Accessory tragus502
ACCESSORY TRAGUS
Histopathology1
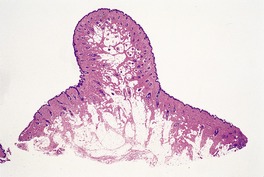
Fig. 19.1
Fig. 19.2
SUPERNUMERARY NIPPLE
Histopathology17
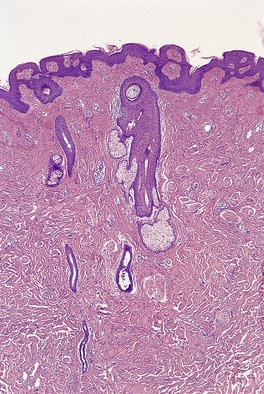
Fig. 19.3
ACCESSORY SCROTUM
ECTOPIC TISSUES
CHEILITIS GLANDULARIS
Histopathology
UMBILICAL LESIONS
Histopathology
RELAPSING POLYCHONDRITIS
Histopathology78
Electron microscopy
ACANTHOSIS NIGRICANS
Paraneoplastic
Carcinomas of alimentary tract, liver, kidney, bladder, lung, cervix and lymphomas
Endocrine and congenital
Insulin resistance syndrome, hyperinsulinemia, lipoatrophy, obesity, Prader–Willi syndrome, leprechaunism, pineal hyperplasia
Familial
Inherited disorder, Down syndrome, ectodermal dysplasia (Lelis’ syndrome), SADDAN, and Crouzon syndromes, FGFR3 mutations
Drugs
Somatotropin, corticosteroids, oral contraceptives, diethylstilbestrol (stilbestrol), methyltestosterone, niacin (nicotinic acid), triazinate, gemfibrozil, amprenavir, palifermin, topical fusidic acid
Histopathology
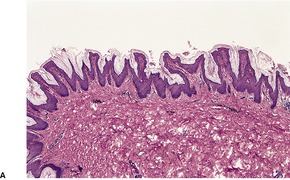
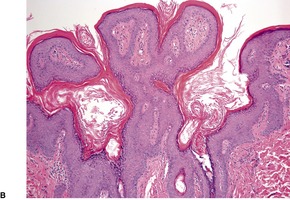
Fig. 19.4
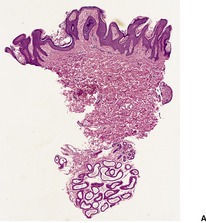
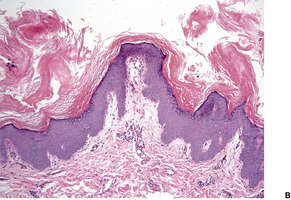
Fig. 19.5
CONFLUENT AND RETICULATED PAPILLOMATOSIS
Histopathology197. and 229.
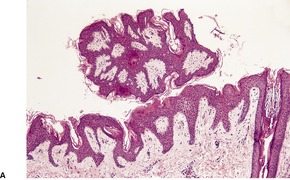
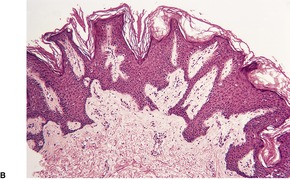
Fig. 19.6
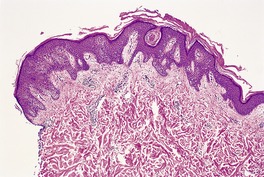
Fig. 19.7
ACROKERATOSIS PARANEOPLASTICA
Histopathology247
ERYTHRODERMA
Pre-existing dermatosis
Psoriasis, atopic dermatitis, seborrheic dermatitis, allergic contact dermatitis, photosensitivity syndromes, pityriasis rubra pilaris, stasis dermatitis, pemphigus foliaceus, bullous pemphigoid, HIV infection
Drugs
Phenytoin, penicillin, isoniazid, trimethoprim, sulfonamides, antimalarials, thiazides, gold, chlorpromazine, calcium carbimide (cyanamide), nifedipine, roxatidine, imatinib, allopurinol, NSAIDs, timolol maleate eye drops, hypericum (St John’s wort), recombinant cytokines
Lymphoma
Cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome), extracutaneous lymphoma, rarely with solid tumors
Idiopathic
Histopathology299
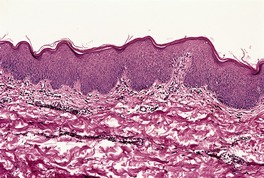
Fig. 19.8
PAPULOERYTHRODERMA
Histopathology
SCALP DYSESTHESIA
CUTANEOUS EMPHYSEMA
Histopathology
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Miscellaneous conditions
Supernumerary nipple502
Accessory scrotum503
Ectopic tissues503
Cheilitis glandularis503
Umbilical lesions504
Relapsing polychondritis504
Acanthosis nigricans504
Confluent and reticulated papillomatosis505
Acrokeratosis paraneoplastica506
Erythroderma507
Papuloerythroderma508
Scalp dysesthesia509
Cutaneous emphysema509
Accessory tragi are usually found as solitary, dome-shaped papules and nodules, present at birth, usually in the preauricular region but sometimes in the neck, anterior to the sternomastoid muscle.1.2.3. and 4. The cervical lesions have been regarded by some as a discrete but closely related entity, also of branchial origin: 5 they have been reported as cervical auricles, ‘wattles’, and congenital cartilaginous rests.6.7. and 8.
