Physical symptoms and signs
Psychological
Systemic
Skin and hair
Hot flushes
Anxiety
Atherosclerosis
Dryness
Headache
Humor alterations
Cardiovascular disease
Decreased collagen content
Nocturnal wet
Sadness
Osteoporosis and osteopenia
Reduction in elasticity
Vaginal dryness
Loneliness sense
Increased body fat, mainly abdominal
Reduction of epidermal an dermal thickness
Dyspareunia
Sleep disturbances
Fragility and poor healing
Muscular and articular ache
Memory disturbances
Alopecia
Genitourinary changes
Concentration disturbances
Hirsutism
In fact, associations between estrogen deprivation and skin changes such as dryness, the reduction of epidermal and dermal thickness, decreased collagen content, a reduction in elasticity, fragility, and poor healing have been shown in postmenopausal women [1, 2]. Additionally, changes in hair are commonly observed. Hirsutism (i.e., the appearance of unwanted facial hair), alopecia, skin atrophy, and weakness of facial skin beginning at this stage of a woman’s life are results of the reduction in progesterone and the increasing impact of free and unopposed circulating androgens on the sebaceous glands and hair follicles [3, 4].
Many authors link the decline in estrogen to distinct cellular aging mechanisms involving oxidative damage and cellular senescence. This theory has been proposed mainly because estrogens function as sex hormones and antioxidant molecules that counterbalance oxidative damage, which is also called oxidative stress.
In this chapter, we review the general aspects of postmenopause that are linked to oxidative stress and the relationship of oxidative stress with skin aging.
4.1 Menopause and Aging
Ovarian aging is caused by a gradual decline in the quantity and quality of oocytes that are present in the ovarian tissue until the menstrual cycle reaches its end (i.e., last menstrual bleeding). This process is characterized by the gradual and erratic decrease of estrogen secretion and a consequent changes in gonadotrophins (FSH and LH) due to the failure of feedback signaling by estrogen and possible alterations of hypothalamic pituitary feedback mechanism [5, 6]. Estrogen depletion affects many tissues of the body and produces a wide range of signs and symptoms. However, in the Study of Woman’s Health Across the Nation (SWAN), it has been observed that the symptoms reported frequently as being part of a menopausal syndrome such as hot flashes, night sweats, menstrual irregularities, vaginal dryness, depression, nervous tension, palpitations, headaches, insomnia, lack of energy, difficulty concentrating, and dizzy spells are not inherent manifestations to this process [7]. Indeed, several of these symptoms may be linked to the biological changes seen in aging; although, it is known that the aging is individualized and is determined by genetic and cultural factors [8–10]. It also greatly impacts female fertility related to aging. In fact, ovarian tissue ages much more rapidly than do the tissues of other body systems [5, 11].
In this context, the age-associated malfunction of human cells results from the physiological accumulation of irreparable damage to biomolecules, which is an unavoidable side effect of normal aerobic metabolism. As noted above, this process is termed oxidative stress (OS).
During aging, oxidative stress increases [12] and causes many of the normal and pathological characteristics that are found in the elderly. Moreover, symptoms and pathological findings that are associated with postmenopause may be related, at least in part, to OS, mainly because of estrogen deficiency; therefore, it is possible to say that this stage of life represents the beginning of the aging process in women.
From the cellular point of view, some researchers have found that the luteinizing granulosa cells in women aged >38 years contain higher numbers of mitochondrial DNA (mtDNA) deletions and exhibit reduced expression of antioxidant enzymes compared with those of younger women [11], which suggests oxidative damage. Other studies have shown that age-related changes in the female ovary could be due to a lowering of enzymatic antioxidant defenses and an increase in oxidant molecules production of possible multiple cellular origins (i.e., mitochondria, cytoplasm) [13].
Recently, it has been noted that the age at natural menopause reflects a complex interrelation of health and socioeconomic factors and might be a marker of aging and general health [14]. Similar to aging, premature and early menopause, either spontaneous or induced, are associated with long-term health risks such as premature death, cardiovascular disease, neurologic disease, osteoporosis, optic nerve aging, and glaucomatous neurodegeneration [15, 16]. However, some of these adverse outcomes may be prevented by hormone therapy with estrogens initiated after the onset of menopause. These effects were observed in women exposed to estrogen therapy and are most likely explained by a positive effect on oxidative damage [17]. Moreover, menopausal symptoms are associated with low biological antioxidant potential [18].
