Fig. 22.1
The distinct stages of tissue IRI. The ischemic injury, a local process of metabolic disturbances, results from glycogen consumption, lack of oxygen supply, and ATP depletion. The cell death released DAMPs (alarmins), activation of complement, and oxygenation-triggered mitochondrial ROS production, all contribute to liver-immune activation after reperfusion. The process involves multiple types of nonparenchymal cells, including macrophages, dendritic cells, T cells, NK cells and neutrophils. This pro-inflammatory immune response in IR-stressed organ sustains itself by recruiting circulating peripheral immune cells from the circulation and is responsible for the ultimate reperfusion injury. DAMPS danger-aassociated molecular patterns, DC dendritic cells, NK natural killer cells, PMN polymorphonuclear cells, ROS reactive oxygen species, ATP adenosine triphosphate, IR ischemia reperfusion, IRI ischemia–reperfusion injury
IR Triggers Toll-Like Receptor Signaling
Based on Dr. Polly Matzinger’s concept that the principal goal of the immune system is to detect and protect the host from “danger” signals resulting from cell/tissue damage [13], Professor Walter Land introduced the “injury hypothesis” in the field of transplantation [14]. Accordingly, post-IRI activates an array of pro-inflammatory immune responses in the transplant itself, which trigger and exacerbate host adaptive immunity, ultimately progressing to graft dysfunction and ultimately rejection. All vertebrates utilize the same sentinel innate immune receptor systems, PRRs, in response to tissue damage even in the absence of infections [15–19]. Four different classes of PRRs have been recognized: Toll-like receptors (TLRs) and C-type lectin receptors (CLRs) are transmembrane proteins; Retinoic acid-inducible gene, (RIG)-I-like receptors (RLRs), and nucleotide-binding domain (NOD)-like receptors (NLRs) are cytoplasmic proteins. These PRRs, expressed primarily by activated macrophages and dendritic cells (DC), function by upregulating pro-inflammatory gene transcription programs [20].
TLRs were discovered in 1998, in mice displaying endotoxin resistance in parallel with a high susceptibility to gram-negative bacterial infections [21]. TLRs are an evolutionarily conserved group of transmembrane proteins of which, to date, 11 have been identified in humans and 13 in mice (Table 22.1; Ref. [22]). These innate receptors are central in promoting immunity against pathogens by virtue of their ability to transduce signals in response to ligation of distinctive molecular motifs, termed PAMPs. They are a major group of PRRs and are ubiquitous, being expressed on a host of both immune and nonimmune cells [23]. TLR–PAMP interactions lead to downstream cytokine and chemokine release and augmentation of co-stimulatory T cell molecule expression [24]. As TLRs are expressed on parenchyma cells, at least some of their functions are unrelated to immune-mediated pathogen destruction. Indeed, it is now apparent that endogenous, cell-derived ligands (DAMPs) from both intracellular and extracellular sources during inflammation and tissue damage do bind and facilitate TLR signaling [25]. During homeostasis, DAMPs are not expressed and remain invisible to the host immune system. However, DAMPs become released from cells are displayed on their surfaces following cellular injury, such as hypoxia. A variety of endogenous DAMPs have been described that readily engage TLRs (Table 22.1), such as heat-shock proteins [26], purines, heparan sulfate, and fibronectin degradation product, the extra domain A (EDA) domain [27].