Accessory tragi may be multiple, including a linear arrangement, 9 and sometimes bilateral. 10 Rarely, they are associated with other syndromes of the first branchial arch, such as hemifacial microsomia (oculo-auriculo-vertebral spectrum (OAVS) or Goldenhar’s syndrome (OMIM 164210)), in which a myriad of congenital anomalies have been described.9. and 11. In one recent case a deletion in the region of 22q11.2 has led to speculation that this is a candidate gene for the syndrome. 12 It is currently thought that the gene maps to 14q32. Clinically, accessory tragi, including the lower cervical variants, are usually diagnosed as skin tags.
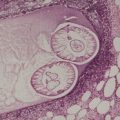
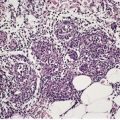
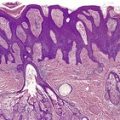
The lesions are polypoid elevations with a fibrovascular zone beneath the epidermis containing numerous hair follicles with small sebaceous glands sometimes attached (Fig. 19.1 and Fig. 19.2). Some of the follicles are of vellus type. Beneath this area, there is a zone of adipose tissue and usually a central core of cartilage. There is a prominent connective tissue framework in the fat, irrespective of the presence of cartilage. 13 Eccrine glands are often present and occasionally there are large nerve fibers and even Pacinian corpuscles.
Accessory tragus. In this case there is adipose tissue but no cartilage in the core. (H & E)
Accessory tragus. Numerous hair follicles, some of vellus type, are present. Sebaceous glands are not well developed. (H & E)
In the cervical lesions there is mature cartilage embedded in fibrous tissue. Striated muscle in continuity with the underlying platysma muscle may also be present in the core. Telogen hairs are not always present in cervical lesions or at least their presence is not mentioned in the reports. Histological features of both hair follicle nevus (see p. 758) and accessory tragus can coexist in a single lesion. As a rule, the accessory tragus has abundant fat cells. 14
The rare condition described as dermatorynchus geneae is a related first arch abnormality in which large amounts of striated muscle and sometimes bone form the central core of the elongated polypoid lesion. 15 The lesions reported as striated muscle hamartomas are closely related16 morphologically; they were not in the preauricular region (see p. 863).
Polythelia, as the presence of supernumerary nipples is sometimes called, is a developmental abnormality found in approximately 1% of the population. 17 Much less common is polymastia, the term used to describe the presence of two or more breasts. This rare phenomenon occurs mostly in the axilla, but other sites have been recorded.18. and 19. Polymastia refers to the presence of nipple, areola, and glandular tissue and if areola is not present, the term supernumerary glandular tissue is more appropriate.20. and 21. There is a predilection for females in both supernumerary nipple and polymastia. Familial cases of supernumerary nipple have been recorded. 22 The supernumerary structure is usually a solitary, asymptomatic, slightly pigmented, nodular lesion, often with a small, central, nipple-like elevation. It may occur anywhere along the pathway of the embryonic milk line, particularly on the anterior aspect of the chest or the upper abdomen. Rarely, it is outside this line.23. and 24. Clinically, it resembles a nevus or fibroma. There is said to be an increased incidence of renal abnormalities in patients with a supernumerary nipple, 18 although recent studies have failed to confirm this association.25.26.27. and 28. Rarely, a small patch of hairs may be the only marker of underlying accessory breast tissue (‘polythelia pilosa’). 29 Accessory breast tissue is rarely seen in a Becker’s nevus. 30
Dermoscopy of one case showed a central white area, central streak, and a very faint pigmented network at the periphery. 31
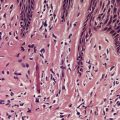
The appearances resemble those seen in the normal nipple and include epidermal thickening with mild papillomatosis and basal hyperpigmentation, the presence of pilosebaceous structures, variable amounts of smooth muscle, and mammary ducts which open into pilosebaceous ducts or enter the epidermis (Fig. 19.3). There may be some underlying breast tissue but, as already stated, complete supernumerary breasts are very rare. 18
Supernumerary (accessory) nipple. There is smooth muscle and a ductal structure. (H & E)
Clear cells of Toker, found in approximately 10% of normal nipples by routine H & E, but in up to 83% by immunohistochemistry, have been demonstrated in 65% of accessory nipples using CK7 staining. 32
Accessory scrotum is the presence of scrotal skin outside of its normal location, but without testicular tissue. It is usually found in the perineal or inguinal region. 33 A normal scrotum is also present. An accessory scrotum has been reported in a patient with a Becker’s nevus (see p. 294).
Agenesis of the scrotum is an exceedingly rare event. 34
There are isolated reports of the occurrence in the skin of ectopic tissue. Most cases represent embryological vestiges, but trauma (surgery or a gun-shot injury) is the explanation for splenic tissue in the subcutaneous tissues of the abdomen.35.36. and 37. Ectopic bowel mucosa may follow congenital eversion or implantation after an –ostomy involving bowel. 38 Rarely, nephrogenic rests are found in the lumbosacral region, either with or without associated spinal dysraphism. 39
Other ectopic tissues reported in the skin have included thymus, 40 respiratory mucosa, salivary gland, 41 thyroid, 42 brain, thymus, 43 and pancreas. There is one report of a subcutaneous thymoma, but it probably spread from its more usual location. 44 Ectopic nail (onychoheterotopia) is a rare condition in which nail-like tissue occurs in a location other than the nail bed.45. and 46. Fewer than 50 cases have been recorded. It may be congenital or acquired, the latter usually following acute trauma or repeated minor injuries to the nail unit.45. and 46.