A biological model of premature aging is Down syndrome (DS). Individuals with this pathology experience premature aging and dementia with alterations in the oxidative metabolism of neutrophils, which contribute to alterations of physiological functions [19]. It is reported that persons with DS have excess Cu/Zn superoxide dismutase (SOD) activity, which causes an imbalance between the activity of antioxidant enzymes and leads to increased oxidative molecule levels and cellular senescence [19, 20]. In fact, women with DS experience the menopause at an earlier age than women in the general population [21], and this finding has health implications because early menopause is a risk factor for several age-related diseases, as we described previously.
Therefore, it is important to understand the oxidative process and place it in the context of perimenopause and the changes that estrogen deprivation causes in women.
4.2 Oxidative Stress
Oxidative stress, or redox imbalance, occurs when the balance between oxidant reactive species, free radicals (FRs), non-radicals, and antioxidants is disrupted because of an accumulation of reactive species or the depletion of antioxidants; this in turn leads to oxidative damage to biomolecules [22, 23]. This biochemical process was first described by Rebeca Gerschman et al. in 1954 [24], who noted that FRs are toxic agents that cause disease.
By 1956, Denham Harman [25] had linked FRs to aging and the most prevalent chronic conditions that plague this stage of life. This proposal was later reinforced by the discovery of the enzyme SOD by McCord and Fridovich in 1969 [26]. This enzyme lies at the axis of the antioxidant system. At present, it is recognized that the production of reactive species and oxidative stress occurs intracellularly and within the extracellular space. Oxidative damage can compromise cell metabolism, differentiation, proliferation, survival, and reproduction, and the long-term effects of oxidative damage are implicated in skin aging, cancer, and inflammation [27].
To better understand the biological process of oxidative stress, we will divide the topic into its two components: oxidant and antioxidant process.
4.2.1 Oxidant Process
Reactive species can be separated into four groups based in their main atom: oxygen (ROS), nitrogen (RNS), sulfur (RSS), and chlorine (RCS), with ROS being the most abundant [23, 28–30].
ROS are intermediate metabolites that are produced through the univalent reduction of oxygen in normal aerobic metabolism and are part of a group of FRs and non-radicals with highly oxidizing structures. FRs are chemical species that possess one unpaired electron in their last orbital; therefore, they are able to extract an electron from a neighboring atom or molecule to complete its orbit. When two FRs share their unpaired electrons, non-radical molecules are formed [22, 28, 30].
In living organisms, ROS are produced by various metabolic processes, such as the mitochondrial respiratory chain, phagocytosis, detoxification reactions in cytochrome P450, and prostaglandin synthesis, as well as various inflammatory and nonenzymatic reactions between oxygen and organic compounds that include nicotine adenine dinucleotide phosphate (NADPH) oxidase. ROS can be created by exposure to ionizing radiation but are formed mainly by oxidation-reduction (redox) reactions. It is known that ROS can interfere with cell differentiation, aging, and apoptosis [28, 31–34].
