndrome include multiple NF1-like café-au-lait macules, axillary freckling, and learning problems, but contrasting with NF1 the patients have multiple lipomas, a Noonan-like facial appearance, and macrocephaly, whereas neurofibromas are absent [50]. The disorder is caused by mutations in SPRED1, a gene involved in MAPK (mitogen-activated protein kinase) signalling. The fact that neurofibromin is connected with the same pathway offers an explanation for the overlapping clinical features. In their first report, Brems et al. [50] have inadvertently documented a type 2 segmental manifestation in the form of a large, blocklike café-au-lait hyperpigmentation being sharply demarcated in the midline (Fig. 10.16).
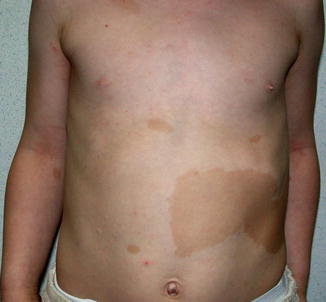
Fig. 10.16
Type 2 segmental manifestation of Legius syndrome [50] (Reprinted with permission from Nature Publishing Group)
10.1.17 Leiomyomatosis
Cutaneous leiomyomatosis is caused by mutations of the fumarate hydratase gene [437]. In some families the trait is associated with an increased risk to develop renal cancer [262, 390]. A type 1 segmental involvement has very rarely been reported [153, 255].
By contrast, type 2 segmental leiomyomatosis (Fig. 10.17) occurs with a strikingly high frequency [5, 21, 80, 177, 182, 322, 333, 369, 448, 474]. Alam et al. [4] found a combination of segmental and disseminated leiomyomas in 19/59 patients (32 %). This may reflect an increased proneness of the fumarate hydratase gene to postzygotic recombination [189]. As a characteristic feature, the superimposed segmental lesions are extremely painful after pressure, exposure to cold, or emotional stress [1, 5, 57, 62, 159, 224, 254, 270, 278, 281, 333, 409, 463].
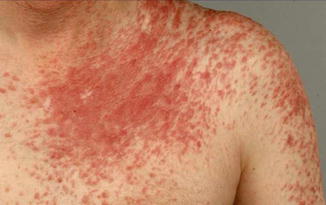
Fig. 10.17
Type 2 segmental leiomyomatosis [21] (Reprinted with permission from Elsevier Limited, Oxford, UK)
Presumably, most cases of segmental leiomyoma without presence of nonsegmental skin lesions in the patient and his family [200, 217, 240, 243, 342, 428] represent examples of type 2 mosaicism. This view is supported by the description of symptom-free gene carriers in families of such patients [293]. On the other hand, female gene carriers may have uterine leiomyoma alone [293, 342, 390, 413].
10.1.17.1 Familial Occurrence of Type 2 Segmental Involvement
10.1.18 Gorlin Syndrome
Major features of this trait are multiple basal cell carcinomas, jaw cysts, palmoplantar pitting, calcification of the falx cerebri, and various skeletal abnormalities. The disorder is caused by PTCH1 mutations.
10.1.18.1 Type 1 Segmental Involvement
Several cases suggesting a type 1 mosaicism have been reported [63, 285, 404]. This particular manifestation has often been equated with, but should today be distinguished from, segmentally arranged multiple basaloid follicular hamartomas (see Sects. 10.1.4 and 12.2.1.3).
10.1.18.2 Type 2 Segmental Involvement
Molecular proof of type 2 mosaicism was provided in a 12-year-old girl with pronounced unilateral lesions of Gorlin syndrome including congenital basal cell carcinomas, an odontogenic cyst, and rather large palmoplantar pits arranged along Blaschko’s lines (Fig. 10.18) [441]. She had inherited a PTCH1 mutation from her father, and biopsies obtained from her unilateral skin lesions revealed an additional postzygotic mutation involving the corresponding PTCH1 allele, thus giving rise to compound heterozygosity. Another impressive case suggesting a type 2 segmental involvement was documented by Gutierrez and Mora [165]. A 46-year-old man had numerous typical features including frontal bossing, maxillary prognathism, multiple basal cell carcinomas, scoliosis, bilateral thumb deformities, dense calcification of the falx cerebri, as well as coloboma and blindness of the right eye and contralateral cataract. The left side of his face was mildly affected with some basal cell carcinomas, whereas on the right side numerous, aggressively growing tumors formed tightly packed groups “with normal appearing skin interspersed.” The pronounced lesions extended from the ear to the posterior midline of the neck and to the anterior aspect of the chest. Moreover, the right hand and foot showed a linear arrangement of atrophic depressions measuring up to 1 cm. The authors described these lesions as being “atypical and unlike pits in other patients with the syndrome.” Hearing loss was likewise localized on the right side. Computed axial tomography showed “moderate atrophy, along with an enlarged right lateral ventricle with a probable area of porencephaly communicating with the frontal horns of the right ventricle.” Hence, a rather severe unilateral involvement was undoubtedly superimposed on mild nonsegmental lesions of Gorlin syndrome.
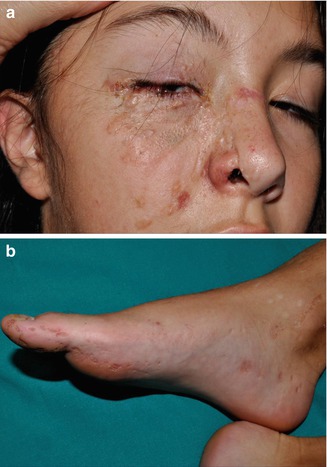
Fig. 10.18
Type 2 segmental Gorlin syndrome. (a) A 12-year-old girl with congenital basal cell carcinomas and linear atrophic lesions involving the right side of her face; (b) linear arrangement of rather large pits on the right foot [441] (a: Reprinted with permission from John Wiley & Sons, USA; b: Courtesy of Dr. Antonio Torrelo, Madrid, Spain)
10.1.19 Hereditary Nonsyndromic Multiple Basal Cell Carcinoma
This autosomal dominant trait (OMIM 605462) should be distinguished from Gorlin syndrome [178]. It is characterized by rather superficial basal cell carcinomas.