Table 22.1
TLRs—their microbial, endogenous ligands and cellular distribution
Receptor | Microbial ligand(s) | Endogenous ligands | Cellular expression |
|---|---|---|---|
TLR1 | Triacyl lipopeptides | – | B cells, monocytes, macrophages, and certain dendritic cells |
TLR2 | Peptodoglyca, zymosan, lipoteichoic acid, and glycolipids | HSP60, HSP70, hyaluronan, HMGB1 | Monocytes and macrophages, mast cells, and myeloid dendritic cells |
TLR3 | Double-stranded RNA, poly I:C | Messenger RNA (mRNA) | B cells, dendritic cells, and fibroblasts |
TLR4 | Lipopolysaccharides (LPS) | Fibrinogen, HSPs, surfactant proten A, b-defensin 2, hyaluronan, fibronectin extra domain A, heparin sulfate, HMGB-1 | Monocytes and macrophages, mast cells, certain dendritic cells, B cells; intestinal epithelium and hepatocytes (low) |
TLR5 | Flagellin | – | Monocytes and macrophages, subset of dendritic cells; intestine |
TLR6 | Multiple diacyl lipopeptides on mycoplasma | – | B cells, mast cells, and macrophages |
TLR7 | Single-stranded RNA imidazoquinolines | RNA and protein complexes | Plasmacytoid dendritic cells, monocytesa and macrophages; B cells |
TLR8 | Single-stranded RNA imidazoquinolines and small synthetic compounds | – | Monocytes and macrophages; subset of dendritic cells; mast cells |
TLR9 | CpG oligodeoxynucleotide DNA | – | Monocytes, macrophages, and plasmacytoid dendritic cells |
TLR10 | Undefined | – | B cells, monocytes, and regulatory T cells |
TLR11 | Profilin | – | Kidney and urinary bladder epithelium |
TLR12 | Profilin | – | Macrophages, neurons, and dendritic cells |
TLR13 | Conserved bacterial 23S ribosomal RNA (rRNA) sequence | – | Monoctyes, macrophages, and dendritic cells |
TLR4 was the first innate immune receptor studied in organ IRI. Indeed, using murine partial hepatic warm ischemia models, data from three separate laboratories demonstrated that TLR4-deficient mice were protected from hepatic damage in liver-warm ischemia model, evidenced by markedly depressed in situ IR inflammation in the absence of TLR4 signaling [28–30]. The functional role of TLR4-specific activation in triggering IRI pathology was also confirmed in a clinically relevant orthotopic liver transplantation model, which comprises both warm and cold IR tissue damage [31] and in a steatotic liver IRI model [32]. Interestingly, donor TLR4 deficiency alone was both necessary and sufficient to confer hepatoprotection in the transplant model, and TLR4 signaling on nonparenchyma rather than parenchyma cells seems more relevant for IRI [30], although a recent study implies a unique role of TLR4 on liver parenchyma cells at the late stage of the disease process [33]. Of note, although TLR2 signaling was dispensable in the development of liver IRI [28], it was found essential in both kidneys [34] and heart [35] IRI models. In the context of solid-organ transplantation, both donor and recipient cells have the capacity to express TLR2. Notably, selective chemical ablation of the recipient TLR2 conferred protection against transplantation-associated ischemic damage in a murine renal isograft model [36], suggesting that leukocyte TLR2 not only functions in the disease pathogenesis but may also constitute a viable therapeutic target against renal IRI in transplant recipients.
All TLRs mediate signal transduction via the adapter molecule myeloid differentiation factor 88 (MyD88), apart from TLR3, which is dependent on the adapter molecule Toll/IL-1R domain-containing adapter-inducing IFN-β (TRIF) and TLR4 through which signaling is dependent on both TRIF and MyD88 [22]. Indeed, MyD88-deficient animals not only developed hepatocellular IR-damage comparable with wild-type (WT) controls, but their “signature” pro-inflammatory cytokines (IL-1, IL-6, type-I IFN) and chemokine (CXCL-10) were largely unaffected [28]. As the MyD88-independent, TRIF-dependent signaling pathway of TLR4 triggers a delayed nuclear factor (NF)-kβ activation, it seems that the MyD88-mediated early-phase NF-kβ activation is not necessary for pro-inflammatory immune response in liver IR. Indeed, this is very different from renal and heart IRI models in which either MyD88 and TRIF or only MyD88 were found operational [37–40]. The fact that severity of liver IRI peaks at 6 h of reperfusion and that of kidney and heart injury last for days may explain this discrepancy. Moreover, the liver is unique in TLR4 activation in such a way that gut-derived endotoxin may have already tolerized the hepatic innate immune system, which has been shown to target more towards the MyD88-dependent pathway [41, 42]. Both pro- and anti-inflammatory gene programs become readily induced by TLR4 activation in macrophages in vitro and in vivo. Recently, Gsk3b, a serine/threonine kinase, was shown to differentially regulate these two programs [43], and a chemical Gsk3 inhibitor selectively inhibited pro-inflammatory, while simultaneously sparing immune-regulatory IL-10 gene program in IR-stressed organs [44].