The main generator of ROS is the mitochondria, as a side effect of the electron transport system of the respiratory chain, whose principal function is ATP production. In this process, electrons are transferred by the tetravalent reduction of oxygen to form water (H2O), but approximately 1–3 % of the electrons are lost in the system, producing the superoxide anion (O2 •−), the first FR molecule that is formed by the addition of one electron to oxygen, mediated by NADPH oxidase [23]. As a cascade of electron transfer reactions occurs, O2 •− is produced in the union sites of the ubiquinone in the I (NADH:ubiquinone oxidoreductase) and III (ubiquinol:cytochrome c oxidoreductase) complexes. The O2 •− is then converted to hydrogen peroxide (H2O2), which is a non-radical molecule with strong oxidant power, by the action of superoxide dismutase (SOD, EC 1.15.1.1); H2O2, in turn, is acted upon by catalases (EC 1.11.1.6), peroxidases, and peroxiredoxins. Glutathione peroxidase (GPx, EC 1.11.1.9) is the main enzyme that is responsible for producing H2O. In the glutathione peroxidase reaction, glutathione is oxidized to glutathione disulfide, which can be reconverted to glutathione by glutathione reductase in an NADPH-consuming process [23, 35–37]. In the presence of transition metal cations such as iron (Fe2+) and copper (Cu2+), H2O2 is converted to a hydroxyl radical (•OH), which is the most reactive and dangerous FR, and the metal is reduced through the Fenton reaction during cellular metabolism. Moreover, O2 •− can oxidize Fe3+. This integrated reaction when produced by iron is named the Haber-Weiss reaction [38, 39]:
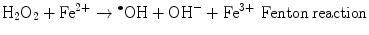
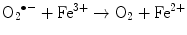
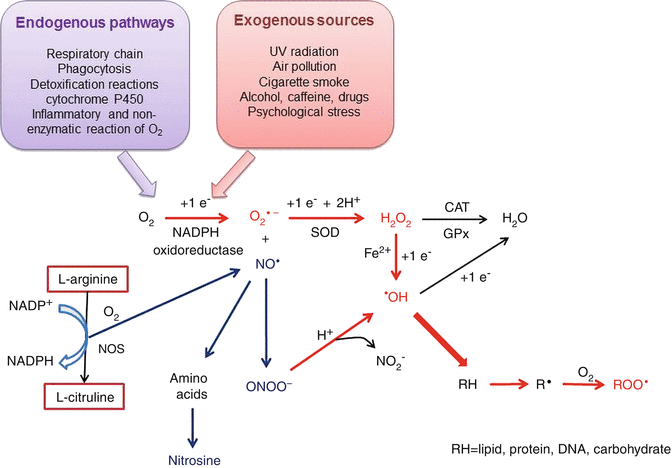
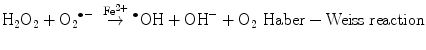 To balance the oxidation reactions, each oxygen atom generates one molecule of H2O, but during intermediary reactions, three ROS are formed by the addition of four electrons (Fig. 4.1):
To balance the oxidation reactions, each oxygen atom generates one molecule of H2O, but during intermediary reactions, three ROS are formed by the addition of four electrons (Fig. 4.1):
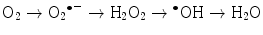 Importantly, it has been documented that •OH can also be formed under reducing conditions, i.e., molecules that function as true antioxidants (reducers) may increase rather than decrease free radical stress [40].
Importantly, it has been documented that •OH can also be formed under reducing conditions, i.e., molecules that function as true antioxidants (reducers) may increase rather than decrease free radical stress [40].
Fig. 4.1
Chain of reactions to produce ROS and RNS, both inside and outside of cells. The reactions begin with the addition of an electron to an oxygen atom either by the metabolic pathways (in purple) or exogenous sources (pink). The ROS formation is in red and the RNS in blue. See corresponding text to explain. SOD superoxide dismutase, CAT catalase, GPx glutathione peroxidase, NOS nitric oxide synthase
Hydroxyl radicals react with biomolecules such as lipids, carbohydrates, proteins, and DNA to produce peroxyl radicals (ROO•−), which conduct a series of chain reactions that result in hydrogen abstraction during biomolecule oxidation. However, O2 •− and H2O2 do not react with most biological molecules.
Recently, it has been noted that the production of ROS in mitochondria is based on its high membrane potential (mtΔΨ) or high amounts of NADH/NAD+. The mechanism that has been proposed is based on results from animal models and suggests a switch of oxidative phosphorylation between high efficiency at low mtΔΨ and low ROS formation, through a second mechanism of respiratory control (RC2 on) and the activated state with maximum rates of ATP synthesis (maximum output power) at lower efficiency accompanied by high mtΔΨ and ROS formation (RC2 off). This hypothesis proposes that health and long life in animals are, in part, dependent on RC2 to maintain low mtΔΨ values and low ROS formation [37].
ROS can also act through indirect mechanisms, and the cascade of effects reflects the changes in the level and function of redox-sensitive transcription factors and signaling proteins such as Nrf2, NF-κB, AP-1, p53, and MAP kinases. These mechanisms are complex and go beyond the scope of this review.
ROS production and release can be influenced by environmental factors such as UV radiation, air pollution, and exogenous toxins such as nitric oxide, cigarette smoke, alcohol intake, caffeine consumption, and exposure to drugs.