Cheilitis glandularis is a chronic inflammatory disorder affecting mucous glands and ducts of the lower lip. It appears to be a heterogeneous entity, which has been attributed in the past to hyperplasia of labial salivary glands.47.48. and 49. However, a critical review of reported cases does not support this explanation50 except in a few cases. 51 It has been suggested that the condition includes cases of factitious cheilitis,52.53. and 54. premature and exaggerated actinic cheilitis, and cases with a coexisting atopic diathesis in which mouth breathing may play a role. 50 A case has been reported of cheilitis glandularis superimposed on oral lichen planus. 55 It has also been reported in albinos. 56 It should be noted that only mild swelling of the lip is needed to produce eversion, which in itself exaggerates the appearance of swelling. 50
The patients present with macrocheilia, usually confined to the lower lip. 57 There is often crusting and fissuring with a mucoid discharge. Salivary duct orifices may be prominent. Clinically, the lesions need to be distinguished from plasma cell cheilitis, 58 cheilitis granulomatosis (Melkersson–Rosenthal syndrome – see p. 188), and Ascher’s syndrome, in which there is acute swelling of the lip and eyelids (usually the upper) resulting from edema, inflammation, and a possible increase in size of labial and lacrimal glands respectively.48.59.60.61. and 62.
Squamous cell carcinoma may sometimes supervene, further evidence that many cases have an actinic etiology.47.49. and 63.
Treatment involves elimination of aggravating factors, reinstating oral hygiene, and the use of lip balms and sunscreens. Topical or intralesional corticosteroids, antibiotics, topical 5-fluorouracil cream, cryotherapy, and surgical excision have all been used. In the patient referred to above with concurrent oral lichen planus, treatment with topical tacrolimus and pimecrolimus was successful. 55
There is usually hyperkeratosis, focal parakeratosis, and sometimes inflammatory crusting. There is underlying edema, variable but usually mild chronic inflammation, and variable solar elastosis. 50 Although the minor salivary glands are usually said to be hyperplastic, one study showed no increase in their size or appearance when compared to controls. Notwithstanding, enlargement of salivary glands with dilated ducts and some chronic inflammation have been present in some cases reported as cheilitis glandularis. 51
The umbilicus is an important embryological structure, into which the vitelline (omphalomesenteric) duct and urachus enter. Remnants of either structure may give rise to lesions at the umbilicus and, rarely, vestiges of both may coexist.64. and 65.
The omphalomesenteric duct normally becomes obliterated early in embryonic life but remnants may persist, producing an enteric fistula, an umbilical sinus, a subcutaneous cyst, or an umbilical polyp.66. and 67. The latter presents as a bright red polyp or fleshy nodule, 0.5–2 cm in diameter; 68 it may discharge a mucoid secretion. A distinctive umbilical polyp, devoid of any epithelial component, and termed a fibrous umbilical polyp is discussed further below.
Urachal anomalies usually present at or shortly after birth. 64 They may present as periumbilical dermatitis. 69 A patent urachus will result in the passage of urine from the umbilicus. Urachal sinuses and deeper cysts result from partial obliteration of the urachus, with small persistent areas. 70
Other lesions presenting at the umbilicus include endometriosis (see p. 455), primary71 and secondary tumors (see p. 928), 72 and inflammatory granulomas. 73 The granulomas result from inflammatory changes associated with persistent epithelialized tracts or simply from accumulation of debris in a deep umbilicus with resulting ulceration and inflammation. 64 Sometimes a pilonidal sinus is present. 74
The fibrous umbilical polyp appears to be a distinctive lesion of early childhood with an uncertain pathogenesis. 75 It has a marked predilection for boys. A series of 19 cases from one institution suggests that it is not rare. They ranged in size from 0.4 to 1.2 cm in diameter.
Umbilical (omphalomesenteric duct) polyps are covered by epithelium which is usually of small bowel or colonic type, but occasionally of gastric type. Ectopic pancreatic tissue has also been described. 66 There is usually an abrupt transition from epidermis to the intestinal or gastric type of epithelium. Urachal remnants are lined by transitional epithelium; 64 sometimes smooth muscle bundles are present in their wall. There may be a mild inflammatory cell infiltrate both in umbilical polyps and in urachal remnants.
Umbilical granulomas show variable inflammatory changes ranging from abscess formation to granulomatous areas. 64 Sometimes hair shafts or debris are present with associated foreign body giant cells. There is variable fibrosis.