The high-mobility group box 1 protein (HMGB1) represents the key endogenous TLR4 ligand responsible for IR-mediated immune activation [45]. HMGB1, released from damaged cells, may stimulate non-parenchyma cells, including macrophages and DC, through TLR4 signaling (Fig. 22.2). Hypoxic cells release HMGB1 through an active process facilitated by TLR4-dependent ROS production. In turn, ROS induces HMGB1 release through a Calcium/calmodulin-dependent protein kinase (CaMK)-dependent mechanism, and such a positive HMGB1–TLR4 signaling promotes a sustained inflammation in IR-stressed tissue [45]. In addition to HMGB1, other DAMPs released from damaged or necrotic cells may also activate innate immune cells via an array of receptors, including S100 proteins via TLR4, RNA via TLR3, or DNA via TLR9. TLR9 was found to function in bone marrow-derived cells, particularly neutrophils in IR-stressed tissues to boost production of pro-inflammatory cytokines/chemokines. Furthermore, the inhibition of TLR9 exerted additive protective effects with concomitant HMGB1 neutralization [46]. Nuclear histone proteins were recently identified as important endogenous TLR9 ligands [47]. Thus, liver IR insult resulted in increased levels of circulating histones, whereas their neutralization was cytoprotective. Extracellular histones enhanced DNA-mediated TLR9 activation, while their infusion exacerbated IRI via TLR9 signaling. Recently, TLR3, which recognizes necrotic cell-derived RNA products, has also been shown to sustain local IR inflammation [48].
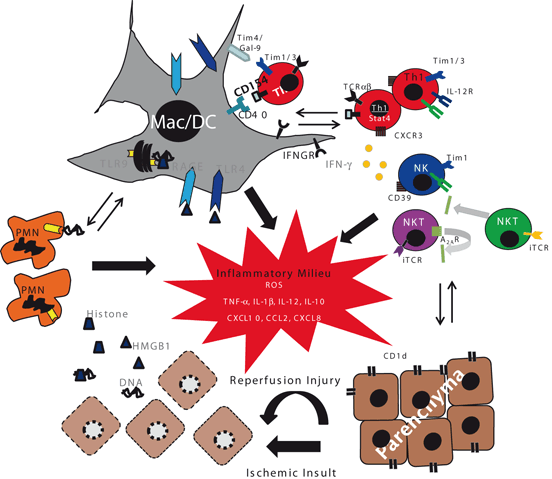
Fig. 22.2
A mechanistic scheme of immune activation in IR-stressed tissue. The ischemia insult induces necrotic cell death, which provide “danger” molecules, such as HMGB1 and DNA fragments to activate innate TLR4, RAGE, and TLR9 signaling pathways on macrophages/DC and neutrophils. CD4 + Th1 effectors may also facilitate local innate immune activation via CD154–CD40 pathway, whereas IFN-γ produced by T cells, NKT, and NK cells enhances innate immune activation. In parallel, CD1d and CD39 activate NKT and NK cells, respectively. This immune activation progresses via both positive and negative regulatory loops. Pro-inflammatory TNF-α, IL-1β, IL-6, IL-12, CXCL10, CCL2, CXCL8, and ROS milieu, further activate local and recruits migrating immune cells to promote cytotoxicity against parenchymal cells. Such a sustained pro-inflammatory activation may be counter-regulated by IL-10, whereas NKT cell activation may be inhibited by adenosine receptor 2A. By inhibiting pro-inflammatory type I NKT cells, type II NKT cells may also downregulate IFN-γ production. IR Ischemia reperfusion, HMGB1 high-mobility group box 1, DC dendritic cells, NK natural killer cells, NKT natural killer T cells, ROS reactive oxygen species, TLR toll-like receptor, IL interleukin, TNF tumor necrosis factor, Th T helper, RAGE receptor for advance glycation end products, IFN interferon
Thus, different TLR signaling pathways may function at distinct stages and in different cell types in IR-stressed solid organs. This is of importance for designing future experiments on innate activation in histologically and immune-divergent VCA tissues.