The second major group of reactive species in the oxidative stress process is nitrogen species (RNS), which are also formed during intracellular metabolism. The most abundant RNS is the nitric oxide radical (NO•), which is a vasodilator with anti-inflammatory and anticoagulant properties. NO• is a gaseous FR derivate that results from the oxidation of the terminal guanido-nitrogen atom of L-arginine in a reaction that consumes molecular oxygen and reduces equivalents of nicotine adenine dinucleotide phosphate reduced coenzyme (NADPH) to form L-citrulline. This reaction is catalyzed by nitric oxide synthases (NOS). NO• can be rapidly inactivated by reacting with O2 •− to form the strong oxidant peroxynitrite (ONOO−), which, in an acidic environment, is converted to peroxynitric acid and subsequently decomposed to •OH and nitrite (NO2 −) [36, 41, 42]. Additionally, under certain conditions, NO• can react with amino acid residues to produce nitrosine, which is a biochemical compound that inactivates enzymes [43]. It is important to note that NO• and its derivatives (RNS) inhibit mitochondrial respiration reactions and either stimulate or inhibit cell death depending on the prevailing conditions [44].
Reactive species of sulfur (RSS) form the third group, and these species have stressor properties that are similar to those found in ROS. Thiols and disulfides are easily oxidized to sulfur species such as thiyl radicals, disulfides, sulfenic acids, and disulfide-S-oxides. RSS are in turn oxidized and inhibit thiol proteins and enzymes, collaborating to exacerbate oxidative stress [30].
The last group of reactive species is composed of chlorine molecules (RCS) that are formed by the action of myeloperoxidase at the expense of H2O2 in a chloride ion-dependent reaction during phagocytosis. The reaction product, the hypochlorite ion (OCl−), is in balance with its protonated form, hypochlorous acid (HOCl). Under physiological conditions, both compounds are potent oxidizing agents that may attack biomolecules and produce ROS and RNS, as does •OH and ONOO− [28, 45].
4.2.2 Antioxidant Process
Cells have effective mechanisms for the defense against oxidative damage. Because the main reactions involving reactive species take place in the intracellular compartment, these defense mechanisms are usually carried out by enzymes; however, in the extracellular compartment, antioxidant molecules and mechanisms of a different chemical nature are found. The antioxidant scavenging capability of cells varies according to the type of oxidants that attack them, and the ability of antioxidants to scavenge peroxyl radicals might be considerably different for other oxidants. Hence, the system of antioxidant protection occurs at different levels, and these levels are divided into three groups: (a) first level, the actions are directed to prevent the formation of reactive species or to intercept these molecules when they are formed; (b) second level, the mechanisms are directed to trap the reactive species that are formed, thus terminating or preventing the initiation of an oxidative chain reaction; and (c) third level, a group of enzymes that directly restore native biomolecules and other catabolic molecules that can specifically degrade nonfunctional molecules. This degradation can serve to remove oxidized molecules from the cytosol or to fill up the pool of precursors for resynthesis (Fig. 4.2) [36, 46–48].
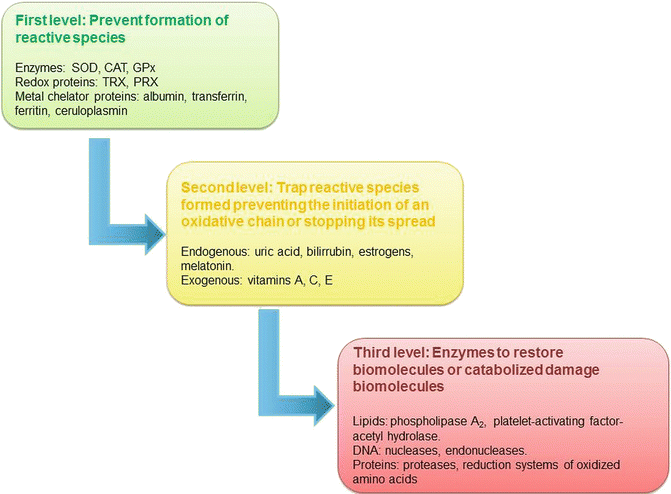
Fig. 4.2
Antioxidant systems by level of action versus oxidative damage. SOD superoxide dismutase, CAT catalase, GPx glutathione peroxidase, TRX thioredoxin, PRX peroxyredoxin
4.2.2.1 First Level of Antioxidant Defense
At this level, we find the major antioxidant enzymes, SOD (EC 1.15.1.11), catalase (EC 1.11.1.6), and GPx (EC 1.11.1.9), and to complete this line of defense, we can add other enzymes, including hemooxygenase-1 (EC 1.14.99.3) and redox proteins such as thioredoxins (TRXs, EC 1.8.4.10) and peroxiredoxins (PRXs, EC 1.11.1.15). All of these molecules are responsible for maintaining a redox steady state [28].