The fibrous umbilical polyp is a dome-shaped lesion with a stromal proliferation of moderately cellular fibrous tissue without significant inflammation. 75 Fibroblastic cells are plump to elongate with abundant pale pink cytoplasm. Some cells show enlarged stellate nuclei or a ganglion cell appearance. Collagen is sparse to moderate in amount. The overlying epidermis shows a loss of rete ridges and basket-weave hyperkeratosis. Some cases show focal staining for muscle-specific actin and desmin but no staining for cytokeratin, CD34, S100, or epithelial membrane antigen. 75
Primary tumors of the umbilicus may take the form of adenocarcinoma, sarcoma, melanoma, squamous cell carcinoma or, rarely, basal cell carcinoma. 72 Rarely, umbilical adenocarcinomas assume a papillary pattern with psammoma bodies. 76 A clear cell acanthoma-like lesion of the umbilicus has been reported in an infant. 77
Relapsing polychondritis is a rare multisystem disorder of unknown etiology manifesting with recurrent inflammation of the cartilaginous tissues of different organs, particularly the ear, but also the nasal septum and tracheobronchial cartilage.78.79.80.81. and 82. A frequent presentation is with tenderness and reddening of one or both ears resembling an infectious cellulitis.83.84. and 85. Onset is usually in middle age. Pediatric cases are uncommon. 86 Other manifestations include polyarthritis, ocular inflammation, audiovestibular damage, cardiac lesions, and a vasculitis.78.87. and 88. Relapsing polychondritis has been reported in association with psoriasis, 89 aseptic abscesses, 90 myelodysplastic syndromes,91. and 92. malignant lymphoma,93. and 94. acute febrile neutrophilic dermatosis (Sweet’s syndrome), 95 human immunodeficiency virus infection, 96 and, in one case, pseudocyst of the auricle. 97 Its association with Behçet’s disease has been called ‘MAGIC syndrome’ (mouth and genital ulcers with inflamed cartilage). 98 Relapsing polychondritis needs to be distinguished from Wegener’s granulomatosis. 99 The course of the disease is variable, but there is usually progressive destruction of cartilage, with consequent deformities. Death ensues in up to 25% of patients as a result of respiratory and cardiovascular complications.
There are pointers to an immunological pathogenesis. These include the detection of cell-mediated immunity to cartilage, the presence of antibodies to type II collagen, 100 and the demonstration by direct immunofluorescence of immunoglobulins and C3 in the chondrofibrous junction and around chondrocytes.97. and 101. A case thought to have been precipitated by the use of the drug goserelin (Zoladex) for the treatment of prostatic adenocarcinoma has been reported. 102
Colchicine,103. and 104. dapsone,105. and 106. and corticosteroids have been used in the management.
The initial changes are a decrease in the basophilia of the involved cartilage, degeneration of marginal chondrocytes which become vacuolated with pyknotic nuclei, and a florid perichondritis with obscuring of the chondrofibrous interface. The inflammatory cell infiltrate initially contains many neutrophils, but there are progressively more lymphocytes, plasma cells, and histiocytes in the infiltrate, with occasional eosinophils. A vasculitic component is sometimes present. With time there is derangement of the cartilaginous matrix and its replacement by fibrous tissue. 107 Calcification and even metaplastic bone may develop in the late stages when only a scattering of chronic inflammatory cells remain.
A large number of dense granules and vesicles, compatible with matrix vesicles or lysosomes, surround the affected chondrocytes. 108
Acanthosis nigricans is a cutaneous manifestation of a diverse group of diseases which include internal cancers and various endocrine and congenital syndromes (Table 19.1).109.110.111. and 112. It may occur as an inherited disorder and in association with Down syndrome. 113 It may also be related to the ingestion of certain drugs. In all these circumstances, acanthosis nigricans presents as symmetrical, pigmented, velvety plaques and verrucous excrescences confined usually to the flexural areas of the body, particularly the axillae.109. and 110. It may also involve the back of the neck, the periumbilical and anogenital regions, and the face.114.115. and 116. Rarely, there is generalized involvement of the skin.117.118.119. and 120. The oral mucosa, particularly that of the lips and tongue, is affected also in 25% or more of cases. 121 Involvement of the esophagus has been reported. 122 Hyperkeratotic lesions may develop on the palms, soles, and knuckles.123.124. and 125. Such cases should be distinguished from the condition called acral acanthotic anomaly or acral acanthosis nigricans in which velvety, hyperpigmented plaques occur on the elbows, knees, knuckles, and the dorsal surfaces of both feet in individuals with a dark complexion. 126. and 127.
The paraneoplastic type of acanthosis nigricans is a rare manifestation of an internal cancer, usually an adenocarcinoma of the stomach or other part of the alimentary tract.128.129.130.131.132.133. and 134. Lymphomas, 135 renal carcinoma, 136 endometrioid carcinoma of the parametrium, 137 lung carcinoma, 138 bladder carcinoma, 139 liver tumors, 140 and squamous cell carcinomas141 are occasionally associated. Acanthosis nigricans may precede or follow the diagnosis of the cancer but in most instances the two are diagnosed simultaneously.129.137. and 142. The sign of Leser–Trélat, another paraneoplastic phenomenon, may occur in association with acanthosis nigricans. 134 Florid cutaneous papillomatosis may also accompany this variant of acanthosis nigricans. 143 There are some reports of the reversibility of the skin lesions upon removal or treatment of the accompanying malignant disease.129. and 142.
