Miscellaneous Craniofacial Conditions: Fibrous Dysplasia, Moebius Syndrome, Romberg Syndrome, Treacher Collins Syndrome, Dermoid Cyst, and Neurofibromatosis
Robert J. Havlik
This chapter describes several disorders that do not “fit” in with other conditions in plastic surgery, or for that matter, with other conditions in medicine. With the exception of neurofibromatosis, these conditions are distinctly uncommon. With the rapid developments in molecular biology, two of these disorders have been shown to be caused by disorders in intracellular second messenger systems—fibrous dysplasia and neurofibromatosis 1 (NF-1).
FIBROUS DYSPLASIA
Fibrous dysplasia is a benign disorder of the bone that affects both the axial and the craniomaxillofacial skeleton. Fibrous dysplasia is not “classically” a congenital disorder, since it is not usually evident at birth, but becomes clinically evident during late childhood or adolescence. It occurs sporadically and genetic transmission has not been documented. Fibrous dysplasia has been traditionally divided into three main categories: monostotic (or monocystic), polyostotic, and the McCune-Albright syndrome. The majority of patients (∽70% to 80%) present with a single area of bony involvement (monostotic or monocystic fibrous dysplasia).1 Of the patients with polyostotic fibrous dysplasia (20% to 30%), approximately 3% present with a triad of polyostotic fibrous dysplasia, precocious puberty, and skin pigmentation known as the McCune-Albright syndrome.1 This skin pigmentation presents as “café-au-lait” spots with irregular borders, described as being similar to the coastline of Maine. In addition to this classic triad, McCune-Albright syndrome is also associated with several different endocrine disorders, all caused by autonomous hormonal overproduction, such as growth-hormone producing pituitary adenomas, hyperthyroid goiters, and adrenal hyperplasia.
The craniomaxillofacial structures are involved in approximately 25% of cases with monostotic fibrous dysplasia and up to 50% of cases with polyostotic involvement. The most common presentation in the craniofacial skeleton is that of a painless, enlarging mass of bone. The maxilla is the bone most often involved, followed in frequency by the frontal bone, but all bones of the craniomaxillofacial skeleton may show involvement. The clinical manifestations of fibrous dysplasia include expansive growth leading to aesthetic and functional compromise. Maxillary lesions can lead to dental malocclusions, tilting of the occlusal plane, or significant facial deformity and asymmetry. In lesions with orbital involvement, visual disturbance, ocular proptosis, and orbital dystopia can occur. In lesions with sphenoid involvement, blindness may occur as a result of impingement on the optic nerve.
The difficulty in the diagnosis of fibrous dysplasia varies with the extent of presentation of the disease. The polyostotic and McCune-Albright forms of fibrous dysplasia are relatively easily diagnosed based on clinical and radiologic investigation, whereas establishing the diagnosis of the monostotic form is more difficult because of the number of other important lesions that are included in the differential diagnosis. In the axial skeleton the lesions frequently appear as well-circumscribed radiolucent lesions with a thin sclerotic periphery. In contrast, the lesions of the craniofacial skeleton are more poorly defined and more radiopaque. Bone biopsy in many areas of the axial skeleton in fibrous dysplasia is generally avoided, especially where the risk of pathologic fracture is high. However, in the mandible, where monostotic involvement is most frequent in the craniomaxillofacial skeleton and therefore there is the greatest difficulty differentiating this from other solitary bony lesions, bone biopsy has not been reported to cause a pathologic fracture in fibrous dysplasia. Malignant degeneration of fibrous dysplasia has been reported to occur in 0.5% of cases with monostotic involvement, and up to 4% of cases with McCune-Albright syndrome. Notably, the most frequent site for sarcomatous degeneration is the craniofacial skeleton.
Pathogenesis
This disorder is centered around a structural and functional change in the cellular transduction mechanism involving G-proteins.1 The G-protein is a membrane-bound intracellular signaling mechanism that carries the message of extracellular hormone binding into the cell to create an effect. The G-proteins themselves have an intrinsic activity that causes hydrolysis of GTP to GDP. In fibrous dysplasia, the G-protein has a decreased ability to hydrolyze GTP, resulting in the G-protein remaining in an activated state, leading to continued stimulation of cAMP and multiple other effects.1 Significantly, many of the adrenal, hypophyseal, thyroid, and gonadal cells of patients with McCune-Albright syndrome show the same mutations, thereby leading to increased “on” activity, and constitutively increased hormone production.