The superoxide dismutases are a group of tetrameric oxidoreductases that catalyze the dismutation of O2 •− to H2O2 and O2. Three isoforms that are located in different subcellular locations but catalyze the same reaction are included in this group: SOD1 (CuZnSOD) in the cytosol, SOD2 (MnSOD) in the mitochondrial matrix, and SOD3 (ecSOD) in the extracellular fluid. SOD2 is of particular interest because the mitochondrial matrix represents the first line of antioxidant defense against the O2 •−that is produced as a byproduct of oxidative phosphorylation [28, 41]. Moreover, it has been reported that SOD1 has a CO2-dependent peroxidase weak activity; this CO2 dependence occurs by generating a strong oxidant in the copper site of the enzyme due to two sequential reactions with H2O2, followed by the oxidation of CO2 to a carbonate radical, which is responsible of several oxidation reactions subsequent [49].
The H2O2 product of the dismutation reaction is more stable than O2 •−. It can traverse membranes and has been shown to be an essential signaling molecule in a variety of signaling cascades and cell matrix interactions [50]. This molecule, in turn, is converted into H2O by the action of catalases, peroxiredoxins, and peroxidases. Catalase is a tetrameric hemoprotein with four heme groups at its active site and is located in the cytosol and peroxisomes. Its catalytic action resides in the iron within its structure that is actively involved in coordinately binding water or forming a complex with an atom of O2 during the reaction. Likewise, catalase can also bind NADPH, which prevents its oxidative inactivation or the formation of complex II by the action of H2O2 because it is reduced to water [28, 51].
Glutathione peroxidases form a group of tetrameric enzymes that contain a molecule of selenium-cysteine in their active sites and use glutathione (GSSG) and other thiols to reduce H2O2 or organic hydroperoxides to water or alcohols, respectively. Four glutathione peroxidase isoforms have been identified in mammals: GPx1 (in the cytosol and mitochondria), GPx2 (in the intestinal epithelium), GPx3 (in the extracellular space), and GPx4 (in cellular membranes). The mechanism for reducing glutathione (GSH) or a thiol compound begins by reducing selenium. The reduced enzyme then reacts with H2O2, and H2O is released. Subsequently, a second GSH molecule undergoes thiol-disulfide exchange, which releases GSSG and regenerates the enzyme [28, 52].
Metal chelator proteins are also considered part of the first level of defense against oxidation, and the principal proteins include albumin, α-lactalbumin, transferrin, ferritin, ceruloplasmin, β-lactoglobulin, and immunoglobulins. These enzymes act mainly to remove FR from the extracellular space, and their antioxidant mechanism involves amino acids such as tyrosine and cysteine, due to their structures, and the chelation of transition metals [53–55]. Albumin is the most abundant of the serum-circulating proteins, and its antioxidant function involves more than one mechanism of action. It is noted that more than 70 % of the serum FR-trapping activity is due to this protein [54]. The antioxidant capability of albumin can be attributed to two mechanisms: its ability to bind multiple ligands through its multiple binding sites and its propensity for FR trapping. However, this protein is also susceptible to oxidation, which diminishes its antioxidant activity [55]. Its FR trapping ability resides in its cysteine (Cys34) and methionine (Met) residues, but cysteine plays a more important role in this process [56].
Iron-binding proteins such as transferrin and ferritin are capable of preventing the Fenton reaction because they are iron chelators. This antioxidant action is possible because they bind iron, and once bound, they are not available to stimulate FR reactions or form •OH, which is an important part of the extracellular antioxidant defense system [53, 57, 58]. Another enzyme that avoids the Fenton reaction is ceruloplasmin. This enzyme has ferroxidase action, which inhibits the formation of •OH iron and is dependent on H2O2. This enzyme also has a synergic activity with ferritin, which inhibits superoxide-dependent iron release and limits its biodisponibility in vivo to reinstate iron to the enzyme when it is released [53, 59].