The various endocrine disorders and congenital syndromes which may be complicated by acanthosis nigricans appear to have in common a resistance of the tissues to the action of insulin;112.144.145.146.147.148.149.150.151. and 152. hyperinsulinemia may be present.153. and 154. Insulin resistance is present in the case of lipoatrophy (lipodystrophy),155.156.157.158. and 159. the Prader–Willi syndrome, 160 leprechaunism, 156 pineal hyperplasia, and the so-called ‘type A and B insulin resistance syndromes’ (Rabson–Mendenhall syndrome – OMIM 262190).144.145.161.162. and 163. Obesity is often present.164. and 165. Acanthosis nigricans has been found in 19%, 23%, and 4% of African American, Hispanic, and non-Hispanic white youth, respectively, who have a body mass index ≥98th percentile. 166 Alterations in leptin levels have also been implicated.126. and 159. A combination of acanthosis nigricans and insulin resistance is found in approximately 5% of hyperandrogenic females, often in association with polycystic ovaries.145.148.167. and 168. Acanthosis nigricans localized to a plaque of scleredema has been reported in a diabetic patient. 169 Its occurrence in pregnancy, not complicated by diabetes mellitus, is difficult to explain. 170
Onset of the rare familial cases is in early childhood.171.172. and 173. There may be accentuation of symptoms at puberty. This variant is inherited as an autosomal dominant trait, although there may be variable phenotypic expression. 171 The association of acanthosis nigricans with ectodermal dysplasia has been called Lelis’ syndrome (OMIM 608290). 174 Other syndromes associated with acanthosis nigricans include the severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN syndrome – OMIM 187600), and Crouzon syndrome with acanthosis nigricans (OMIM 612247). They result from mutations in the fibroblast growth factor receptor 3 (FGFR3) gene on chromosome 4p16.3.175. and 176. Sometimes familial acanthosis nigricans occurs with mutations in this gene, but there are no associated dysmorphic features. 177 Rare cases, distributed along Blaschko’s lines, have variously been called nevoid acanthosis nigricans or epidermal nevus of the acanthosis nigricans type. 178 In a series of four cases, the presence of typical acanthosis nigricans elsewhere in one case suggests the possibility that the unilateral component along Blaschko’s line may be an example of type 2 mosaicism. 179 Of further interest is the finding that a large proportion of epidermal nevi are caused by mutations in the gene controlling activating fibroblast growth factor receptor 3 (FGFR3) in the epidermis, 180 a finding also made in some cases of acanthosis nigricans (see above) and in some seborrheic keratoses. 181
Drugs that have been incriminated in the causation of acanthosis nigricans include somatotropin, 182 corticosteroids, 183 niacin (nicotinic acid),184. and 185. oral contraceptives, diethylstilbestrol (stilbestrol), the folic acid antagonist triazinate, 186 gemfibrozil, amprenavir, 187 palifermin, 188 and methyltestosterone. 189 Acanthosis nigricans-like lesions have developed in ichthyotic skin following the topical application of fusidic acid. 190 Its occurrence at the site of repeated insulin injections is rare. 191
The molecular basis of acanthosis nigricans is becoming increasingly clearer; it appears to represent an abnormal epidermal proliferation in response to various factors.158. and 192. Members of the tyrosine kinase receptor (TKR) superfamily, including epidermal growth factor receptor (EGFR), insulin growth factor receptor 1 (IGFR1), and fibroblast growth factor receptors (FGFRs), particularly FGFR3, are some of the factors involved. 192 They have both mitogenic and antiapoptotic effects on keratinocytes. In the case of the paraneoplastic group, the factor may be a tumor-produced peptide, such as epidermal growth factor (EGF) or transforming growth factor (TGF-α),138. and 193. while in the group related to tissue insulin resistance, the tissue growth factors include insulin itself, which may be increased in some of these conditions.148.158. and 194. Alterations in insulin growth factor receptors also occur. 192 The characteristic flexural localization remains an enigma. The rare keratins, 18 and 19, have been reported in basal keratinocytes in acanthosis nigricans. 195
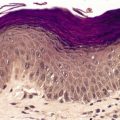
There is hyperkeratosis and papillomatosis but only mild acanthosis (Fig. 19.4). 110 The papillomatosis results from the upward projection of finger-like dermal papillae which are covered by thinned epidermis. 121 In the ‘valleys’ between these papillary projections the epithelium shows mild acanthosis with overlying hyperkeratosis (Fig. 19.5). 121 There may be some hyperpigmentation of the basal layer, but it should be noted that the pigmentation of the lesions noted clinically results largely from the hyperkeratosis. In some instances, there is hypertrophy of all layers of the epidermis and the pattern resembles that seen in epidermal nevi (see above). A resemblance to seborrheic keratoses has been noted in some cases. There is usually no dermal inflammation. There is a close histological resemblance to the lesions of confluent and reticulated papillomatosis (see below).