It has been postulated that the timing at which this mutation occurs in embryologic development may determine the clinical extent of the disease. In other words, a mutation that occurs late in embryologic development or even a postnatal one will lead to a decreased cellular complement with the mutation and therefore lead to the development of monostotic bone involvement. In contrast, a somatic mutation that occurs in early embryologic development would lead to a larger portion of afflicted cells within the individual, and this leads to multicentric involvement (polyostotic fibrous dysplasia). If the mutation occurs early enough in development, it may also lead to the involvement of additional tissues (endocrine disorders, McCune-Albright syndrome, etc.). As noted above, there have been no documented cases of genetic transmission of fibrous dysplasia, it is believed to be a lethal mutation.
Treatment
Treatment of fibrous dysplasia in the craniofacial skeleton is determined by the functional or aesthetic problems created by the disease process. The mere existence of fibrous dysplasia does not mandate treatment. In bones of the axial skeleton, the expansile process, coupled with cortical resorption, can lead to decreased structural strength and pathologic fracture. This is seldom the case in the craniomaxillofacial skeleton, and indications for treatment are more frequently related to aesthetic imbalance, facial disfigurement, distortion of the functional occlusion, ocular proptosis, and impingement on neural foramina. Impingement on the optic nerve has led to visual disturbance and blindness.
Treatment recommendations for fibrous dysplasia have occasionally been made based on the clinical observation that the disease will “burn out” in the post-pubertal adolescent state as skeletal maturity is reached. Unfortunately, there are no data to support this contention.
Surgical treatment is often designed to counter the effects of mass expansion and the consequent deformity that occurs in the facial skeleton. Therefore, in most cases, surgery of the craniofacial skeleton will consist of either a contour reduction of the afflicted area, or resection and replacement of the affected bone. Contour reduction is a more limited operation, but the lesion always recurs. A decision is made between contour reduction versus resection and replacement based upon the rate of tumor growth, or the “aggressiveness” of this benign process, and the location and potential complications of continued growth and expansion. Resection is followed by reconstruction with either prosthetic materials or bone autograft.
In the cranial vault, bone involvement is frequently both expansile and hyperostotic. The frontal bone is the most frequently involved bone, followed by the sphenoid bone. In the case illustrated in Figure 28.1 both resection and contour reduction techniques were used for hyperostotic fibrous dysplasia in the right frontoorbital region and the right parietal region. The resection and cranial bone autograft reconstruction were performed in the right orbit and frontal bone, where the potential problems with recurrent tumor and repeat resection would be more complicated. In contrast, the parietal bone was completely removed, and contour reduction was performed down to the level of the cortical plate. When fibrous dysplasia involves the skull base, surgical resection is not possible.
Fibrous dysplasia of the orbit raises several special considerations. First, the mass effect of bone growth can lead to dystopia and visual disturbances. The potential problems of recurrence in this area, particularly with the potentially more difficult surgery in the recurrent field from scarring, often swing the balance toward resection of the afflicted bone and replacement. Second, specific to the orbit is the concern that growth of fibrous dysplasia can lead to optic nerve compression, subsequently leading to visual change and blindness. Visual loss has been cited as the most common neurologic complication of fibrous dysplasia involving the skull.
Although optic nerve decompression in patients with fibrous dysplasia of the sphenoid surrounding the optic canal and with documented changes in vision is widely accepted, “prophylactic” decompression of the optic nerve is not recommended. Furthermore, decompression after visual decrement or visual loss will not restore the visual deficit.2 The risks involved with surgical decompression of the nerve include a lack of improvement in vision, which occurs in anywhere from 5% to 33% of cases, and blindness resulting from the surgery.2
Fibrous dysplasia of the mandible presents as a mass lesion with cortical expansion. Because the presentation with mandibular disease is frequently monostotic, bone biopsy may be indicated to establish or confirm the diagnosis. As noted previously, biopsy of the mandible can be safely accomplished in fibrous dysplasia without a significant risk of fracture. Depending on the severity of the involvement, either contour reduction or resection can be performed. In lesions involving the ramus in which the temporomandibular joint (TMJ) is spared, every effort should be made to plan the resection and reconstruction maintaining the existing joint. In larger lesions, resection of the tumor with free fibula reconstruction is a reasonable approach.