Inflammasomes in IR Innate Immune Activation
The role of other PRRs in the mechanism of tissue IRI has only recently started to be elucidated. In addition to TLRs, the necrotic cells can be sensed by the inflammasome, a caspase-1 activation platform, which regulates the secretion of L-1β, IL-18 pro-inflammatory mediators. One member of NLR family, NLRP3 (NLR family, pyrin domain containing 3) was found essential in the mechanism of polymorphonuclear neutrophil (PMN) recruitment to sites of focal hepatic necrosis in a model of sterile in vivo inflammation [49]. Indeed, gene silencing of NACHT, LRR and PYD domains-containing protein 3 (NALP3) attenuated tissue damage in association with reduced IL-1β, IL-18, Tumor necrosis factor α (TNF-α), and interleukin (IL)-6 levels, diminished HMGB1, and decreased local inflammatory cell infiltration [50].
Apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) plays a critical role in the activation of inflammasomes as an adaptor protein that bridges procaspase-1 and inflammasome receptors, such as NLRP3 and absent in melanoma 2 (AIM2) [51–53]. Indeed, ASC contributes to immune response through the assembly of inflammasome complexes that activate downstream effector cysteine protease caspase-1, resulting in the generation of active IL-1β and IL-18 from inactive pro-IL-1β and pro-IL-18 precursors (Fig. 22.3). Although under normal conditions ASC-associated inflammasomes are autorepressed, they become activated by a wide range of pathogen stimuli, oxidative stress, ischemia, and damage signals. The molecular mechanisms of ASC/Caspase-1/IL-1β signaling to program pro-inflammatory phenotype might involve activation of multiple intercellular pathways. We found disruption of ASC-inhibited HMGB1/TLR4 expression, leading to depressed induction of inflammatory mediators, suggesting ASC/Caspase-1/IL-1β plays an important role in triggering local inflammation in IR-stressed organ [54]. In fact, the adaptor ASC was initially believed to exert its effects by bridging the interaction between NLRs and caspase-1 in inflammasome complexes [55]. Activation of ASC within inflammasomes leads to the maturation of caspase-1 and processing of its IL-1β and IL-18 substrates, whereas knockout (KO) of ASC decreased caspase-1 activity and IL-1β/IL-18 production, implying the role of ASC in caspase-1/IL-1β-mediated inflammation. Although the ASC/caspase-1/IL-1β axis seems essential for the initiation of IR-inflammatory response, the molecular pathways involved in cross talk with HMGB1 remain unclear. Of note, treatment of ASC KO mice with recombinant HMGB1 increased IR tissue damage, whereas disruption of ASC without exogenous HMGB1 prevented local inflammatory development. Hence, ASC-mediated caspase-1/IL-1β axis promotes HMGB1 to produce TLR4-dependent inflammatory phenotype, leading to IR tissue inflammation and subsequent injury.