(A) Acanthosis nigricans. (B) The papillomatosis and intervening ‘valleys’ are conspicuous in this case. (H & E)
Acanthosis nigricans. (A) This biopsy taken from the axilla shows a less regular appearance than the previous case. (B) Another atypical case illustrating the variability that this condition can show. (H & E)
Oral lesions differ from the cutaneous ones by showing marked thickening of the epithelium with papillary hyperplasia and acanthosis. 121 There is a superficial resemblance to the lesions of condyloma acuminatum. There is usually mild chronic inflammation in the submucosal tissues. 121
Finally, it is worth noting that a pattern resembling acanthosis nigricans has been reported at the site of repeated insulin injections. 196
Confluent and reticulated papillomatosis (of Gougerot and Carteaud) is a rare form of papillomatosis characterized by the development of asymptomatic, small red to brown, slightly verrucous papules with a tendency to central confluence and a reticulate pattern peripherally.197.198.199. and 200. Uncommonly the lesions are non-pigmented with a fine white scale. 201 Pruritus has been reported. 202 It involves particularly the upper part of the chest and the intermammary region and back; the neck, chin, upper parts of the arms, and the axillae may also be involved. It has a predilection for younger individuals, with the mean age of onset 15 years in one series of 39 patients. 203 Familial occurrence has been documented.204.205. and 206.
It has been regarded as a variant of acanthosis nigricans, as a genodermatosis, as an unusual response to ultraviolet light207 or to Pityrosporum orbiculare infection,208.209.210. and 211. and as a result of some unidentified endocrine imbalance.197. and 212. In 2005, a previously unknown Dietzia strain (an actinomycete) was isolated from the skin scrapings of a case. 213 This remains to be confirmed. Its coexistence with acanthosis nigricans has been reported. 214
Response to calcipotriene (calcipotriol)215. and 216. and retinoids217. and 218. favors the theory that it is an abnormality of keratinization. On the other hand, response to various antibiotics, particularly minocycline, has also been reported.203.219.220.221.222.223.224.225.226. and 227. Minocycline is now the treatment of choice. It has also been successfully treated with topical mupirocin ointment. 228
The epidermis is undulating with hyperkeratosis, low papillomatosis, and some acanthotic downgrowths from the bases of the ‘valleys’ between the papillomatous areas (Fig. 19.6). Papillomatosis may be absent or subtle in early or late lesions of this condition, but it has been suggested that the presence of basket-weave hyperkeratosis invaginating through the epidermis may be a ‘clue’ in such cases. 230 There may also be mild basal hyperpigmentation and focal atrophy of the malpighian layer. These changes resemble acanthosis nigricans, 231 although they are not usually as well developed as in this condition (Fig. 19.7). However, there may be mild dilatation of superficial dermal blood vessels and sometimes beading of elastic fibers, changes not usually attributed to acanthosis nigricans.
(A) Confluent and reticulated papillomatosis with papilloma formation. (B) The epidermis is undulating with acanthosis. (H & E)
Another case of confluent and reticulated papillomatosis with less papillomatosis. (H & E)
Some cases have a resemblance to seborrheic keratosis but the interface between the adjacent normal skin and lesional skin is better defined in seborrheic keratosis. 232
Acrokeratosis paraneoplastica (Bazex’s syndrome) is a rare paraneoplastic dermatosis associated with cancers which are usually supradiaphragmatic234.235.236.237.238. and 239. in origin, although tumors at other sites have also been incriminated.240. and 241. Sarcomas and lymphomas have also been implicated in the etiology.242. and 243. It commences with violaceous erythema and psoriasiform scaling on the hands and feet with later extension to the ears and nose.244. and 245. Violaceous keratoderma of the hands and feet develops, and ill-defined psoriasiform lesions eventually form at other sites on the arms and legs. 235 Bullous lesions are rare. 246 These changes in the skin usually precede the onset of symptoms related to the associated cancer.247.248. and 249. Removal of the neoplasm leads to a remission of the condition in 95% of cases and recurrence triggers a relapse. 243 This syndrome has been reported in association with acquired ichthyosis, another paraneoplastic disorder. 250 The pathogenesis of the condition is unknown, although transforming growth factor-α, produced by tumor cells, may play a role. 250
The changes are somewhat variable and not diagnostically specific. There is hyperkeratosis, focal parakeratosis, and acanthosis which may be psoriasiform, or mild and irregular in format. Variable epidermal changes include spongiosis with associated exocytosis of lymphocytes, basal vacuolar change, and scattered apoptotic keratinocytes at all levels of the epidermis.234. and 235. Rarely, intraepidermal or subepidermal blisters have occurred. 251 There is a mild perivascular lymphocytic infiltrate in the papillary dermis. Fibrinoid degeneration of small vessels and scattered ‘pyknotic’ neutrophils have been described in some reports, but specifically excluded in others.234.247. and 252.
Erythroderma (exfoliative dermatitis) is a cutaneous reaction pattern that can occur in a wide variety of benign and malignant diseases.16.253.254. and 255. It is uncommon, with an incidence of 1–2 per 100 000 of the population.253. and 256. Clinically, it is characterized by erythema and exfoliation which involve all or most of the skin surface. 254 Distressing pruritus is often present. 253 Other clinical features include fever, malaise, keratoderma, alopecia, and a mild, generalized lymphadenopathy. Laboratory findings include blood eosinophilia, elevated levels of IgE and, in some, a polyclonal gammopathy.253. and 257. The mean age at onset is approximately 60 years, although cases in infancy, often associated with ichthyosiform dermatoses (see p. 251) or immunodeficiency, have been reported.258.259.260.261. and 262. Omenn’s syndrome (OMIM 603554) combines erythroderma and combined immunodeficiency. 263 There is a male predominance, particularly in the idiopathic group (see below).
