Malignant atrophic papulosis
Specific investigations
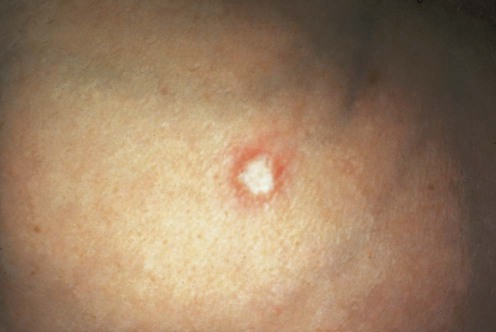
 Tissue biopsy (skin primarily, but also tissue from the intestines and other internal organs)
Tissue biopsy (skin primarily, but also tissue from the intestines and other internal organs)
 Protein C, protein S and factor V Lieden, antithrombin III, homocysteine levels
Protein C, protein S and factor V Lieden, antithrombin III, homocysteine levels
 Anticardiolipin antibody titer
Anticardiolipin antibody titer
 Antiphospholipid antibody titer
Antiphospholipid antibody titer
 Endoscopy of the gastrointestinal tract (i.e., stomach, esophagus, duodenum, colon, rectum)
Endoscopy of the gastrointestinal tract (i.e., stomach, esophagus, duodenum, colon, rectum)
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Malignant atrophic papulosis