4.2.2.2 Second Level of Antioxidant Defense
When the first level of antioxidant defense is exceeded, organisms have a second level of protection. The role of these antioxidants is to “trap” FR once it is formed, thus terminating or preventing the initiation of an oxidative chain reaction. Exogenous and endogenous molecules are included in this group. Serum compound such as uric acid and bilirubin, and hormones such as estrogens and melatonin, are all endogens secondary antioxidants; vitamins A, C and E; poliphenolic compounds, and aromatic amines are all exogenous antioxidant compounds. An important characteristic of exogenous antioxidants is that they have the ability to become radical species; therefore, they are potentially prooxidants [39, 47].
Estrogens are linked to menopause; therefore, they will be emphasized here. Estrogens and their metabolites possess antioxidant capabilities due to the presence of an OH at C3 of the phenolic ring in the A position. The antioxidant activities of estrogen are carried out in different ways: estrogen molecules act as free radical scavengers, neutralize excess ROS, and increase the amount of antioxidant molecules such as thioredoxin and SOD [17, 60, 61]. As FR scavengers, estrogens can be considered polycyclic phenolic compounds, which are molecules that are capable of intercalating into the cellular and mitochondrial membranes, where they directly interrupt lipid peroxidation (lipid oxidation) or through poorly defined lipophilic derivatives [62]. Indeed, several in vivo and in vitro studies indicated that the oxidation of LDL isolated from postmenopausal women was differentially inhibited by various estrogens, and the most active were the unique, ring B unsaturated estrogens [63]. Moreover, estrogens can act in trapping transition metals, such as Fe2+ and Cu+, reducing and maintaining low levels of these metals, and preventing them from acting as oxidants again. Thus, estrogens attenuate the ROS that are created by the Fenton reaction in vitro [64]. Because of its capability for incrementing antioxidant molecules, estradiol increases the transcription, expression, and activity of SOD2 and SOD3 without affecting GPx and catalase [65].
Additionally, estrogens can act as prooxidants because they are oxidized to 4-hydroxy-estrogens or catechol-estrogens by the action of a wide range of oxidative enzymes, such as cytochrome P450, and metallic ions in the presence of molecular O2. The cytochrome P450 pathway generates reactive electrophilic estrogen o-quinones and ROS, through the redox cycling of the o-quinones, mainly by the TCDD-inducible P450 isozymes, P4501A1/1A2 and P4501B1, which selectively catalyze hydroxylation at the 2 and 4 positions of estrone and 17β-estradiol, respectively. In this redox cycling, the 4-hydroxy-estrogens are oxidized to semiquinones and quinones, which can be reduced by cytochome P450 NADPH-dependent reductase to generate O2 •−, which in turn reduces Fe3+ to Fe2+ by the Fenton reaction to produce •OH. These reactions are dependent on the levels of metallic ions in the given environment. Importantly, catechol-estrogen metabolites are likely contributors to the development of cancer [66, 67].
Antioxidant vitamins are also relevant during menopause. Recent research has focused on the antioxidant activity and possible anticancer properties of vitamins A, C, and E, which have been commonly thought to prevent or dampen the effect of chronic degenerative diseases. These vitamins have been effective antioxidants in vitro and in vivo [68, 69], and several studies have shown that vitamins C and E, when administered at different concentrations, exert antioxidant effects that prevent or reduce oxidative stress. Vitamin E or α-tocopherol is the major membrane-bound antioxidant in the cell. Vitamin E donates an electron to a peroxyl radical, which is produced during lipid peroxidation and acts as a chain-breaking antioxidant by scavenging chain-initiation and -propagation radicals. Vitamin E also reduces the consequences of oxidized LDL by decreasing the ability of monocytes to bind to endothelial cells [70–72]. Under such circumstances, vitamin E is considered a suppressor of LDL lipid oxidation because it is the main antioxidant molecule in human lipoproteins [73]. Vitamin C, when in its L-ascorbic acid form, acts as an antioxidant in the aqueous conditions of the plasma and cytoplasm and is converted to dehydroascorbic acid in the presence of oxidants. Ascorbic acid crosses the cytoplasmic membrane, directly scavenges ROS through the major antioxidant pathway and acts as an electron-donor by maintaining iron in its ferrous state. This ability to donate one or two electrons makes ascorbate an excellent reducing and antioxidant agent [74, 75]. Additionally, vitamin C acts synergistically with vitamin E because vitamin E protects membranes from lipid peroxidation by transforming itself into an α-tocopheroxyl radical once it captures free ROS in the membrane. Ascorbic acid reduces this radical form and makes it reactive for new, reducing lipid oxidation, which consequentially leads to a decrease in oxidative stress. The interaction between vitamins E and C has led to the idea of “vitamin E recycling,” which is when the antioxidant function of oxidized vitamin E is continuously restored by other antioxidants, mainly vitamin C [76, 77]. Under normal physiological conditions, the α-tocopherol/ascorbic acid pair is the major antioxidant system that suppresses excess LDL oxidation in the plasma [78]. Another observed effect of vitamin C is in the modulation of DNA repair, although the impact of vitamin C on DNA damage is dependent on the background values of vitamin C in the individual and on the level of oxidative stress [79]. Moreover, vitamin C affects NO bioavailability due to its ability to prevent eNOS-related superoxide production by a mechanism that is not entirely known [77].