Erythroderma is most often seen as an exacerbation of a pre-existing dermatological condition but it may also be drug related or be associated with cutaneous T-cell lymphoma or some other malignant tumors. Approximately 15% of cases are idiopathic (Table 19.2). In one retrospective study, 62.5% were related to an underlying dermatosis, 16% were due to drug reactions, and 12.5% to cutaneous T-cell lymphoma. 264 In a more recent study, there was a pre-existing dermatosis in 74.4%, 14.6% were idiopathic, and drugs and malignancy each accounted for 5.5% of cases. 265
A pre-existing dermatosis is present in more than 50% of cases. 256 Psoriasis is the most common of these.266.267. and 268. Various factors, including the use of systemic steroid therapy or retinoids, have been incriminated in precipitating an erythrodermic crisis in psoriasis.266. and 269. Other underlying dermatoses include atopic dermatitis, seborrheic dermatitis, allergic contact dermatitis, photosensitivity syndromes, 270 pityriasis rubra pilaris and, rarely, stasis dermatitis, dermatophytosis, pemphigus foliaceus, and even bullous pemphigoid.265.271.272.273. and 274. In some countries, there is a significant association with HIV infection; 275 in contrast, pulmonary tuberculosis is a very rare association. 276
Drug-induced cases may follow topical sensitization to neomycin, ethylenediamine, or clioquinol (Vioform). 277 Usually, erythroderma follows the ingestion of a drug such as phenytoin, penicillin, isoniazid, trimethoprim and sulfonamides, antimalarials, 278 thiazide diuretics, gold, chlorpromazine, calcium carbimide (cyanamide), 279 nifedipine, 280 roxatidine, 281 escitalopram, 282 imatinib, 283 or allopurinol.271. and 284. There are a few case reports of erythroderma following the use of certain non-steroidal anti-inflammatory drugs such as sulindac, meclofenamate sodium, and phenylbutazone. 285 One case followed the use of beta-blocker eye drops (timolol maleate) 286 and, another, the use of hypericum (St John’s wort). 287 The use of recombinant cytokine therapy may precipitate erythroderma (see p. 521).288. and 289. Drug-related cases usually have a rapid onset and relatively quick resolution over 2–6 weeks, in contrast to the more prolonged course of the idiopathic and lymphoma-related cases. 271
Approximately 10% of cases are associated with a cutaneous T-cell lymphoma, in the form of the Sézary syndrome or erythrodermic mycosis fungoides. 290 The erythroderma may precede or occur concurrently with the diagnosis of the cancer.254. and 291. Uncommonly, erythroderma is associated with an extracutaneous lymphoma or some other tumor.292. and 293.
The idiopathic group, also known as ‘the red man syndrome’, is associated more often with keratoderma and dermatopathic lymphadenitis than the other groups. 294 It is also more likely to persist than some of the other types. Some cases may progress to mycosis fungoides after many years.294. and 295.
The pathogenetic mechanisms involved in erythroderma are not known. The erythema results from vascular dilatation and proliferation and it has been suggested that interactions between lymphocytes and endothelium may play a role. 296 Circulating adhesion molecules are detectable in erythroderma but their values are not of differential diagnostic use. 297 A lymphocytopenia involving CD4+ T cells is sometimes found, probably a consequence of the sequestration of these cells in the skin. 298
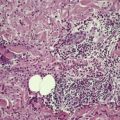
Skin biopsies in erythroderma have been regarded as ‘largely unrewarding’, 284‘of variable usefulness’, 271‘of little value’300 and ‘misleading’. 300 However, in one series, an etiological diagnosis was made on the skin biopsy in 53% of cases301 and, in another, in 66% of cases302 (Fig. 19.8). In this latter series, it was found that diagnostic histopathological features of the underlying disease were retained in the majority of cases. 302 The diagnostic accuracy increases if multiple biopsies are taken simultaneously. 301 Biopsies are most often diagnostic in erythroderma associated with cutaneous T-cell lymphoma and, to a lesser extent, psoriasis and spongiotic dermatitis. 301 Nevertheless, one-third of biopsies from patients with established erythrodermic cutaneous T-cell lymphoma are non-diagnostic. 303 In these cases, an aberrant T-cell immunophenotype may be found; a clonal population of T cells is usually present.303. and 304.
Erythroderma. In this case of atopic dermatitis there is little spongiosis. The epidermal thickening with only mild psoriasiform folding is a feature of atopic dermatitis. (H & E)
Usually, there is variable parakeratosis and hypogranulosis. The epidermis shows moderate acanthosis and, at times, there is psoriasiform hyperplasia, but this finding does not always correlate with the presence of underlying psoriasis. 300 Mild spongiosis is quite common even when there is underlying psoriasis. Other features of psoriatic erythroderma resemble those seen in early lesions of psoriasis with only mild epidermal hyperplasia, mounds of parakeratosis with few neutrophils, and red cell extravasation in the papillary dermis. 305 Blood vessels in the upper dermis are usually dilated and sometimes there is endothelial swelling. 299
There is a moderately heavy chronic inflammatory cell infiltrate in the upper dermis: this is sometimes perivascular in distribution and at other times more diffuse. 306 Atypical cells with cerebriform nuclei are present in the infiltrate in cases of erythroderma related to cutaneous T-cell lymphomas. Eosinophils may be present in the infiltrate and occasionally they are plentiful. 307 Exocytosis of lymphocytes is a frequent finding.