Type 1 mosaicism in the form of strictly unilateral involvement (Fig. 10.19) has been documented in several reports [46, 239, 261, 310]. Sometimes it was mistaken for a unilateral manifestation of Gorlin syndrome [403]. A case suggesting type 2 mosaicism was so far described only once [160].
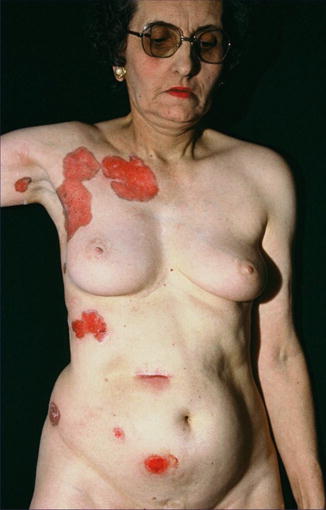
Fig. 10.19
Type 1 segmental manifestation of hereditary nonsyndromic multiple basal cell carcinoma (Courtesy of Dr. Georges Moulin, Lyon, France)
10.1.20 PTEN Hamartoma Syndrome (Cowden Disease Included)
PTEN hamartoma syndrome comprises two major clinical variants in the form of Cowden syndrome (OMIM 158350) and Bannayan-Riley-Ruvalcaba syndrome (OMIM 153480) [334]. Molecular analysis has shown that both variants can be caused by the same PTEN mutation and that both phenotypes may occur within the same family [201, 436], which is why Marsh et al. [289] proposed the new unifying term “PTEN hamartoma tumor syndrome.” This designation appears to be redundant, because the name “PTEN hamartoma syndrome” contains all of the distinguishing information. On the other hand, the bold theory that cases of Proteus syndrome may be categorized as a particular form of PTEN hamartoma syndrome [349, 492] has turned out to be wrong (see below).
10.1.20.1 Cowden Variant of PTEN Hamartoma Syndrome
The Cowden variant is characterized by macrocephaly, multiple facial trichilemmomas, acral keratotic papules, and proneness to develop malignancies of breast, thyroid, and colon. The phenotype is predominantly reported in women [201].
10.1.20.2 Bannayan-Riley-Ruvalcaba Variant of PTEN Hamartoma Syndrome
The phenotype includes macrocephaly, mental deficiciency, pigmented macules of the penis, lipomatosis, hemangiomas, and intestinal polyps. Cancer proneness of various organs is present but appears to be less pronounced when compared to the Cowden variant [201]. This clinical variant is preponderantly described in males.
10.1.20.3 Lhermitte-Duclos Variant of PTEN Hamartoma Syndrome
10.1.20.4 Type 2 Segmental PTEN Hamartoma Syndrome
During the first decade of this century, type 2 segmental manifestations of PTEN hamartoma syndrome were often misdiagnosed as Proteus syndrome. The investigators erroneously asserted that some cases of Proteus syndrome were caused by a PTEN germline mutation [7, 119, 274, 290, 336, 414, 465, 492, 493], whereas other authors argued that this could not be true [84, 183]. Today this controversy has come to an end because Proteus syndrome was shown to originate from an AKT1 mutation [269], whereas all cases of alleged “Proteus syndrome” caused by PTEN germline mutations can be categorized as examples of type 2 segmental PTEN hamartoma syndrome [186].
In 2007, the term “type 2 segmental Cowden disease” was proposed to denote a syndrome reflecting early postzygotic loss of the corresponding wild-type allele at the PTEN locus [186] (see also Table 3.4). A cutaneous hallmark is linear PTEN nevus that tends to be thicker and more papillomatous than common keratinocytic nevi (Fig. 10.20) [187] (see Sect. 7.3.1.4). Large connective tissue or vascular nevi, lipoblastomatosis, polyps of jejunum or colon, and focal segmental glomerulosclerosis may be associated, whereas the macrocephaly apparently reflects heterozygosity for the PTEN germline mutation [191]. As a synonym, the term “SOLAMEN syndrome” (segmental overgrowth, lipomatosis, arteriovenous malformation, epidermal nevus) was proposed by Caux et al. [69].