As described previously for estrogens, antioxidant vitamins also act as prooxidant agents. The α-tocopheroxyl radical is relatively nonreactive, although it can exert prooxidant activities in the absence of ascorbic acid and reduced coenzyme Q10, thus acting on LDL as a chain-transfer agent rather than as a radical trap and oxidizing LDL via a free radical chain [73, 78]. Recent studies in yeast have suggested the potential prooxidant action of α-tocopherol and coenzyme Q10, warning against antioxidant supplementation as an approach to increase longevity [72].
In the same way, ascorbic acid can cause serious oxidative damage because it reacts with free iron and produces the ascorbyl radical, and it can undergo pH-dependent autoxidation to form H2O2. The ascorbyl radical in turn reduces iron when it appears as a free ferric cation in biological fluids, thus accelerating the auto-oxidation reaction and favoring the Fenton reaction to produce an •OH, which causes oxidative damage to biomolecules [74, 75]. Additionally, it is feasible that the release of iron and the presence of vitamin C during acute inflammation could lead to the generation of •OH, ascorbyl, and thiyl radicals [80].
4.2.3 Third Level of Antioxidant Defense
There are some circumstances in which biomolecules are oxidized, necessitating a third level of antioxidant defense including repair enzymes such as phospholipases, nucleases, reductases, and proteases, which repair oxidative damage, remove the oxidized products, and reconstitute the lost function. Phospholipases, such as phospholipase A2 or platelet-activating factor-acetyl hydrolase, remove oxidized fatty acids because they must be in a nonesterified form to be detoxified [81, 82].
Redoxy-endonucleases endogenously repair DNA damage that is generated by exposure to ionizing radiation [83]. The action of these enzymes is mediated by nucleotide excision repair (NER), which is a pathway of DNA repair that is responsible for eliminating diverse oxidative lesions. Approximately 30 proteins participate in this mechanism, but ERCC1-XPF and XPG are the major structure-specific endonucleases that make 5′ and 3′ excisions into damaged DNA, respectively, and release the damaged oligonucleotide [84]. Other smaller base adducts, such as hydroxyl or alkyl groups at different positions, and single-strand breaks serve as substrates for DNA base excision repair (BER) [85].
Finally, most oxidized proteins cannot be repaired by cells. An alternative method for preventing further damage is the catabolism of oxidatively modified proteins via proteases that are generated in latent proteolytic systems, which are activated when cells are challenged with ROS [54, 86, 87]. Two key proteases have been identified: a cytosolic, proteolytic system known as the 20S proteasome and a mitochondrial protease called the Lon protease, which is involved in the degradation of oxidized aconitase [88].
Together with degradation, specific enzyme systems contain proteins that reduce certain amino acid oxidation products that contain sulfur, cysteine, and methionine and are most susceptible to oxidation. The glutaredoxin/glutathione/glutathione reductase and thioredoxin/thioredoxin reductase systems reverse the oxidation of cysteine disulfide bonds and sulfonic acid, while the methionine sulfoxide reductase system (MSR) reduces methionine sulfoxide to methionine [88]. If FR damage to DNA is not repaired, it may lead to genetic instability, which has been proposed as the leading cause of disease processes such as carcinogenesis [89].