Drug-related cases may sometimes simulate the picture of mycosis fungoides, with prominent exocytosis and scattered atypical cells with cerebriform nuclei in the infiltrate. 307 In contrast to mycosis fungoides, however, Pautrier microabscesses are not present in the benign erythrodermas. Eosinophils are not always present in drug-related cases, as might be expected. 307 A very occasional apoptotic keratinocyte may be a clue to the drug etiology. Rarely, the picture is frankly lichenoid in type.280. and 308.
Papuloerythroderma of Ofuji is a rare entity, first reported from Japan, featuring widespread erythematous, flat-topped papules with a striking sparing of body folds (the so-called ‘deck-chair’ sign).309.310.311. and 312. Eosinophilia and lymphopenia are sometimes present. It occurs most commonly in elderly males. It is often associated with underlying lymphoma or cancer.313.314. and 315. It has developed in patients with atopic erythroderma, 316 HIV infection, 317 hepatitis C infection, 318 and in one with biliary sepsis. 319 Aspirin and furosemide (frusemide) have also been suggested as precipitants.320. and 321. Papuloerythroderma appears to be a distinct clinical entity, but its etiology is unknown. It has been suggested recently that it may be an early variant of mycosis fungoides.322.323.324.325.326. and 327. Most cases run a chronic course. Several treatment regimens have been used with varying success. 328
The epidermis is usually normal, although it may show slight acanthosis, spongiosis, and parakeratosis. There is a dense, predominantly perivascular infiltrate of lymphocytes, plasma cells, and eosinophils in the upper and mid dermis.
S100-positive dendritic cells are abundant in the dermis. 311
Scalp dysesthesia is characterized by symptoms of burning, stinging, or itching, which is often associated with psychological stress. The author has received biopsies (all normal) from at least 10 cases, and coined the term ‘burning scalp syndrome’. Sometimes telogen effluvium may accompany this condition. Patients have benefited from low-dose antidepressants. 329
Cutaneous and soft tissue emphysema is rarely encountered in clinical dermatology. 330 It may follow head and neck surgery, including various dental treatments, trauma, cutaneous cryotherapy,331. and 332. intermittent positive pressure ventilation, and lung disease associated with alveolar rupture. 330 It is usually confined to the cervicofacial region but involvement of the elbow region has been described. 333 Clinically it is most often confused with angioedema. 330 It resolves in a few days.
Collagen bundles in the dermis are attenuated and separated by clear spaces. There are no mucin deposits or inflammation. Adipose tissue has been reported to show ‘fragmentation of cell membranes’. 330
1. Brownstein, MH; Wanger, N; Helwig, EB, Accessory tragi, Arch Dermatol 104 (1971) 625–631.
2. Sebben, JE, The accessory tragus – no ordinary skin tag, J Dermatol Surg Oncol 15 (1989) 304–307.
3. Schissel, DJ; Sartori, C, An unusual papule, Arch Dermatol 134 (1998) 499–504.
4. Jansen, T; Romiti, R; Altmeyer, P, Accessory tragus: report of two cases and review of the literature, Pediatr Dermatol 17 (2000) 391–394.
5. Hsueh, S; Santa Cruz, DJ, Cartilaginous lesions of the skin and superficial soft tissue, J Cutan Pathol 9 (1982) 405–416.
6. Sperling, LC, Congenital cartilaginous rests of the neck, Int J Dermatol 25 (1986) 186–187.
7. Hogan, D; Wilkinson, RD; Williams, A, Congenital anomalies of the head and neck, Int J Dermatol 19 (1980) 479–486.
8. Clarke, JA, Are wattles of auricular or branchial origin? Br J Plast Surg 29 (1976) 238–244.
9. Miller, TD; Metry, D, Multiple accessory tragi as a clue to the diagnosis of the oculo-auriculo-vertebral (Goldenhar) syndrome, J Am Acad Dermatol 50 (2004) S11–S13.
10. Tadini, G; Cambiaghi, S; Scarabelli, G; et al., Familial occurrence of isolated accessory tragi, Pediatr Dermatol 10 (1993) 26–28.
11. Resnick, KI; Soltani, K; Bernstein, JE; Fathizadeh, A, Accessory tragi and associated syndromes involving the first branchial arch, J Dermatol Surg Oncol 7 (1981) 39–41.
12. Xu, J; Fan, YS; Siu, VM, A child with features of Goldenhar syndrome and a novel 1.12 Mb deletion in 22q11.2 by cytogenetics and oligonucleotide array CGH: Is this a candidate region for the syndrome? Am J Med Genet 146A (2008) 1886–1889.
13. Satoh, T; Tokura, Y; Katsumata, M; et al., Histological diagnostic criteria for accessory tragi, J Cutan Pathol 17 (1990) 206–210.
14. Ban, M; Kamiya, H; Yamada, T; Kitajima, Y, Hair follicle nevi and accessory tragi: variable quantity of adipose tissue in connective tissue framework, Pediatr Dermatol 14 (1997) 433–436.