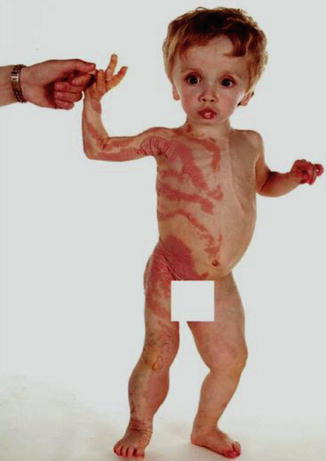
Fig. 10.20
Type 2 segmental PTEN hamartoma syndrome that was initially mistaken as “Proteus syndrome” [274]. Macrocephaly reflects heterozygosity for the PTEN mutation, whereas the unilateral linear PTEN nevus originated from early loss of heterozygosity (Reprinted with permission from John Wiley & Sons, USA)
An impressive case of type 2 segmental PTEN hamartoma syndrome has repeatedly been reported under various diagnostic terms such as “Proteus-like syndrome” [493], “arteriolovenular anomaly and epidermoid nevus involving right lower limb, scrotum, and penis” [426], or “PTEN hamartoma of soft tissue” [258]. This boy had giant overgrowth of his right leg that was covered by a linear epidermal nevus (Fig. 10.21). A R335X germline mutation was found in exon 5, whereas the lesional tissue of his right leg showed compound heterozygosity in the form of an additional R130X mutation involving exon 8 of the opposite allele [493]. Another case was reported under the name “hemigalencephaly as part of Jadassohn nevus sebaceus syndrome” [304]. The authors believed that this “may be another phenotypic finding associated with germline PTEN mutation.” For obvious reasons, the correct classification of the associated epidermal nevus is not nevus sebaceus but linear PTEN nevus (see Sect. 7.3.1.4). Tan et al. [426] described arteriovenous fistulas in 26 cases of PTEN hamartoma syndrome. Most, if not all, of these cases can today be categorized as examples of type 2 segmental involvement. The patients had lumps or “mass lesions” giving rise to cutaneous discoloration, swelling, or pain. In the involved segment of the body, these vascular anomalies tended to disrupt the architecture of neighboring muscles, and they were usually associated with variable amounts of ectopic fatty tissue. The high frequency of such complex vascular anomalies as noted by Tan et al. [426] may reflect a bias of ascertainment because all of the cases were documented in a specialized referral center for vascular anomalies. Iacobas et al. [219] described a boy affected with the Bannayan-Riley-Ruvalcaba variant who had a pronounced segmental involvement of his left arm with arteriovenous malformation, fatty infiltration of the muscles, and increased fibrous tissue, resulting in pain and loss of function of this arm.
10.1.21 Cutaneous Mastocytosis
This disorder often affects several members of a family [10, 26, 82] and is presently considered to be inherited as an autosomal dominant trait (OMIM 154800 [334]). Familial childhood-onset mastocytosis can occur in both presence or absence of c-KIT mutations [37], whereas sporadic adult-onset mastocytosis is usually caused by gain-of-function mutations of c-KIT [276].
A type 1 mosaic involvement has been described in the form of linear or otherwise segmental arrangement of telangiectasia macularis eruptiva perstans (Fig. 10.22) [136, 152, 156, 249, 326, 344, 462].
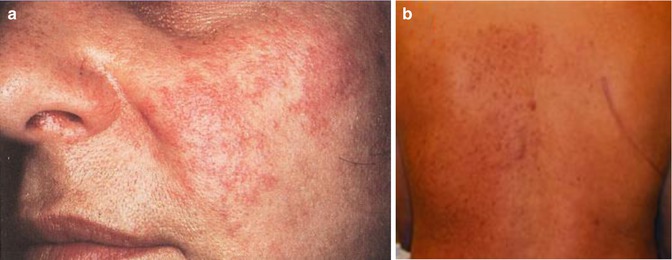
Fig. 10.22
Unilateral telangiectasia eruptiva perstans. (a) Facial lesions in a 36-year-old woman [152]; (b) dorsal lesions with strict midline separation in a 58-year-old man [326] (a: Reprinted with permission from John Wiley and Sons, USA; b: Reprinted with permission from John Libbey Eurotext, Montrouge, France)
10.2 Disorders of Keratinization
Examples of both type 1 and type 2 mosaicism were documented in various autosomal dominant disorders of keratinization.
10.2.1 Epidermolytic Ichthyosis of Brocq
A type 1 segmental involvement occurs rather frequently and is usually diagnosed as an “epidermal nevus of the epidermolytic type.” Affected individuals run an increased risk to give birth to a child with diffuse epidermolytic ichthyosis of Brocq [31, 42, 98, 172, 207, 277, 323, 327, 362]. Molecular proof was provided by Paller et al. [341].
10.2.2 Darier Disease
The lesions of mosaic Darier disease are always arranged along Blaschko’s lines. Many examples of type 1 segmental involvement (Fig. 10.23) have been reported [49, 67, 99, 146, 378, 420, 488]. Molecular evidence of mosaicism was documented in some cases [381, 464]. Theoretically, one would expect that individuals with type 1 segmental Darier disease may sometimes give birth to a child with the nonsegmental form of the disorder (see Sect. 3.1.1.3). Surprisingly, however, no such case has so far been reported. Presumably it will take a long time until this riddle can be solved.
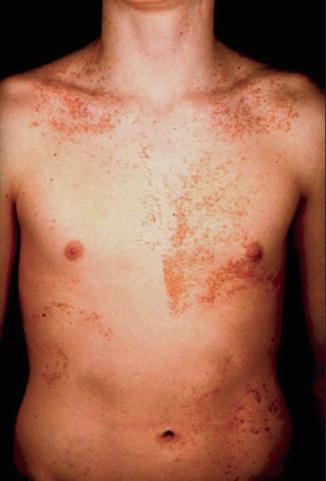
Fig. 10.23
Type 1 segmental Darier disease
Several case reports can be categorized as examples of type 2 segmental Darier disease [76, 129, 194]. In one of these cases (Fig. 10.24), molecular proof of the postulated genetic mechanism has been provided [129]. Interestingly, cases of didymosis in the form of paired linear areas of either excessive or absent involvement were likewise reported (see Sect. 8.1.4). So far, such cases have not been investigated at the molecular level.
