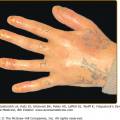
Inherited Palmoplantar Keratodermas: Introduction
|
Epidemiology
Inherited palmoplantar keratodermas are individually rare; the prevalence of epidermolytic keratoderma in Northern Ireland was 4.4 per 100,000.9 Autosomal recessive keratodermas may occur with locally high prevalence in sequestered populations or communities amongst whom consanguineous union is common.
Etiology and Pathogenesis
Palmoplantar skin is structurally specialized,10 with absence of hair and increased epidermal thickness and rugosity, features necessary to cope with increased friction and mechanical stress. Dermatoglyphics together with eccrine sweat also enhance grip. Localized palmoplantar hypertrophy (callus) is a physiological response to sustained friction, for example, from ill-fitting footwear or manual work. In the inherited PPKs, excessive epidermal thickening is the result of a broad range of pathogenic pathways.
Many keratodermas are associated with defects of keratinocyte structure. The major structural component of keratinocytes is the 10-nm (intermediate) filament cytoskeleton. Keratins are a family of rod-like proteins, expressed in pairs in a tissue and differentiation specific manner, which initially dimerize then assemble to form multimeric intermediate filaments.7 Defects in individual keratins affect skin in a distribution corresponding to the expression pattern of the particular keratin.3 Keratin 9 (K9) is specific to palmoplantar skin, although its likely partner in these sites, keratin 1, is also expressed in hair-bearing skin. Other keratins constitutively or facultatively expressed in palmoplantar skin include K6, K16 (also found in mucosa, hair follicles, and nail bed), and K17 (hair follicles and nail bed11). There are multiple K6 isoforms,12,13 and defects in K6a, K6b, and K6c as well as all the other keratins named above can result in keratoderma. The majority of pathogenic mutations in keratins occur in the highly conserved boundary peptides of the α-helical rod domain regions, which are thought vital for end-to-end overlap interactions during the elongation phase of filament assembly.3 Typically, keratin defects result in a disrupted intermediate filament cytoskeleton. The intermediate filament network is attached to desmosomes, intercellular junctions which in turn form paired plaques with the corresponding structures in adjacent keratinocytes. Defects in desmosomal proteins such as desmoglein 1, desmoplakin 1, plakoglobin, and plakophilin 1 also cause PPK.6 Structural weakness due to keratin and desmosome defects has the potential to cause epidermolysis or acantholysis of keratinocytes, of which hyperkeratosis may be an indirect consequence, but nonmechanical mechanisms are also probable. Keratins, for example, are also involved in the regulation of proliferation, apoptosis, and skin pigmentation.14,15 Moreover, cell stress as a nonspecific response to an accumulation of misfolded proteins may contribute to pathogenesis. Mechanically stressed keratinocytes containing mutant keratins of the type which cause severe epidermolysis bullosa simplex show greater resistance to apoptosis than wild-type keratinocytes; the increased resistance was dependant on an increase in extracellular signal-regulated kinase (ERK) and Akt signaling.16 Keratoderma in tyrosinemia type II may be also due to tonofilament accumulation secondary to excessive intracellular tyrosine.17
Another large group of PPK syndromes are due to defects in connexins, the proteins that make up gap junction communication channels between cells.5 Gap junctions are assembled in plaques containing multiple connexon units, each of which consists of a pair of hemichannels with a central channel through which small molecules (<1 kDa) can pass between the cytoplasm of adjacent cells. Each hemichannel in turn contains six homomeric or heteromeric connexin proteins. The 21 human connexin proteins, like keratins, are expressed in a tissue and differentiation specific manner and the phenotype of gap junction diseases in part reflects their expression pattern. For example, most mutations in the gene (GJB2) encoding connexin 26 (Cx26) cause impaired hearing because the gene is expressed in the inner ear where it is necessary for the circulation of endolymph. Cx26 is also expressed in skin, and some Cx26 defects also cause a cutaneous phenotype, such as PPK. PPK is also found in syndromes due to mutations affecting connexin 30 (Cx30, associated with hidrotic ectodermal dysplasia18,19) and connexin 43 (Cx43; oculo-dental digital dysplasia20). However, point mutations in a single connexin can cause a range of phenotypes depending on the specific amino acid affected. While some pathogenic mutations interfere with the formation of functional gap junctions, others exhibit defects in trafficking to the cell membrane and the connexins instead accumulate within organelles.21 Recent evidence in erythrokeratoderma variabilis (EKV) suggests that accumulation of mutant proteins causes the unfolded protein response.22 In the case of connexin mutations associated with palmoplantar keratoderma, this endoplasmic reticulum stress may be responsible for hyperkeratosis and inflammation.