Until recently, surgical treatment was the only option for the treatment of fibrous dysplasia. Recently, several small series have been published using medical therapy with pamidronate, an aminobisphosphonate.3 This treatment has resulted in an increase in bone mineral density and radiologic signs of healing with the thickening of the cortical bone in some cases. In many cases, there has also been a significant decrease in pain with pamidronate therapy. Bisphosphonate therapy can be complicated by the formation of bony sequestra. Figure 28.2 shows a bony sequestrum of the maxilla that eroded through the palate. Radiation therapy is contraindicated because of an increased propensity for malignant transformation.
MOEBIUS SYNDROME
Moebius syndrome is a rare disorder characterized by absence of certain cranial nerves. Classically, Moebius syndrome is defined by the absence of the sixth and seventh cranial nerves resulting in masklike facies, incapable of animation, and an inability to laterally deviate the eyes (abducens palsy). The inability to show a facial response to verbal and nonverbal communication is a devastating deficiency. In the United Kingdom, there were approximately 90 cases of Moebius syndrome in a population of 50 million people, yielding a prevalence of 1 in 550,000. By extrapolation, in the United States there would be expected to be approximately 500 cases. In some publications, Moebius syndrome is defined more broadly, including patients with additional facial nerve palsies. Involvement of nearly all of the facial nerves has been documented, but the third, ninth, tenth and twelfth nerves are most commonly involved.4 In addition to the characteristic facies associated with the sixth and seventh nerve palsies, ptosis, nystagmus, or strabismus may be present, and epicanthal folds are frequent (Figure 28.3). The nose typically has a high, broad nasal bridge, and this increased breadth extends down-ward to the nasal tip. The mouth opening is typically small. In addition, there can be hypoplasia of the tongue, either unilaterally or bilaterally. There frequently is poor palatal mobility, poor suck, inefficient swallowing, and drooling. The mandible tends to be hypoplastic. These factors can contribute to difficulty feeding during the first year of life, frequently leading to poor growth. A coarse voice and speech impairment can be present, although hearing is usually normal. As the child grows, the ability to open the mouth and feed improves slowly and significantly. The facial paralysis and masklike facies may tend to bias early estimates of psychomotor activity, which tend to be low. Despite the perception created by the lack of facial expression, only 10% to 15% are mentally retarded.4
Etiology and Pathogenesis
The etiology of this disorder has not been clearly elucidated. It occurs sporadically, and in cases of “classic Moebius syndrome” involving only sixth and seventh cranial nerves, genetic transmission is rare. For Moebius syndrome, four separate and distinct, though not mutually exclusive, pathogenetic mechanisms have been advanced: (1) aplasia/hypoplasia of the cranial nerve nuclei; (2) destruction of the cranial nerve nuclei; (3) peripheral nerve abnormalities; and, (4) primary myopathy. Operative findings and postmortem examinations have supported each of these four mechanisms.4,5 In those patients with cranial nerve nuclei deficits, as with other central deficits, “downstream” changes in nerve and muscle would be expected to occur.
