and Emir Q. Haxhija2
(1)
Department of Plastic Surgery and Burns, Institute for Mother and Child Health Care of Republic Serbia, University of Belgrade, New Belgrade, Serbia
(2)
Department of Pediatric and Adolescent Surgery, Medical University Graz, Graz, Austria
Keywords
Malignant soft tissue tumorsChildrenTreatment12.1 Introduction
Malignant skin tumors in pediatric population are extremely rare (1–2% of all skin tumors excised); however the possibility of a malignant soft tissue lesion must be systematically considered [1–7]. Worrisome mass have following risk factors: rapid growth, ulceration, fixation or deep localization on fascia, rough texture, hard structure, size larger than 3 (5) cm, onset in neonate, and high vascularity [1, 5–7].
The most common skin malignancies that affect children are rhabdomyosarcoma (RMS), fibrosarcoma (FS), synovial sarcoma (SS), neuroblastoma (NB), malignant peripheral nerve sheath tumor, and cutaneous lymphomas [1, 2, 6, 7, 8]. Pediatric plastic surgeon can be involved in managing of cervical teratoma, which is mostly histologically benign lesion, but represents significant challenge for treatment [9–11].
12.2 Rhabdomyosarcoma
Rhabdomyosarcoma (RMS) is a rapidly growing malignant soft tissue tumor of mesenchymal origin (cells committed to becoming skeletal muscle) that rarely involves the skin [2–5, 8, 14–21]. Cutaneous appearance is secondary to extension from the soft tissue into the dermis [5]. It is the most common of the pediatric soft tissue sarcoma, and it accounts for 4–8% of all malignancies in children less than 15 years of age (the peak incidence of RMS is between 1–4 and 2–6 years) [2, 3, 5, 7, 8, 14, 17, 19, 21]. Congenital RMS is extremely rare, and only 0.4% of patients are under 1 month of life [17, 19]. The lesion is in near 40% of cases located in head and neck region, followed by genitourinary tract, and extremities [2, 3, 17, 21]. The clinical appearance varies from small cutaneous nodule to an extensive fast-growing tumor (it is often overlooked or misdiagnosed for infection, lymphatic malformation, or hemangioma) [4, 5, 16–18]. There are four groups of RMS according to the Intergroup Rhabdomyosarcoma Study based on whether tumor is localized or metastatic and whether or not it is resectable [2].
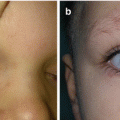
Fine needle biopsy, core needle biopsy, and incisional or excisional biopsy are used as diagnostic tools for rhabdomyosarcoma, followed by light microscopic examination, immunohistochemistry, electron microscopy, and cytogenetic analysis [5, 17–19]. Magnetic resonance imaging (MRI) and computerized tomography (CT) are used for tumor and lymph node evaluation [3, 8, 20, 21]. There are three histologic types of RMS: embryonal, alveolar, and undifferentiated (pleomorphic) [2, 3, 8, 17]. Embryonal tumors are found in 80–85% of cases and they mostly occur at birth, and alveolar occurs at adolescent period (worse prognosis) (Fig. 12.1a, b) [2, 3, 15]. RMS can be present in syndromes such as Li-Fraumeni and Beckwith-Wiedemann syndrome [3, 18]. Metastasis of RMS is primarily by hematogenous route [17, 19].
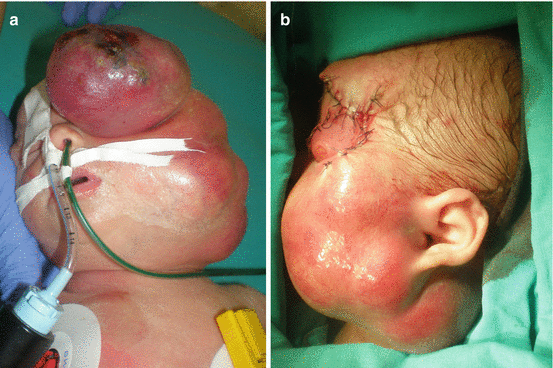
Fig. 12.1
Rhabdomyosarcoma of the head: (a) intraoperative view; (b) tumor biopsy
Treatment of RMS includes chemotherapy, radiation therapy, and surgery [2, 3, 5, 8]. Radiotherapy and chemotherapy are treatment of choice for local control of the primary lesion, regression of tumor size, and for unresectable tumors [2, 3, 8, 17]. Primary excision should be attempted only if complete excision can be accomplished without significant consequences, and secondary excision may be considered after chemotherapy [2–4]. Poor prognosis occurs if the diagnosis of RMS is established during infancy or adolescence, if there is alveolar histology, and if there is metastatic disease at the time of presentation [3–5, 8].