Other mechanisms of PPK are extremely varied. In loricrin keratoderma, a barrier abnormality is associated with a defective CE scaffold that results in increased extracellular permeability defect.23 The C-terminal peptide of the mutant loricrin includes polybasic nuclear recognition signals which cause the aberrant protein to accumulate in the nucleus, which is likely to interfere with terminal differentiation.24–26 In Papillon–Lefèvre syndrome (PLS), in which PPK is associated with predisposition to pyogenic infection, the cysteine protease cathepsin C is inactive.27 This lysosomal enzyme is important in intracellular protein degradation28 and in the activation of neutrophil serine proteases.29 Its absence may have consequences for the regulation of inflammation,30 but the mechanism of keratoderma may also relate to aberrant desmosomal cleavage. In Mal de Meleda, a secreted nicotinic acetylcholine receptor ligand, SLURP-1, is deficient. The normally expressed protein may act by modulating keratinocyte behavior or inflammatory responses.31–33 Keratoderma is a feature of rare neurodevelopmental syndromes due to defective intracellular vesicle trafficking,34,35 and of keratosis linearis with ichthyosis congenita and sclerosing keratoderma (KLICK) syndrome where there is a defect in proteasome production.36
Presentation (Syndrome) | Inheritance | Gene(s)/Locus | Protein(s) |
|---|---|---|---|
Epidermolytic PPK (Vörner) | AD | KRT9 | Keratin 9 |
Transgredient epidermolytic PPK | AD | KRT1 | Keratin 1 |
Diffuse NEPPK (includes Unna–Thost) | AD | KRT1 | Keratin 1 (V1 domain) |
Ichthyosis hystrix | AD | KRT1 | Keratin 1 |
Focal nonepidermolytic PPK | AD | KRT16, KRT6c | Keratin 6c, 16 |
Pachyonychia congenita type 1 | AD | KRT6a, KRT16 | Keratin 6a, 16 |
Pachyonychia congenita type 2 | AD | KRT6b, KRT17 | Keratin 6b, 17 |
Ectodermal dysplasia/skin fragility syndrome | AR | PKP1 | Plakophilin 1 |
Striate PPK | AD | DSG1, DSP, KRT1 | Desmoglein 1, Desmoplakin 1, Keratin 1 |
Keratoderma with cardiomyopathy and wooly hair (Carvajal–Huerta and others) | AR/AD | DSP | Desmoplakin 1 |
Keratoderma with ARVC and wooly hair (Naxos) | AR | JUP | Plakoglobin |
Keratoderma with hearing loss (Vohwinkel, Bart–Pumphrey, and others) | AD | GJB2, GJB6 | Connexin 26, 30 |
Keratitis/Hystrix, ichthyosis, and deafness (KID/HID) | AD | GJB2 | Connexin 26 |
Hidrotic ectodermal dysplasia (Clouston) | AD | GJB6 | Connexin 30 |
Erythrokeratoderma variabilis | AD/AR | GJB3, GJB4 | Connexin 31, 30.3 |
Oculo-dento-digital dysplasia (of the face, eyes, skeletal system, heart, and dentition) | AD | GJA1 | Connexin 43 |
Mitochondrial keratoderma with hearing loss | Mito | MSST1 | Serine transfer RNA |
Loricrin keratoderma | AD | LOR | Insertion mutation in Loricrin |
Keratoderma and periodontitis (Papillon–Lefèvre and Haim–Munk) | AR | CTSC | Cathepsin C |
Mal de Meleda | AR | ARSB | SLURP-1 |
Tylosis with Oesophageal carcinoma (Howel-Evans) | AD | RHBDF2 | Inhibitor of active rhomboid protease RHBDL2 |
Odonto-onycho-dermal dysplasia (includes Schöpf–Schultz–Passarge) | AR | WNT10a | Signaling molecule implicated in development |
Tyrosinemia type 2 (Richner–Hanhart) | AR | TYR1 | Tyrosinase |
KLICK | AR | POMP | Proteasome maturation protein |
CEDNIK | AR | SNAP29 | SNARE protein involved in vesicle fusion |
MEDNIK | AR | AP1S1 | Subunit σ1A of adaptor protein-1 complex |
Mapped Disorders | |||
Sclerotylosis (Huriez) | AD | 4q23 | |
Diffuse NEPPK (includes Unna–Thost) | AD | 12q11–13 | |
Punctate PPK | AD | 15q22.2–15q22.31 |
Clinical Findings