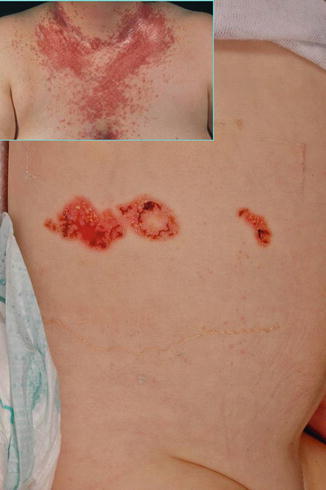
Fig. 10.24
Type 2 segmental Darier disease in a 1-month-old boy. Inset: Nonsegmental Darier disease in the boy’s grandmother [129] (Reprinted with permission from John Wiley & Sons)
10.2.3 Hailey-Hailey Disease
A type 1 segmental manifestation was described by Hwang et al. [218]. On the other hand, the theory of early loss of heterozygosity giving rise to type 2 segmental Hailey-Hailey disease [177] was proven by molecular analysis of the ATP2C1 gene in a patient showing both linear and nonlinear involvement (Fig. 10.25) [354]. Quite understandably, the pronounced linear lesions tend to show a rather poor therapeutic response to dermabrasion as compared to the nonsegmental lesions of the disorder [252].
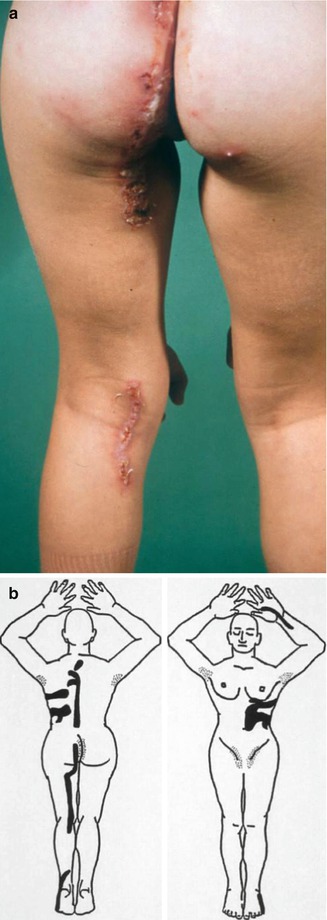
Type 2 segmental Hailey-Hailey disease has sometimes been described as “relapsing linear acantholytic dermatosis,” a term coined by Vakilzadeh and Kolde [454] who thought that this was a distinct entity. More recently, Arora et al. [16] proposed this diagnosis in a 4-year-old boy who had, since the age of 6 months, recurrent papulovesicles distributed along Blaschko’s lines on one side of his body. Exacerbations occurred during summertime. Histopathological findings were typical of Hailey-Hailey disease. From these features we can conclude that later in life the patient will almost certainly develop, in addition, some nonlinear lesions of the disorder.
10.2.4 Dowling-Degos Disease, Including the Galli-Galli Variant
Dowling-Degos disease is characterized by reticulate or patchy hyperpigmentation with an affinity to the flexural areas. It is caused by KRT5 mutations [30]. According to present knowledge, Galli-Galli disease is merely a variant of the same disorder, showing more pronounced features of acantholysis [418].
Arnold et al. [15] found a KRT5 mutation in a type 1 segmental form of the Galli-Galli variant (Fig. 10.26), whereas in clinically unaffected skin and in the blood only the wild-type allele was present. No type 2 segmental involvement has so far been reported.
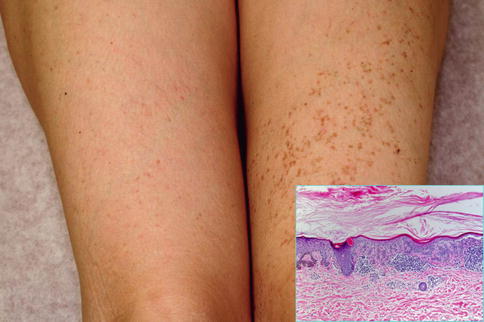
Fig. 10.26
Type 1 segmental Galli-Galli disease. Inset: hyperparakeratosis, acantholysis, hyperpigmentation of basal keratinocytes, and lymphocytic infiltrate in the upper dermis [15]
10.2.5 Acanthosis Nigricans
This disorder can be inherited as an autosomal dominant trait being caused by a K650T mutation in the FGFR3 gene [29]. There are several reports suggesting a type 1 mosaic manifestation of the disorder [86, 121, 226, 257, 348, 412]. Without molecular analysis, however, it seems difficult or even impossible to discriminate such lesions from other types of keratinocytic nevi.
During the years 1936–1976, Helen Ollendorff Curth [92–95] wrote many articles repeatedly describing a young man who had, since the age of 10 years, lesions of acanthosis nigricans showing a symmetrical distribution. In addition, a segmental and rather pronounced involvement was present since birth on one side of his body (Fig. 10.27a, b). During adulthood, the symmetric disorder improved to a large degree, whereas the nevus-like lesion remained unchanged. Ollendorff-Curth was fascinated by this case and wrote: “As the symmetric variety has been recognized as a genodermatosis, it seems justified to conclude that both the unilateral and the symmetric eruptions were caused by the same abnormal gene” [95]. Hence, she was nearby to sort out the problem that today has been solved by the theory of type 2 mosaicism [188]. A case of hystrix-like hyperkeratoses forming “wide stripes” and being superimposed on nonsegmental acanthosis nigricans, as reported by Babalian [20], probably represents another example of type 2 segmental acanthosis nigricans. In 2006, Ersoy-Evans et al. [121] described four cases of “acanthosis nigricans form of epidermal nevus.” One of their patients was a 16-year-old boy who had the linear disorder since childhood and showed, in addition, typical bilateral lesions of acanthosis nigricans. The authors assumed that this represented type 2 mosaicism.