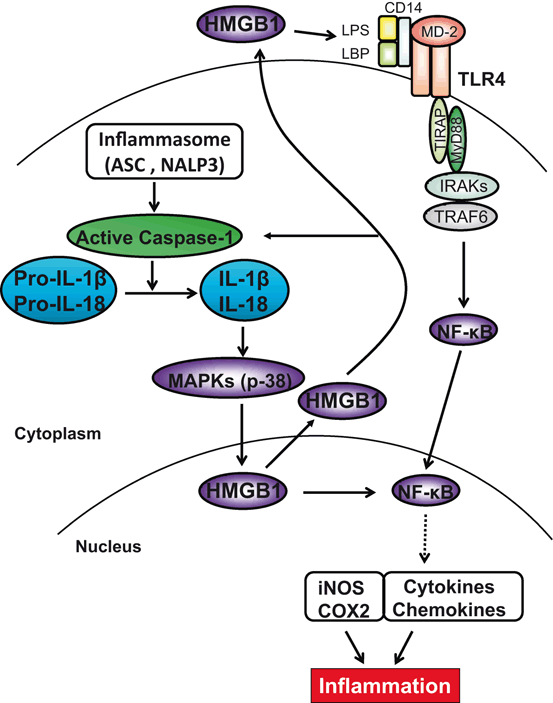
Fig. 22.3
Molecular mechanisms by which ASC/Caspase-1/IL-1β–HMGB1 axis may regulate IR-triggered inflammation. ASC activates inflammasomes, which in turn activates caspase-1 and catalyses pro-IL-1β/pro-IL-18 to mature IL-1β/IL-18. IL-18 is closely related to and shares a similar dimensional structure with IL-1β. ASC/caspase-1/IL-1 promotes HMGB1 induction through activation of p38 MAPK, which triggers TLR4 and NF-kB to program pro-inflammatory mediators. In addition, HMGB1 may provide a positive feedback mechanism to regulate caspase-1 activation. ASC/caspase-1-mediated elaboration of IL-1β and its downstream COX2 are required for the inflammatory development in the course of IRI. ASC Apoptosis-associated speck-like protein containing a caspase recruitment domain, HMGB1 high-mobility group box 1, IL interleukin, MAPK mitogen-activated protein kinases, TLR toll-like receptor, COX2 cyclooxygenase-2, IRI ischemia–reperfusion injury
Although an array of innate PRR-targeting studies have shown promise in different animal models, the caveat is most of these studies focus on “correlation” between genetic deletion and cytoprotection rather than establishing the actual cause of the reduced tissue damage. With limited mechanistic understanding of a successful anti-IRI therapy in VCA settings, exploring multiple PRR pathways with small molecules acting preferably in a synergistic manner and/or selectively targeting positive while simultaneously promoting negative signaling may be required, while keeping in mind their different cellular sources, location specificities, and individual transcriptional kinetics.
IL-10 in IR Innate Immune Activation
Innate immune activation in IR-stressed organ is a self-limiting reaction with active regulatory mechanism by which IL-4, IL-10, and IL-13 may effectively counteract and alleviate local pro-inflammatory phenotype [56–58]. These cytokines, readily expressed in all IR tissues, are often spared or their expression even heightened in IR-resistant animals. Although generally inhibitory to IR-induced pro-inflammatory TNF-α and IL-1β “signature” when administered exogenously, the endogenous role of IL-4, IL-10, and IL-13 may not necessarily be immune regulatory. Indeed, although IL-13-deficient mice suffer from exacerbated liver injury compared with IL-13-proficient (WT) counterparts, IR-induced TNF-α and CXCL8 (MIP-2) production in IL-13 KO and WT mice was comparable in the early post-reperfusion phase [56]. Although IL-13 deficiency alters PMN distribution in IR-stressed tissues, the most profound effect of IL-13 seems to be the direct cytoprotection from ROS-induced cell death. Unlike IL-4 and IL-13, the beneficial role of IL-10 as the key immune regulatory cytokine in tissue IRI has been well documented. Hence, IL-10 neutralization was shown both necessary and sufficient to re-create the pro-inflammatory phenotype in IR-resistant tissues of otherwise immune-suppressed or deficient recipients [59, 60]. Of note, multiple innate immune cell types, including DC, macrophages, and PMNs may all produce IL-10 and exert important autoimmune regulatory functions [61, 62]. The question of which non-parenchyma cells become IL-10 producers in response to IR insult remains to be elucidated. Recently, conventional DC have been shown to exert immune-regulatory functions by producing IL-10 via a TLR9-mediated mechanism [63]. Thus, the very same non-parenchyma cells responsible for initiation of the pro-inflammatory response against IR [64] may also terminate their own early-action function. Such a hypothesis is consistent with in vitro studies in which macrophages (or DC) produced both pro- and anti-inflammatory mediators in response to the very same TLR ligand supplied to the culture.