Clinical findings vary between different genetic forms of PPK. The hyperkeratosis may present as multiple papular lesions (punctate PPK), as callosities localized to points of particular stress (focal/areate, or striate PPK) or may extend over the whole palm or sole (diffuse PPK). The distinction between focal, striate, and diffuse forms is not always easy to make, and the severity of PPK may vary, even within a family. Hyperhidrosis is a common complaint amongst patients with focal or diffuse keratodermas. Corynebacterial overgrowth leading to keratolysis and malodor, and secondary dermatophyte infection, is common. Transgredient keratoderma extends onto the dorsa of the hands and other cutaneous sites; circumferential keratoderma of digits is associated with the formation of constricting bands (“pseudoainhum,” see Chapter 68) and sometimes autoamputation (cicatrizing or mutilating keratoderma). It may also cause tapering of the digits (sclerodactyly) with bony atrophy and nail dystrophy. A number of keratoderma syndromes, in particular Huriez and Olmsted syndromes (see below) are characterized by the frequency of squamous cell carcinoma in affected skin; melanoma has also been reported in various PPKs.
In the history, early age of onset is usual. Punctate keratodermas present in adulthood, but new presentations of diffuse PPK at this age are more likely to be papulosquamous disorders (see Section “Differential Diagnosis”). In inherited PPKs, hyperhidrosis is commonly reported. Pain is a particular feature of focal keratodermas, but in all forms of keratoderma functional, occupational, or social disability should be recorded. A family history, including consanguinity, may be helpful but it is unwise to draw firm conclusions about either clinical type or inheritance patterns without having examined all available members of a pedigree, even if said to be affected. Family history of early onset systemic malignancy or skin cancer should be noted.
The distribution of keratoderma (punctate, focal, striate, or diffuse) is not an absolute guide. Feet tend to be more markedly and diffusely involved, so that in severe cases the underlying pattern may be more obvious on the hands. The appearance of the hyperkeratosis—for example, honeycomb patterned, waxy, or fissured—may be significant. Sharp, livid margins or the presence of transgredient hyperkeratosis on dorsa of hands, feet, and digits should be noted. The possibility of secondary fungal or bacterial infection, and of the development of malignancy, should be considered. It is important to examine the whole skin for signs of other skin diseases such as eczema, psoriasis, and lichen planus. In inherited PPK, mild acral hyperkeratosis may indicate a generalized cutaneous disorder that is locally severe on palms and sole; fissures at the oral commissures and follicular hyperkeratosis are also common. Mild ichthyosis seen in loricrin PPK may be missed if not specifically sought. Many PPKs involve mucosa, including genital mucosa. The presence of other ectodermal abnormalities should be documented, i.e., abnormalities of nails, teeth, hair and hair follicles, sweat glands, and sweating. Finally, specific features associated with syndromic keratoderma should be elicited by history and examination. These include hearing impairment (which may be subtle), neuropathy, and cardiac disease (conduction defects or cardiomyopathy).
A pathway to aid diagnosis (Fig. 50-1) is provided, but is neither comprehensive nor absolute. Individual presentations are discussed in more detail below.
Figure 50-1
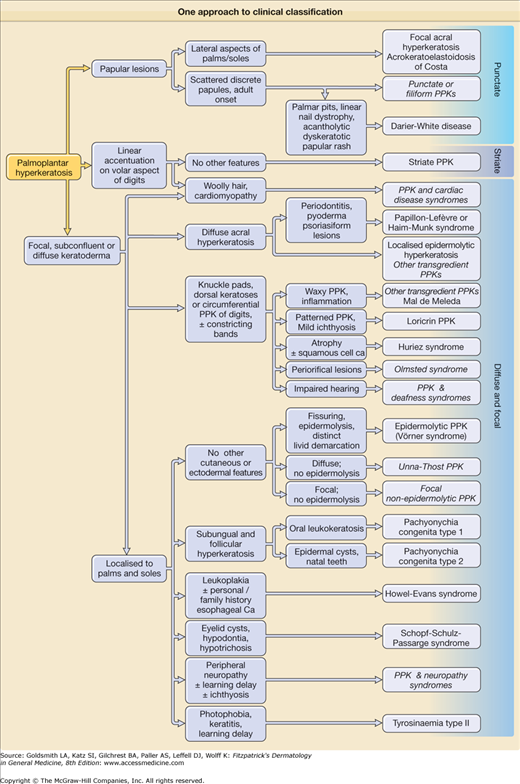
One approach to clinical classification. Diagnoses shown in italics are pragmatic groups rather than genetically defined syndromes (see text). It is unwise to use the pattern of keratoderma (i.e., papular, focal, striate, diffuse) as the primary means of classification, since this can vary even within families. It is a good principle to look for ectodermal and syndromic associations in all new cases.