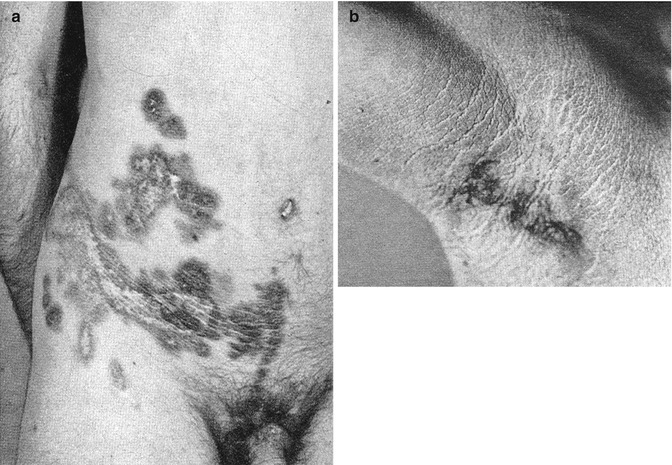
Fig. 10.27
A historical case of type 2 segmental acanthosis nigricans. (a) Pronounced unilateral involvement on the abdomen; (b) mild symmetrical skin changes in the axillary regions [93]
10.2.6 KID Syndrome
The keratitis-ichthyosis-deafness syndrome is an autosomal dominant trait being caused by GJB2 mutations. The disorder is charaterized by corneal epithelial defects, erythrokeratoderma-like hyperkeratosis, and sensorineural hearing loss.
A systematized linear nevus indistuiguishable from porokeratotic eccrine nevus was documented in the mother of a child with KID syndrome [435]. Indeed, porokeratotic eccrine nevus (Fig. 7.38) appears to represent a type 1 segmental manifestation of KID syndrome [112] (see Sect. 7.3.2.6).
In a 13-year-old girl affected with KID syndrome, Restano et al. [365] noted a pronounced unilateral hyperkeratotic lesion arranged on her back in a Blaschko-linear pattern, being superimposed on the diffusely hyperkeratotic and thickened skin as usually noted in this trait. The linear lesion had been present since infancy. The authors inferred that this may represent a type 2 segmental manifestation of the disorder.
10.2.7 Autosomal Dominant Dyskeratosis Congenita
This type should be distinguished from the more common X-linked dyskeratosis congenita (see Sect. 12.1.7) and from an autosomal recessive form. All of these types are characterized by reticular hyperpigmentation, leukoplakia, nail dystrophy, and proneness to develop aplastic anemia. The autosomal dominant form appears to be milder than the other two types, with a lower incidence of aplastic anemia [111].
A type 2 segmental involvement was described in a 17-year-old boy with typical skin lesions of dyskeratosis congenita [25]. In this case, a V-shaped pattern of pronounced reticular hyperpigmentation, being superimposed on a diffuse, less severe dyspigmentation, was noted on his back (Fig. 10.28). The patient was otherwise rather mildly affected and had a normal karyotype 46,XY. The authors considered an autosomal dominant trait with a pronounced linear manifestation, reflecting early loss of heterozygosity, to be the most likely diagnosis.
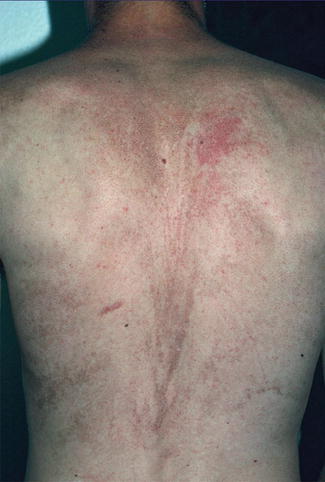
Fig. 10.28
Systematized type 2 segmental manifestation in a patient with autosomal dominant dyskeratosis congenita [25] (Reprinted with permission from John Wiley and Sons, USA)
10.2.8 Pachyonychia Congenita of the Jadassohn-Lewandowsky Type
The disorder, also known as pachyonychia congenita type 1, is caused by keratin 16 mutations [300]. A type 1 segmental manifestation was reported under the diagnosis “unilateral palmoplantar verrucous nevus.” A KRT16 mutation was found to be present in DNA obtained from the affected palm and to be absent in the unaffected palm.
10.2.9 Disseminated Superficial Actinic Porokeratosis
Disseminated superficial actinic porokeratosis (DSAP) is the most frequently occurring form of porokeratosis [109]. Its genetic basis is so far unknown. The disorder is characterized by multiple, small, rather inconspicuous lesions. Their slightly elevated margin corresponds to the histopathological feature of “cornoid lamella.” Both type 1 and type 2 mosaicism have been described.
Type 1 mosaicism was so far noticed only once, which may in part be explained by the fact the lesions of DSAP are rather tiny and easily overlooked. A 68-year-old woman who received immunosuppressive treatment for pemphigus vulgaris developed DSAP exclusively on her right leg (Fig. 10.29) [54]. Remarkably, the lesions were scattered within a large segmental area and did not show any linear arrangement.
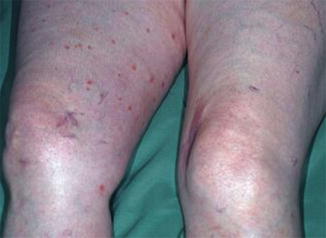
Fig. 10.29
Type 1 segmental manifestation of disseminated superficial actinic porokeratosis [54] (Reprinted with permission from Acta Dermato-Venereologica)
By contrast, cases suggesting a type 2 segmental manifestation have rather frequently been reported (for reviews see [175, 177, 189]). Such reports are often published under the ambiguous diagnosis of “linear porokeratosis” [140]. The linear lesions often appear early in life and sometimes even during infancy [110, 115, 127, 149, 238, 313, 325], or they may be noted at birth [309, 424], whereas the nonsegmental lesions tend to develop later in life (Fig. 10.30) [275, 346, 423]. Apparently, the underlying gene locus is a hotspot for postzygotic recombination [189]. Type 2 segmental DSAP may dramatically worsen when immunosuppressive drugs are given [284].