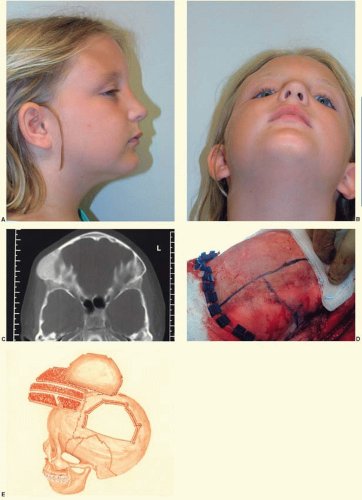 FIGURE 28.1. Fibrous dysplasia. A. Preoperative lateral view of fibrous dysplasia involving right orbit, frontal bone, and parietal bone. B. Preoperative “worm’s eye” view. C. Preoperative axial CT image. D. Intraoperative view showing increased density of fibrous dysplasia involving right frontal and right parietal bones. E. Planned split graft donor site using metallic template fabricated in 1F (illustration by Min Li MD). F. Intraoperative view following completed resection of tumor and reconstruction of orbital roof and supraorbital bar using split cranial bone grafts from left parietal bone and contour reduction of right parietal bone. G. CT scan showing completed reconstruction. H. Three-year postoperative frontal view. I. Three-year postoperative “worm’s eye” view. |
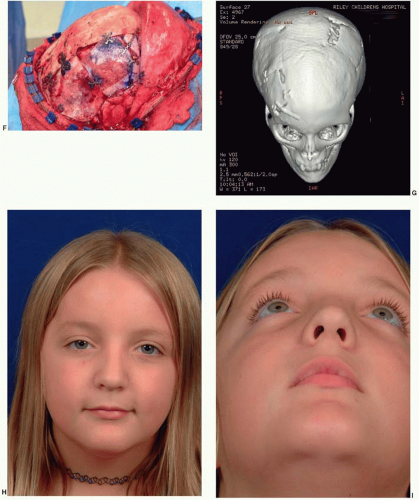 FIGURE 28.1. (Continued) |
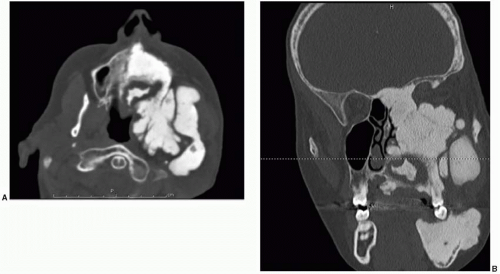 FIGURE 28.2. A. Axial and (B) coronal images of bony sequestrum of maxilla in patient with fibrous dysplasia that eroded through palate. |
Treatment
Treatment has progressed more rapidly than our understanding of this rare disorder. This treatment is focused on alleviating one of the most socially devastating problems of this disorder—the inability to show a facial response. Facial reanimation surgery, which employs microvascular free muscle transfer innervated through the use of a suitable motor nerve, is effective in restoring function in many different etiologies of facial nerve deficits, and this treatment has been extended to Moebius syndrome. Zuker et al. report a series of 10 patients who had an average movement of their oral commissures following bilateral microvascular transfers of 1.37 cm, allowing for meaningful and deliberate facial animation.5 In addition, the children showed improvement with drooling and improved ability to drink. The surgery also has a definite benefit on speech.5
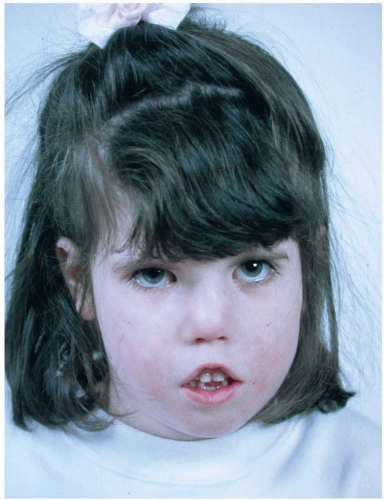 FIGURE 28.3. Child with Moebius syndrome with typical masklike facies and downslanted oral commissures. |
ROMBERG DISEASE (PROGRESSIVE HEMIFACIAL ATROPHY)
Progressive hemifacial atrophy (PHA) is widely known by the eponym Romberg disease. In an era of molecular and genetic analysis, PHA remains the most enigmatic of the craniofacial disorders. The etiology of this disorder remains unknown.