As IR activates pro- and anti-inflammatory gene programs, the question remains as to the mechanisms that determine the nature of immune responses and dictate the outcome of tissue injury. Is it merely the difference in the kinetics of innate immune gene induction or tissue/cell responsiveness to the gene products, in such a way that the pro-inflammatory cytodestructive program precedes the anti-inflammatory cytoprotective pattern, resulting in self-limited tissue damage response? Alternatively, endogenous ligands generated at different IR stages may trigger pro- and anti-inflammatory response sequentially, possibly via distinct TLR pathways and/or in different cell types. One may also envision cell–cell interactions, such as macrophage/DC–T cell, which may dictate the nature of local immune response by engaging additional activation signaling pathways. Addressing these questions in Langerhans cell-rich skin tissue should further our understanding of IRI mechanism in VCAs, and help to identify therapeutic targets to suppress pro-inflammatory without interfering with immune regulatory functions.
T Cells in IR Innate Immune Responses
Although IRI develops in syngeneic grafts, in ex vivo settings, or under sterile conditions, T cells, particularly of CD4 phenotype, are indispensable for the activation and regulation of pro-inflammatory immune sequelae (Fig. 22.2). The original observation that systemic immunosuppression CsA (Cyclosporin A); FK506 (Tacrolimus) attenuated peri-transplant tissue damage provided indirect evidence for T cell involvement in IRI development [65]. Experiments in T cell- (nude) and CD4-deficient mice have documented the pivotal function of CD4 + T cell in the mechanism of tissue damage in several IR models [66–69]. However, the question as to how T cells may function in innate immune-driven response and in the absence of exogenous antigenic stimulation remains unanswered.
The role of T cell costimulation in promoting IRI pathology in the absence of antigen stimulation was originally shown in a study in which CD28 blockade with CTLA-4-Ig-protected rat kidneys from IRI by reducing T cell and macrophage infiltration [70]. Both CD28 and CD154 pathways were in fact essential for the development of local pro-inflammatory milieu critical to IR-mediated organ damage. Indeed, livers in CD154 KO or CD28 KO mice or in WT mice treated with anti-CD154 or CTLA-4-Ig were all IRI resistant [68]. Moreover, T helper type 1 (Th1)-type cells were shown to play a key role in IRI pathogenesis, as Stat4 KO (deficient in Th1 development), but not Stat6 KO, mice were protected from the injury, whereas reconstitution of “nude” mice with T cells from Stat6KO, but not Stat4KO, mice readily restored cardinal features of IRI [71]. Th17 cells have been also implicated in autoimmune inflammatory diseases, and their putative role in IRI has started to be unraveled. Although cellular sources of IL-17 remain to be defined, IL-17A KO mice suffer less severe IRI in parallel with reduced neutrophil infiltration. The impact of IL-17A deficiency was associated with relatively late stages of the disease and with acute IR-damage being unaffected [72]. Indeed, we consistently detect massive CD4 + T cell sequestration into post-ischemic hepatic tissue well before any appreciable local neutrophil sequestration. This may occur via released IL-17, which then acts upon macrophages to produce MIP-2, a known neutrophil chemoattractant.
IL-22, an inducible cytokine of the IL-10 superfamily, is produced by selected T cell subsets (Th17, Th22, γ/δ, NKT) [73]. IL-22 is quite unique because its biological activity, unlike other cytokines, does not serve the communication between immune cells, but rather signals directly to the tissue. Its tissue action is through a heterodimer IL-10R2/IL–22R1 complex. In contrast to IL-10R2, which is ubiquitously expressed and largely dispensable, the expression of IL-22R1 is restricted to epithelial cells including hepatocytes and skin, and has not been detected in cells of the hematopoietic lineage.