12.3 Infantile Fibrosarcoma
Infantile fibrosarcoma (IF) is classified as nonrhabdomyosarcoma malignant mesenchymal tumor (the second most common childhood sarcoma involving the skin), with the incidence of 5 per million infants less than 1 year of age [5, 22, 23].
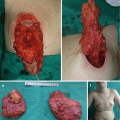
IF affects children primarily before the age of 2, representing 5–10% of all sarcomas in children younger than 1 year of age, and congenital fibrosarcoma has clinical signs before 3 months of age [22, 23]. In most cases the tumor represents as bulging, locally destructive, rapidly growth mass mostly on the lower extremities and trunk with a very low rate of metastases [5, 20, 22–24]. Skin involvement is usually secondary [5].
Differential diagnosis includes RMS, vascular tumors and malformations, and infections [5, 23]. Surgical removal has remained a primary component of the treatment with adjuvant chemotherapy (preoperatively or postoperatively) (Fig. 12.2a–h) [22, 23]. There is small difference in disease-free survival between the patients who had positive tumor margins and those who did not, meaning that “heroic” surgery should be avoided [22]. The overall 10-year surviving rate is 90% [23].
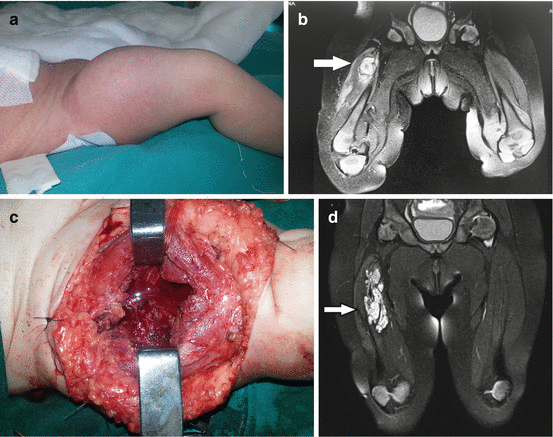
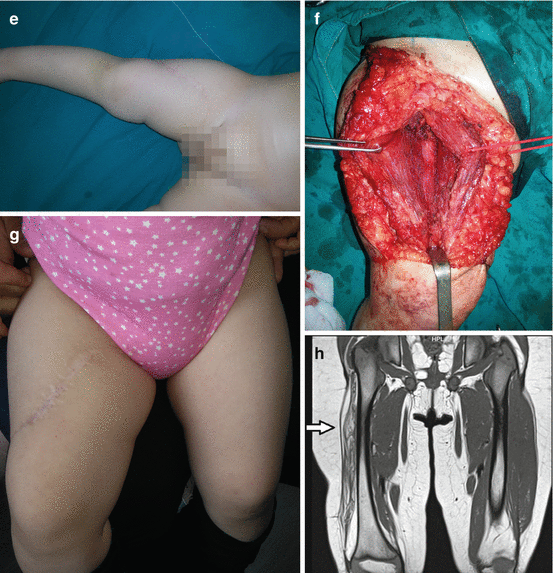
Fig. 12.2
Infantile fibrosarcoma of the right femoral region: (a) preoperative view; (b) magnetic resonance imaging finding; (c) tumor biopsy; (d) magnetic resonance imaging finding prior definitive surgery; (e and f) excision of the tumor; (g) 2-year postoperative finding; (h) magnetic resonance imaging finding revealing no presence of tumor
12.4 Cervical Teratoma
Teratomas are rare and unusual tumors derived from all three germ cell layers (ectoderm, endoderm, and mesoderm) [9–11, 20, 25]. Congenital teratoma is usually found in sacrococcygeal region, and head and neck region is affected in 5% of cases [9–11, 20]. The incidence is reported to be between 1 in 20,000 and 40,000 live births [9, 10, 20]. The tumors may be diagnosed in the antenatal, perinatal, and postnatal period [11, 25]. Antenatal diagnosis is crucial because it optimizes the preparedness of the clinical personnel in attendance at the delivery of these children to execute whatever intervention is required to secure the airway [9–11, 25]. There is maternal polyhydramnios in near one third of prenatally diagnosed cervical teratoma on routine ultrasonography (US) [10, 11, 25]. Although usually histologically benign, the mortality rate for untreated tumors is 80–100% [10, 11]. The major risk posed by large cervical teratomas is that of neonatal airway obstruction [9, 11, 25].