Clinical Findings
PHA may involve any or all of the facial tissues, typically involving skin and subcutaneous tissue, but also potentially muscle, cartilage, and bone. Although there is new evidence that the trigeminal nerve (V) is involved, Pensler et al.6 have reviewed the clinical course in 41 patients and report that the initial presentation included the distribution of V1 in 35% of cases, the distribution of V2 in 45% of cases, and the distribution of V3 in the remaining 20% of cases. Facial involvement is unilateral in 95% of all cases, and either side of the face is equally likely to be involved. The initial presentation typically involves the skin and may be quite subtle, sometimes including pigment changes in which there may be either a brownish or bluish color to the skin, or even hypopigmentation. Alternatively, the disorder may present as a limited area of atrophy of the subcutaneous fat. A striking archetypal presentation often includes a nearly vertical linear depression of the forehead extending into the eyebrow and frontal hairline, known as the coup de sabre, or “cut of the saber” (Figure 28.4). This clinical sign was thought to be
pathognomonic for Romberg disease, but can also be noted in linear scleroderma, a subtype of localized scleroderma, and this has led to a potential overlap of these diagnoses.
pathognomonic for Romberg disease, but can also be noted in linear scleroderma, a subtype of localized scleroderma, and this has led to a potential overlap of these diagnoses.
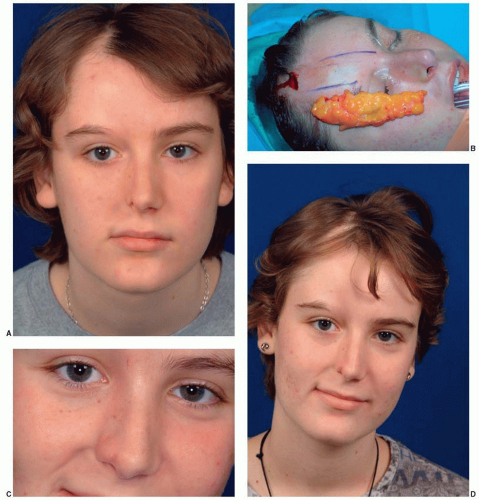 FIGURE 28.4. Progressive hemifacial. A. Frontal view of 14-year-old female with onset at 10 years. There is a large area of alopecia of scalp, mild soft tissue depression, loss of medial eyebrow, and vertical deficiency of right alar rim consistent with a mild “coup de sabre” type deformity. The nose not only shows vertical deficiency, but also thinning and collapse. Radiographic evaluation revealed no evidence of skeletal irregularity. B. Intraoperative view showing dermal fat graft in position for grafting soft tissue deficiency of forehead. An ear cartilage conchal bowl composite graft was used to reconstruct the alar deficiency after the rim was dissected free. C. Initial postoperative result of ala reconstruction. D. Postoperative result. The dermal fat graft initially led to significant overcorrection of the deficiency, but now yields a favorable result. The alar correction was diminished by the late presentation of a Pseudomonal infection of the cartilage. |
PHA is not a congenital disorder, with the typical onset being in the first or second decade of life. The hallmark of the disorder is a slowly progressive course, with an “active phase” of disease characterized by involution, or “wasting away” of the skin, subcutaneous tissue, and muscle. This active phase lasts from 2 to 10 years. The subcutaneous tissue is the most severely involved, followed by substantial involvement of the skin and muscle. The facial musculature undergoes thinning, but usually maintains sufficient power to animate the face. The muscle involvement commonly includes atrophy of the tongue and palatal tissues. Patients with an early age of onset (during facial growth) are much more likely to have significant skeletal involvement. Pensler et al.6 report that 65%
of their patients had osseous involvement, and they found a strong correlation between the age at onset and the degree of bone hypoplasia. However, in their review, they noted no correlation between the other findings of severity of soft tissue atrophy, the duration of the disease, the initial site of skin changes, and the eventual location or magnitude of the skeletal involvement.
of their patients had osseous involvement, and they found a strong correlation between the age at onset and the degree of bone hypoplasia. However, in their review, they noted no correlation between the other findings of severity of soft tissue atrophy, the duration of the disease, the initial site of skin changes, and the eventual location or magnitude of the skeletal involvement.