4.3 Oxidative Damage from Biomolecules
As explained above, the consequence of producing reactive species is the oxidation of biomolecules such as lipids, proteins, DNA, and carbohydrates. In this section, we will address the oxidation of each type of molecule.
4.3.1 Lipid Oxidation
Of all biomolecules that can be attacked by ROS, lipids are the most susceptible. Three different mechanisms for lipid oxidation exist: (1) enzymatic oxidation; (2) nonenzymatic, free radical-mediated oxidation; and (3) nonenzymatic, non-radical oxidation [90, 91].
Lipid oxidation reactions that are mediated by enzymes are carried out as part of the catalytic action of the enzyme; for example, lipoxygenases, cyclooxygenases, and cytochrome c use FRs as intermediaries in the reactions that form endoperoxides and hydroperoxides. In these cases, the FR is bound to the protein active site [43, 92], where it induces structural changes in the cells under different physiopathological conditions [93].
The nonenzymatic reactions are performed primarily in cell membranes because they are rich in polyunsaturated fatty acids (PUFA), which are easily oxidized by lipid peroxidation, and their products are referred to as lipoperoxides. This process directly damages the structure of the cell membrane and indirectly damages other cellular components by producing reactive aldehydes. Lipid peroxidation is carried out by five in-chain reactions [90–92]:
1.
A PUFA is attacked by a FR (•OH or ROO•−), which abstracts one hydrogen atom (H•) from a methylene group with an adjacent double bound (—CH—), and this abstraction results in a pentadienyl carbon-centered lipid radical (—•CH—).
2.
The carbon-centered lipid radical undergoes a molecular rearrangement to form a conjugated diene, which then combines with O2 to form a lipidic peroxyl radical (ROO•).
3.
The lipidic peroxyl radical is fragmented to give O2 and a lipid radical (the reverse of reaction 2).
4.
A rearrangement of the peroxyl radical occurs.
5.
A cyclization of the peroxyl radical occurs, but this reaction is only carried out when PUFA has more than three double bonds.
These new peroxyl radicals are capable of extracting the hydrogen atom of other PUFAs and inducing further oxidation. Peroxidation is continued until the substrate is terminated or the sequence is interrupted by an antioxidant [43, 90, 91].
Extensive lipid peroxidation in biological membranes causes fluidity loss, loss of membrane potential, increased permeability to H+ and other ions, and, eventually, the release of cell contents and organelles resulting in cell death, whether programmed (apoptosis) or unprogrammed [43, 92, 93]. Likewise, lipid peroxidation products such as malondialdehyde and unsaturated aldehydes inactivate cellular proteins by forming protein cross-linkages [23].
Lipid peroxidation by a nonenzymatic, non-radical mechanism is induced by singlet oxygen and ozone [90], but this process has not been well studied, and the mechanism is not fully understood.
4.3.2 Proteins Oxidation
Proteins are also direct targets of ROS/RNS because of their high concentrations in living organisms, and different reactions involving proteins occur, such as carboxylation, nitration, loss of sulfhydryls, backbone damage, side-chain damage, fragmentation, unfolding and conformational changes in tertiary and quaternary structures, alteration of electrical charge, oxidation of specific amino acids, spontaneous reactions with glucose (glycosylation), and covalent cross-links [83, 87, 94].
Protein, as well as lipid, oxidation by FRs is initiated by the abstraction of a hydrogen atom from the a-carbon site, resulting in a carbon-centered radical. This reaction can be carried out at the backbone or in the side chain, and in both cases, these species appear to have two major fates: reaction with O2 to give a peroxyl radical or a reaction with another radical. The peroxyl radicals that are formed undergo different subsequent reactions that are dependent on if the reaction is with the backbone or side chain. When the reaction occurs with the backbone, an elimination reaction releases HO2 • and generates an imine, which undergoes further hydrolysis and backbone fragmentation. When the reaction occurs with the side chain, peroxyl radicals undergo a range of radical–radical termination reactions that can generate alcohols and carbonyl compounds or, alternatively, alkoxyl radicals and O2. When the reaction occurs with another radical, the carbon-centered radical is converted into a hydroperoxide, and if this reaction occurs with the backbone, the subsequent decomposition of the hydroperoxide to a radical can result in backbone fragmentation via an alkoxyl-radical-mediated process [85, 95].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