By increasing tissue immunity in barrier organs such as skin, lungs, and the gastrointestinal tract, IL-22 has been associated with a number of human diseases and shown to contribute to the pathogenesis of psoriasis, rheumatoid arthritis, and Crohn’s disease [73–76]. However, parallel studies in murine models of mucosal defense against pulmonary bacterial infection, inflammatory bowel disease, or acute/chronic liver failure indicate that IL-22 may exert immunoregulatory pathologic or protective functions, depending on the context in which it is expressed [77–82]. Thus, advancing our appreciation of the IL-22–IL-22R biology may yield novel therapeutic targets in multiple human diseases. Having documented that administration of rIL-22 exerted cytoprotection via STAT3 activation [83], we favor the concept that IL-22 is well positioned to orchestrate innate–adaptive immune networks by activating cell survival genes, preventing apoptosis, and promoting cell regeneration in IR-stressed organs, a novel idea directly relevant to studies on skin IRI in the emerging VCA field.
Recently discovered T cell Immunoglobulin Mucin (TIM) cell surface proteins have attracted much attention as novel regulators of host immunity [84]. T cell stimulation amplifies TIM-1, a phosphatidylserine (PS) receptor, primarily on CD4 + Th2/Th1 cells, whereas TIM-4, one of the major TIM-1 ligands, is expressed mostly by macrophages and other APCs. Hence, TIM-1–TIM-4 interactions constitute a novel molecular mechanism of T cell—macrophage regulation at the innate–adaptive interface, and may be a therapeutic target. Indeed, treatment with anti-TIM-1 mAb ameliorated liver [85] and renal [86] IR damage and was accompanied by decreased PMN infiltration/activation, inhibition of T lymphocyte/macrophage sequestration and diminished homing of TIM-1 ligand expressing TIM-4 + cells in ischemic tissues. The mechanism by which TIM-1 mediates IR-triggered innate-driven inflammation is shown in Fig. 22.4a. In the “direct” pathway, TIM-1 expressed on activated Th2 cells cross-links TIM-4 to directly activate macrophages. In the “indirect” pathway, TIM-1 on activated Th1 cells triggers interferon (IFN)-γ that results in macrophage activation. Regardless of the pathway, activated macrophages do elaborate cytokine/chemokine programs that facilitate ultimate IR organ damage.
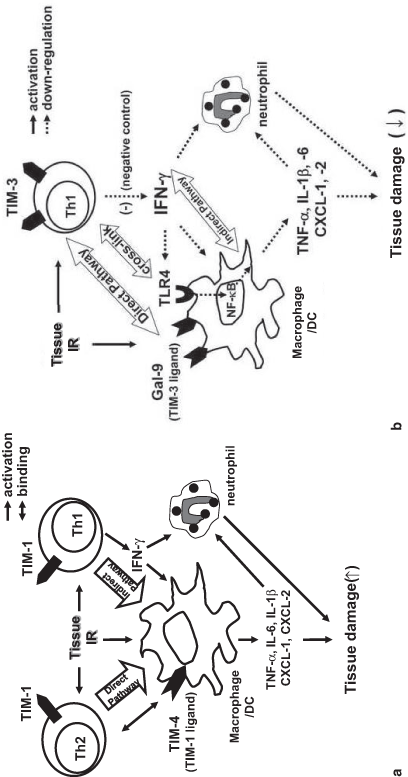
Fig. 22.4
a TIM-1–TIM-4 “positive” T cell costimulation in tissue IRI. Th1 and Th2 cells express TIM-1, whereas macrophages and DC express TIM-4, the TIM-1 ligand. During IR insult, TIM-1 on activated Th2 cells cross-links TIM-4 to directly activate macrophages (“direct pathway”), whereas TIM-1 on activated Th1 cells triggers IFN-g that may also function to further stimulate macrophage activation (“indirect pathway”), evidenced by elaboration of cytokine/chemokine programs that facilitate ultimate tissue damage. TIM T-cell immunoglobulin and mucin, IRI ischemia–reperfusion injury, Th T helper, IR ischemia reperfusion, IFN interferon. b TIM-3– Gal-9 “negative” costimulation in tissue IRI. IR triggers activation of TIM-3 expression by activated macrophages and Th1 cells. TIM-3 signaling negatively regulates Th1 cells by suppressing TLR4-NF-kB pathway via IFN-g, which in turn stimulates Gal-9 and mitigates macrophage activation. Diminished pro-inflammatory cytokine/chemokine programs ameliorate tissue damage and promote homeostasis. TIM T-cell immunoglobulin and mucin, Th T helper, IFN interferon, TLR toll-like receptor, NF necrosis factor, Gal-9 galectin-9