In cases where the disease occurs during the first decade of life, profound skeletal hypoplasia is usually present. This stands in distinct contrast to those cases that present initially in the second decade of life. In these latter cases, there is typically limited, if any, impact on the facial skeleton with the gross morphologic changes being limited to the skin and subcutaneous tissue. The patient in Figure 28.4 had disease onset at 10 years of age. There is clinical involvement of the skin, eyebrow, and periocular tissues. There is clearly hypoplasia or atrophy of the right nasal sidewall and cartilaginous atrophy of the nasal ala. Her dental development and occlusal relations show no signs of involvement. In view of this strong correlation of the severity of this disorder with the age of onset, it is unclear whether the facial skeleton actually undergoes atrophy. More likely, the bone fails to develop fully in the field of overlying atrophy of the skin and subcutaneous tissue. The skeletal “atrophy” in PHA is more accurately termed hypoplasia.
In early onset cases, the skeletal involvement often includes the mandible and midface, with concomitant implications for the occlusal relationships and facial appearance. There can be hypoplasia of the mandible, including significant vertical undergrowth of the ramus and a deficiency in posterior facial height. The mandible may also show significant sagittal undergrowth. The maxilla may also manifest both vertical and sagittal undergrowth in the sagittal plane. Because the involvement is unilateral, profound tilting of the occlusal plane develops. When PHA involves periorbital tissues enophthalmos is a frequent finding. Pensler et al., based on radiographic orbital measurements, suggest that the enophthalmos is not due to a skeletal change in the orbital volume, but that it is related to atrophy of the periorbital soft tissues.6
PHA can be associated with many other findings, including areas of skin and subcutaneous atrophy elsewhere on the body distinct from the face. The disorder is associated with nervous system dysfunction including Horner syndrome, trigeminal neuralgia, and unilateral mydriasis. Central nervous system involvement has been reported in smaller series by several authors, ranging from MRI changes to seizure disorders. However, the relative paucity and inconsistency of data at this point precludes any definitive correlation between these reports.
Etiology
The etiology of PHA is unknown. PHA does not show any genetic predilection, is found in all races, and there is no evidence of a hereditary basis. It does occur more frequently in females in most series. Patients will frequently remember an “initiating event” in PHA and the onset of the disorder is often linked to an episode of trauma or infection. However, it is unclear whether this is simply an event that calls attention to a subtle area of initial clinical involvement, or whether there are true pathogenetic associations. Traditionally, three theories have been advocated for the etiology of Romberg disease: the infection hypothesis, the trigeminal-peripheral neuritis hypothesis, and the sympathetic hypothesis. The infectious hypothesis was historically linked to an irritation of nerves. In the current era of a new understanding of infectious agents (viruses, prions, mad cow disease, chronic wasting disease of deer, etc.), the infectious hypothesis may be remain a tenable etiology until a definitive understanding of this disorder is truly established. The trigeminal-peripheral neuritis hypothesis suggests a neuritis involving the trigeminal nerve, and is supported by episodes of pain in the involved areas prior to the onset of tissue involution. The sympathetic hypothesis is based on an association of Horner syndrome, pilomotor reflex changes, unilateral mydriasis, vasomotor disorders, unilateral migraine, and perspiration disorders. Based on current evidence, no definitive etiology has been established.
The insightful work of Pensler et al. has provided some enhanced understanding.6 First, in their clinical review, they found no evidence of sensory, sympathetic, parasympathetic, or sudomotor dysfunction. Muscles of mastication and facial expression were found to be fully functional. Biopsies revealed epidermal atrophy and a variable perivascular mononuclear cell infiltrate, with morphologic characteristics of lymphocytes and monocytes, that were grouped around dermal neurovascular bundles. Many of the venules were noted to have striking degenerative alteration in the lining epithelium with reduplication of the basal lamina. Significantly, they also noted that elastic fibers were present and morphologically intact (in contrast to linear scleroderma). They interpret these findings as being consistent with a lymphocytic neurovasculitis, and they advance this theory as a pathogenetic mechanism.
Understanding the pathogenesis of this disease is complicated further by the apparent overlap between the disorder of linear scleroderma and PHA. It is very likely that many of the cases that have historically been termed Romberg disease may include cases of linear scleroderma, since differentiating the two clinically is difficult, if not impossible. Linear scleroderma may also show monocytic infiltrates. The only finding that has been reported as useful to differentiate these two disorders is the absence of elastic fibers in the scleroderma group, and their preservation in the PHA group.6 The fact is that these two diagnoses may be describing the same entity.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