Multidisciplinary approach is very important for these children [9–11]. Both ex utero intrapartum treatment (EXIT) and operation on placental support (OOPS) procedures have been performed for cervical teratoma to secure the airway [9, 11, 25]. Preoperatively CT scanning or MRI should be performed in stabile patients [11].
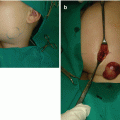
Definitive treatment of these tumors is surgical excision, and it should be performed promptly because of respiratory and other complications (Fig. 12.3a–d) [9–11, 25]. They are pseudo-encapsulated and should be treated easily, and excision of the thyroid cartilage, pharyngeal wall, and thyroid parenchyma is sometimes required [9–11]. Operative mortality rate nowadays is low, with injury of regional nerves reported postoperatively [11].
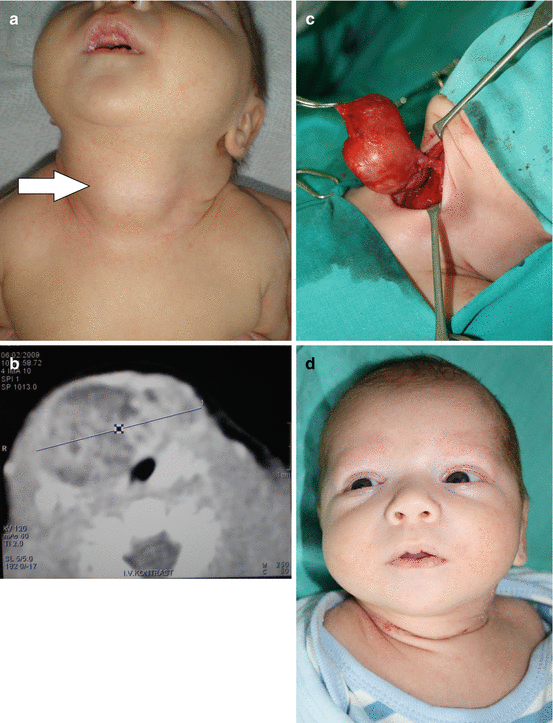
Fig. 12.3
Cervical teratoma: (a) preoperative view; (b) computerized tomography finding; (c) intraoperative view; (d) postoperative scar at lower anterior neck region
12.5 Cutaneous Neuroblastoma
Neuroblastoma (NB) is one of the most common solid malignant tumors in children (50% of malignancies in infants), derived from the primitive neural crest cells of the sympathetic nervous system, with the incidence of 10 per million births [4, 5, 25–27]. NB mostly occurs in the abdomen (80%), and around 5% are primarily located in cervical region [12, 20, 21, 26]. Primary cervical neuroblastoma can be presented with Horner syndrome, cranial nerve palsy IX–XII, heterochromia iridis, and compression of vital cervical organs; diagnosis is made by US, CT, and MRI, and it has favorable prognosis [20]. Primary cutaneous neuroblastoma is extremely rare in children (most commonly there are cutaneous metastases from primary adrenal tumor) presenting as bluish, hard, and painless nodules or papule [1, 5, 12]. After compression there is central pale region “icy blanch” secondary to local catecholamine release [5, 27]. Clinically cervical NB presents as a palpable indolent mass in the lateral neck with compression on vital neck structures [27]. Diagnostic evaluation includes genetic analysis, immunophenotyping, serum and urine catecholamines and lactate dehydrogenase (LDH) levels, and metaiodobenzylguanidine (MIBG) scan (to rule out metastasis) [4, 5, 26, 27]. N-myc amplification with 10 or more copies per haploid genome is considered as highly unfavorable factor [27]. Radiological evaluation including US, MRI, and CT is necessary to evaluate the exact extension of the tumor [5, 21, 26, 27]. Treatment of neuroblastoma includes combination of surgery, chemotherapy, and radiation (Fig. 12.4a, b) [4, 5]. Surgery is the treatment of choice for localized neuroblastoma without N-myc amplification [4, 26, 27].