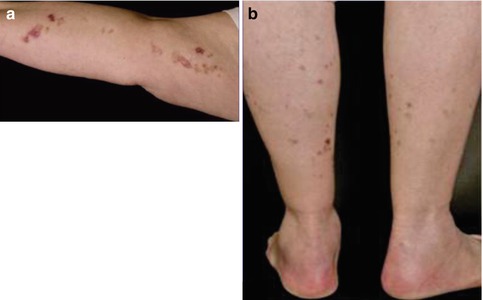
Fig. 10.30
Type 2 segmental manifestation of disseminated superficial actinic porokeratosis. (a) Congenital linear lesions on the right arm; (b) less severe bilateral involvement of legs since the age of 67 years [275] (Reprinted with permission from S. Karger AG, Basel, Switzerland)
As a rule of thumb, if linear porokeratosis is noted at birth [75, 161, 250, 394] or during the first years of life, a type 2 segmental manifestation of either DSAP [151, 206, 295, 340, 396, 429, 460] or porokeratosis of Mibelli [317] (see Sect. 10.2.10) is very likely.
10.2.9.1 Practical Aspect
10.2.10 Plaque-Type Porokeratosis of Mibelli
The designation “porokeratosis of Mibelli” is used by many authors as an umbrella term for all types of porokeratosis, whereas others use it to describe the classical plaque-type porokeratosis. Regarding this problem of ambiguity, I must confess that in several reviews published during the 1990s [175–177], I myself have erroneously subsumed the classical Mibelli type under the term DSAP. Autosomal dominant transmission of the Mibelli type has been documented beyond doubt [305, 384].
A case of type 1 mosaicism was described by Scholl [391]. The 13-year-old girl had scattered plaques involving the face, arm, hand, and foot exclusively on the left side of her body.
An impressive example of type 2 mosaicism was documented by Vittorio Mibelli in his original case report (Fig. 10.31) [305]. The patient had a pronounced linear porokeratosis present since the age of 2 years. Moreover, multiple nonsegmental plaques had first been noticed at the age of 7 years. Two siblings and the father were also affected with nonsegmental porokeratotic lesions. Additional nonambiguous cases of superimposed linear involvement occurring in families with nonsegmental porokeratosis of Mibelli have later been reported [35, 283]. In other reports the systematized linear disorder developed soon after birth [118, 361] or was accompanied in adulthood by “classic lesions of porokeratosis” [361], which is why a type 2 mosaicism is almost certain.
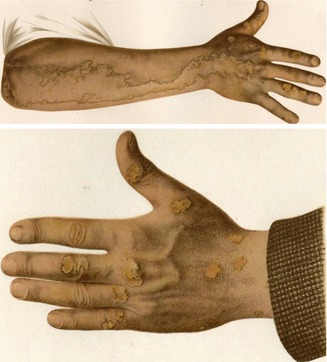
Fig. 10.31
Mibelli’s original report on plaque-like porokeratosis can today be taken as an early documentation of type 2 segmental manifestation. A pronounced linear involvement of the right arm was present since the age of 2 years, whereas nonsegmental lesions as noted on the left hand developed since the age of 7 years [305]
10.2.10.1 Familial Occurrence of Type 2 Segmental Involvement
This story of an unusual familial constellation is set in Pavia in northern Italy, where Mario Truffi [447] described in 1905 a 13-year-old boy who had since early infancy pronounced porokeratotic lesions arranged in a systematized linear pattern on the left side of his body, including the oral mucosa. On the right side some nonsegmental lesions were noted. His mother had some scattered bilateral lesions of porokeratosis of Mibelli. Twenty-three years later Franco Flarer [128] presented a follow-up report with photographs of the conspicuous systematized linear involvement. He mentioned that the patient had developed some additional nonsegmental plaques on the right side of his body. Finally, Marco Gandola [137] presented in 1951 a revised family history. The patient had a younger brother who was less severely affected but, surprisingly, showed systematized lesions involving the right side of his body. This family observation can today be taken as a clinical hint that the underlying gene locus represents a hotspot for somatic recombination.
10.2.11 Porokeratosis Palmaris, Plantaris et Disseminata
This particular form of porokeratosis [164] is characterized by very small disseminated lesions that initially tend to appear on the palms and soles and later affect the entire integument. A type 2 segmental manifestation in the form of pronounced linear lesions involving the left arm of a 33-year-old woman has been documented [350]. Another possible example of superimposed linear porokeratosis palmaris, plantaris et disseminata was reported in a 31-year-old Afro-American woman with end-stage liver disease [214].
10.2.12 Type 2 Segmental Manifestation in Cases of Unclassifiable Porokeratosis
Some authors described linear porokeratosis superimposed on nonlinear lesions, but it is difficult to determine from their report which type of porokeratosis was present [150, 356, 357]. In some cases [142, 358], porokeratosis of Mibelli is probable but not certain, whereas in other reports [105] a diagnosis of DSAP is likely but not definite. A case of congenital linear and giant porokeratosis [280] represents almost certainly an example of type 2 mosaicism but can so far not be categorized further. The same holds true for a boy with systematized linear porokeratosis [387] who, as an adult, developed 14 squamous cell carcinomas on his limbs [132, 386].
10.2.13 Costello Syndrome
Costello syndrome includes a peculiar facial appearance, growth retardation, mental deficiency, musculoskeletal defects, and cardiomyopathy [334]. Cutaneous features comprise redundant skin (especially on the hands), periorificial papillomas, acanthosis nigricans-like hyperkeratosis, palmoplantar keratoderma with deep creases, sparse and curly hair, and dystrophic nails. The disorder is caused by heterozygous HRAS mutations [417].