The TIM-3–Gal-9, on the other hand, constitutes a “negative” costimulation signaling between Th1 and macrophages, and has been shown to promote tolerance in transplant recipients [84]. Interestingly, TIM-3 blockade worsened the IR damage, along with increased IFN-γ but depressed IL-10 expression in IR-stressed organs [87]. One potential mechanism by which TIM-3–Gal-9 pathway controls IRI immune cascade is depicted in Fig. 22.4b. TIM-3 blockade on activated Th1 cells increases their production of IFN-γ, which in turn enhances or prolongs the activation of macrophages, DC, neutrophils, and upregulates TLR4 expression. Activated macrophages elaborate cytokine/chemokine programs through TLR4 pathway, critical to promote organ damage that can be negatively modulated via TIM-3 signaling. We favor the notion that the TIM-3 pathway may exert “protective” function by depressing IFN-γ production, and hence spare the IR-stressed organ in TLR4-dependent manner. However, although the blockade of “positive” TIM-1/TIM-4 or enhancement of “negative” TIM-3/Gal-9 costimulation might be essential, further studies are needed to accurately assess their therapeutic potential, given the opposing effects of TIM-1 and TIM-3 signaling. As PD-1– PD-L1 pathway has also been shown to promote cytoprotection [88, 89], harnessing physiological mechanisms of PD-1 negative T cell costimulation should prove instrumental for organ homeostasis by minimizing local damage and promoting IL-10-dependent cytoprotection.
In addition to “traditional” T cells, NK natural killer cells (NK) and natural killer T cells (NKT) cells may also play distinctive roles in the mechanism of IRI. Although depletion of NK1.1 cells (NK/NKT) fails to affect the severity of tissue IRI at early stages [90], it reduces the cellular damage in the later phase [91]. IR-triggered activation of NKT cells is mediated by CD1d and glycolipid Ags, released possibly by necrotic cells, to NKT cell invariant TCRs. Furthermore, NKT cell subsets may play distinctive roles in vivo, with type II NKT cells shown to prevent IRI when activated by specific glycolipid ligand sulfatide [92
 Bayesian Classifier and Molecular Marker Platforms for Immune Monitoring
Bayesian Classifier and Molecular Marker Platforms for Immune Monitoring
 Immunological Similarities and Differences Between Extremity and Face Transplants
Immunological Similarities and Differences Between Extremity and Face Transplants
 Mesenchymal and Adipose Stem Cell Strategies for Peripheral Nerve Regeneration
Mesenchymal and Adipose Stem Cell Strategies for Peripheral Nerve Regeneration
 Advances in Diagnosing Skin Rejection and Immune Monitoring
Advances in Diagnosing Skin Rejection and Immune Monitoring
 Experimental Models and Clinical Tools to Assess Nerve Regeneration and Functional Outcomes
Experimental Models and Clinical Tools to Assess Nerve Regeneration and Functional Outcomes
 Antibody-Mediated Rejection in Reconstructive Transplantation
Antibody-Mediated Rejection in Reconstructive Transplantation




Related posts:
Bayesian Classifier and Molecular Marker Platforms for Immune Monitoring
Immunological Similarities and Differences Between Extremity and Face Transplants
Mesenchymal and Adipose Stem Cell Strategies for Peripheral Nerve Regeneration
Advances in Diagnosing Skin Rejection and Immune Monitoring
Experimental Models and Clinical Tools to Assess Nerve Regeneration and Functional Outcomes
Antibody-Mediated Rejection in Reconstructive Transplantation
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