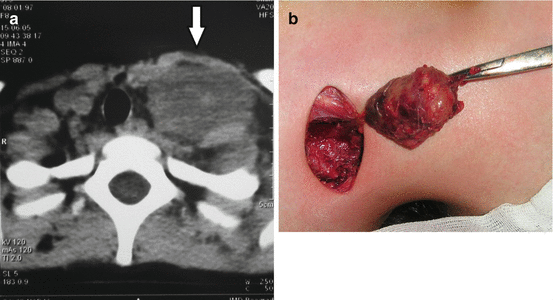
Fig. 12.4
Neuroblastoma metastases, cervical localization: (a) preoperative radiological finding; (b) tumor excision
12.6 Dermatofibrosarcoma Protuberans (DFSP)
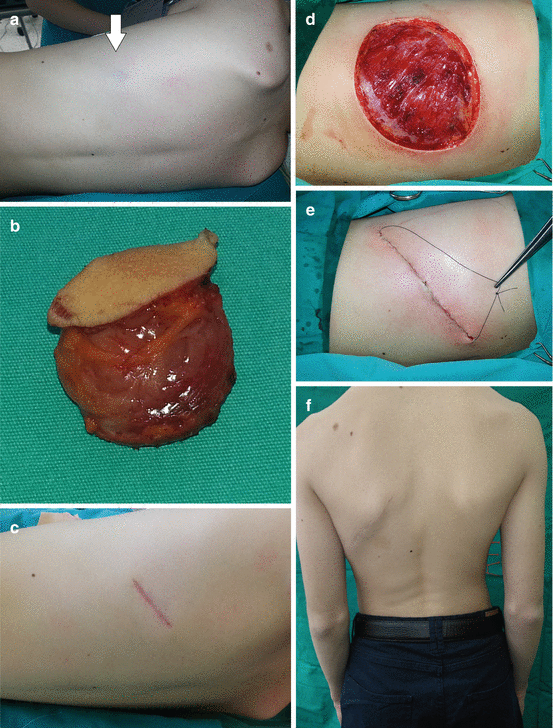
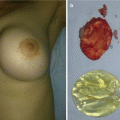
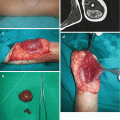
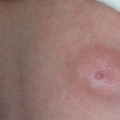
Dermatofibrosarcoma protuberans (DFSP) (also called giant cell fibroblastoma and low-grade sarcoma) is a rare soft tissue tumor with low- to intermediate-grade malignancy, characterized by high rates of local recurrence yet low risk of metastasis [5, 12, 15, 28]. It is in most cases located on the trunk and the proximal portion of the limbs [4, 5, 12]. Pediatric DFSP is reported between 6 and 20% in literature [4, 12]. DFSP in children demonstrated atypical variations and in that way mimics keloids, vascular anomalies, or fibrosarcoma [4, 12, 15]. The initial presentation of the DFSP is of an asymptomatic, nodular plaque, ranging from 1 to 5 cm, which is fixed to the skin [5, 12]. The main characteristic of DFSP is its capacity to invade surrounding tissue to considerable distance from the central focus of the tumor [12].
The treatment of choice for nonmetastatic DFSP is complete surgical resection (classically or by Mohs micrographic surgery) with margin at least 2–5 cm (Fig. 12.5a–f) [4, 5, 12, 15]. After complete excision, the defect can be reconstructed by direct sutures, skin grafts, and local or distant flaps [5, 12]. Dermatofibrosarcoma protuberans is immunoreactive for CD34 (differentiating from dermatofibroma) and S-100 protein-negative (differentiating from neurofibromas) [4, 5]. The recurrence rate ranges from 0 to 30% and grows with narrower excision border [4, 12]. Radiotherapy should be used in cases of microscopic residual disease or as an adjuvant therapy [12].