A type 1 mosaic involvement was documented in a 15-year-old girl [157]. She had segmental acanthosis nigricans-like skin lesions on her abdomen and right arm (Fig. 10.32), and a linear hyperkeratosis on her left sole. An HRAS mutation was found to be present in buccal DNA samples but absent in peripheral blood lymphocytes. Another patient with a mosaic HRAS mutation giving rise to type 1 segmental Costello syndrome, including patches of curly hair in his otherwise straight hair, had a son with nonsegmental Costello syndrome [417]. On the other hand, no clinical features of mosaicsm could be detected in a 1-year-old boy with Costello syndrome and a mosaic HRAS mutation present in blood lymphocytes [147].
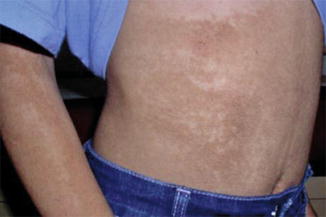
Fig. 10.32
Type 1 segmental manifestation of Costello syndrome. Mosaic arrangement of acanthosis nicgricans-like skin changes on the abdomen [157] (Reprinted with permission from John Wiley and Sons, USA)
10.2.14 Acrokeratoelastoidosis
In this trait multiple small, shiny, keratotic papules are arranged along the margins of hand and feet. They appear in childhood and progress during adolescence. Two cases of type 1 mosaicism in the form of unilateral involvement of both hand and foot have been reported (Fig. 10.33) [245, 311].
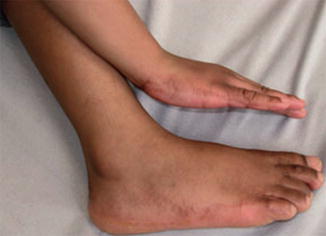
Fig. 10.33
Type 1 segmental acrokeratoelastoidosis [245] (Reprinted with permission from John Wiley and Sons, USA)
10.3 Disorders of Connective Tissue or Bones
In this group of disorders, mosaic manifestations are often noted. In particular, cases suggesting a type 2 segmental involvement have been documented in tuberous sclerosis, Buschke-Ollendorff syndrome, Albright’s hereditary osteodystrophy, and hereditary osteoma cutis.
10.3.1 Tuberous Sclerosis
Tuberous sclerosis (TS) is characterized by multiple hamartomas of the connective tissue involving the brain and skin as well as various other organs such as the eyes, lungs, or kidneys. In most patients, progressive brain involvement gives rise to seizures, learning difficulties, or behavior abnormalities. The longer term “tuberous sclerosis complex (TSC)” has no additional meaning. It was created by geneticists for the simple reason that they needed, for the purpose of classification, a three-letter symbol. The disorder can be caused by two different genes. TSC1 encodes for hamartin, whereas TSC2 is producing tuberin.
Mosaicism is well documented in TS [259, 458]. So far, however, reviews of cases of mosaic TS do not discriminate between type 1 and type 2 mosaicism.
10.3.1.1 Type 1 Segmental TS
This type of mosaic TS has preponderantly been described in the form of unilateral facial angiofibromas (Fig. 10.34) [9, 103, 167, 298, 400, 410, 445]. Other forms of type 1 mosaic involvement may likewise occur. For example, a young man showed unilateral facial angiofibromas with ipsilateral presence of periungual fibromas of fingers and toes, small shagreen plaques, and hypopigmented macules involving the trunk, as well as retinal hamartomas [296]. A patient with postzygotic mosaicism confirmed at the molecular level [459] had multiple dental pits, and brain tomography showed one paraventricular calcification. Two cases of “solitary cortical tubers” [108] may likewise be best explained as type 1 mosaic TS. The nosological significance of a solitary periungual fibroma [491] is so far unclear.
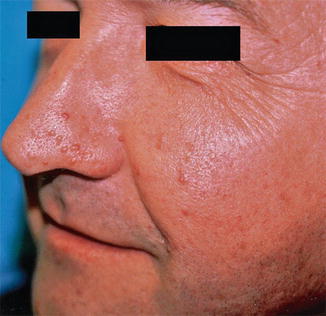
Fig. 10.34
Type 1 segmental tuberous sclerosis in the form of unilateral facial angiofibromas [445] (Reprinted with permission from Elsevier Limited, UK)
10.3.1.2 Type 2 Segmental TS
This type of mosaic manifestation of TS occurs rather frequently but has so far been disregarded, which means that up to now we can find cases of classical type 2 segmental involvement erroneously categorized as a “forme fruste” [138, 139] or as a simple mosaic of TS [44, 64]. Large shagreen patches or cobblestone nevi most likely represent a type 2 segmental involvement [177]. Such lateralized lesions may sometimes show conspicuous growth during adolescence or adulthood (Fig. 10.35). A large linear lesion involving the tongue was explained by the same mechanism (Fig. 10.36) [221]. Impressive examples of type 2 segmental TS with unilateral maxillary and mandibular involvement were inadvertently documented by oral surgeons (Fig. 10.37) [379, 470]. Moreover, all cases of “folliculocystic and collagen hamartoma” as observed in children with TS (Fig. 10.38) [440] may best be explained as a superimposed segmental manifestation of TS. The same holds for large “fibrous hamartomas of infancy” [169, 401, 451] or cases of macrodactyly reported in patients with TS [85, 282, 380, 401, 406, 451], or for overgrowth involving all tissues of a forearm [337, 469].
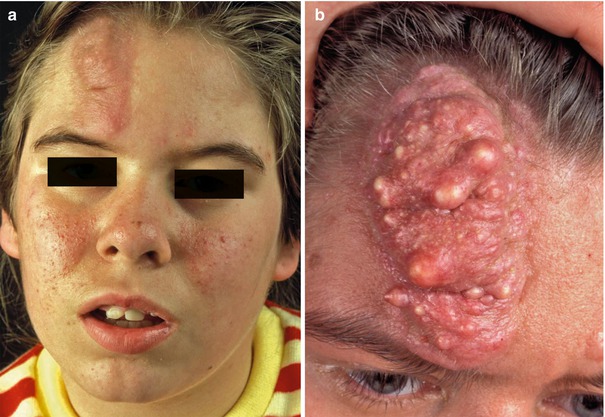
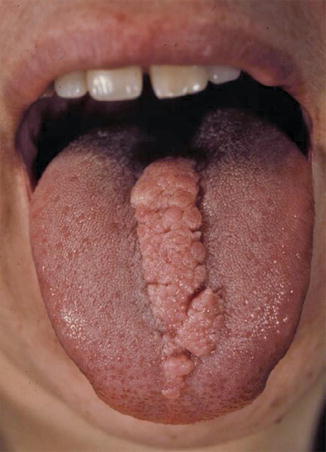
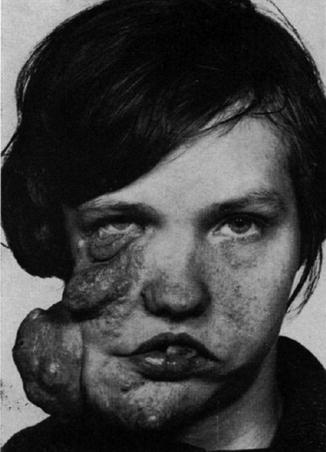
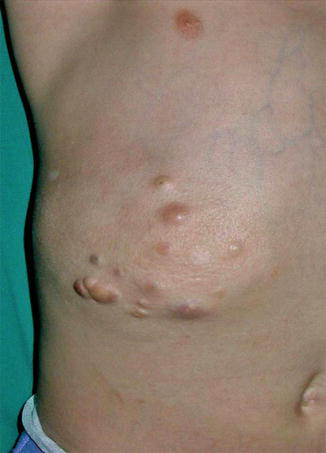
Fig. 10.35
(a) Type 2 segmental tuberous sclerosis involving the forehead of a young girl; (b) the same lesion some years later (Courtesy of Dr. Gerhard Kurlemann, Münster, Germany)
Fig. 10.36
Type 2 segmental involvement of the tongue in a patient with tuberous sclerosis [221] (Reprinted with permission from John Wiley & Sons, USA. Color photograph kindly provided by Dr. Peter H. Itin, Basel, Switzerland)
Fig. 10.37
Type 2 segmental involvement of maxilla and mandible in a patient with tuberous sclerosis [470] (Reprinted with permission from Elsevier Limited, UK)
Fig. 10.38
Type 2 segmental tuberous sclerosis presenting as “folliculocystic and collagen hamartoma” in a 6-year-old boy [440] (Reprinted with permission from Elsevier Limited, Oxford, UK)
10.3.1.3 Cases of Unclassifiable Mosaic TS
Silvestre et al. [410] reported on a 12-year-old boy with multiple unilateral facial angiofibromas that had developed since the age of 5 years. In addition, he had a large hypopigmented macule involving the left side of his abdomen with a sharp midline separation. During the following years, the clinical course of the disorder may have shown whether this boy carried a germline mutation and thus was affected with a type 2 segmental TS.
10.3.1.4 Genetic Counseling
In cases of type 1 segmental TS, siblings and parents of the patient have a normal population risk of carrying a TS mutation, because such mosaicism excludes gonadal mosaicism of one of the parents [458]. For the next generation, the risk of nonsegmental TS is increased to some degree because gonadal mosaicism may be present. Children of patients with type 2 segmental TS have a 50 % risk to be affected with nonsegmental TS. Hence, it is of great practical importance to bear the dichotomy of mosaic TS in mind. For example, in a child with unilateral angiofibromas, it is mandatory to exclude a type 2 mosaic manifestation of TS.
10.3.2 Buschke-Ollendorff Syndrome
The disorder is caused by loss-of-function mutations in LEMD3 [199] and characterized by small disseminated connective tissue nevi of an elastin-rich type in combination with osteopoikilosis.
10.3.2.1 Type 2 Segmental Skin Lesions
In addition to the disseminated, skin-colored or yellowish, firm papules of dermatofibrosis, large plaques showing an asymmetrical arrangement have been called “juvenile elastoma” [170, 260, 471]. These plaques almost certainly represent a type 2 segmental manifestation of Buschke-Ollendorff syndrome [116, 181], especially when they are associated with nonsegmental lesions in the patient or his family members (Fig. 10.39) [17, 59, 180, 392, 481]. An exemplary type 2 segmental involvement has been described as “Buschke-Ollendorff syndrome of the scalp” [444]. Remarkably, such lesions have sometimes been mistaken for a “forme fruste” of Buschke-Ollendorff syndrome [130].
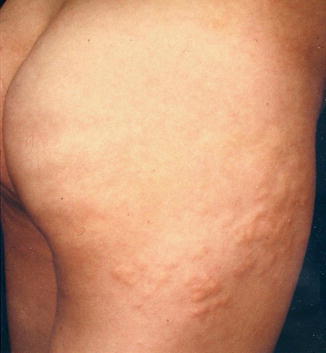
10.3.2.2 Familial Occurrence of Type 2 Segmental Skin Lesions
In Buschke-Ollendorff syndrome, asymmetrically arranged plaques or lumps are very often noted in several members of a family, for example, in a father and two of his children [415], in a father and his son or daughter [18, 59, 308], in two siblings [196, 211, 308], or even in three generations [457]. Because such lesions tend to appear in childhood or may even be noted at birth, they can best be explained as a type 2 mosaicism, which indicates that the underlying LEMD3 locus represents a hotspot for postzygotic recombination.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