Punctate keratoderma (Buschke–Fischer–Brauer syndrome; MIM 146800) is usually inherited as an autosomal dominant trait.37 Unlike most other inherited PPKs, punctate PPK presents in adult life as multiple keratinizing papules (Fig. 50-2A). It may be an incidental finding, although plantar lesions can produce significant disability and also cause secondary focal keratoderma. From the gradual accumulation of discrete individual lesions it may be inferred that there is a defect in one inherited allele, and the disease is expressed when the remaining allele is damaged. An association with malignancy of kidney, stomach, breast, and colon, has been reported in a few pedigrees,38,39 but has not been generally substantiated. In any case, the condition is probably genetically heterogeneous. The size of the papules in different families varies from a few millimeters to tiny filiform lesions (music-box spine keratoderma) (see eFigs. 50-2.1 and 50-2.2). Similarly, histology of the lesions may show orthokeratotic or parakeratotic hyperkeratosis. At least two genetic loci have been suggested40,41 although that at 15q22.2–15q22.31 is most certain. Other clinically similar conditions include punctate keratoderma of the palmar creases, and focal acral hyperkeratosis of the margins of palms and soles, with or without elastoidosis. Papular palmoplantar lesions may be seen in other genodermatoses, notably Darier disease (see Chapter 51), in which the pitted lesions common in younger cases become papular with age. Acquired causes of punctate palmoplantar lesions include dioxin toxicity and chronic arsenicism.
Figure 50-2
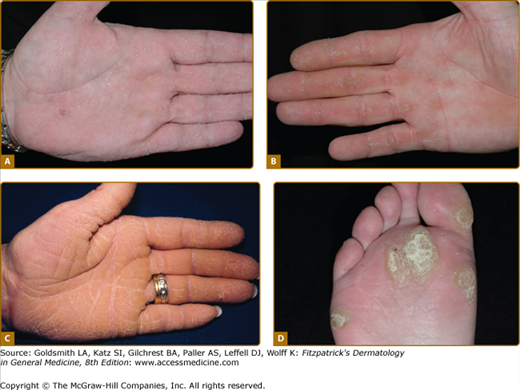
Patterns of familial keratoderma. A. Punctate, which usually does not appear until adulthood. The lesions have been accentuated by immersion in water for a few minutes. B. Striate, often due to desmosomal disorders. C. Diffuse, in this case with fissuring and the sharp demarcation typical of keratin 9 defects. D. Focal, seen as an isolated finding due to keratin 6c mutations. In practice the distinction between these patterns may not be clear, especially on plantar skin.
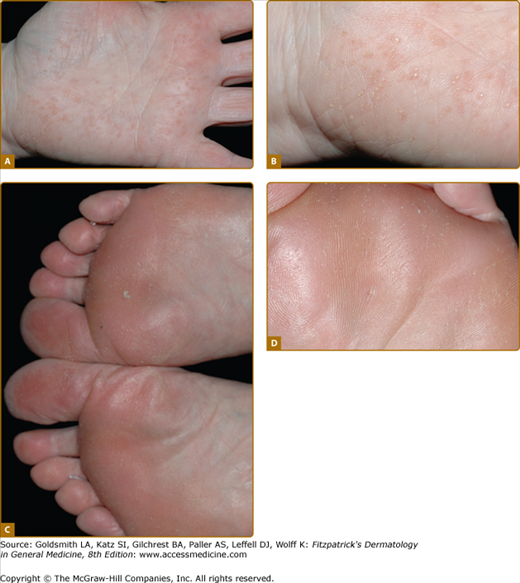
An example of uncomplicated diffuse keratoderma is epidermolytic PPK (EPPK; Vörner syndrome; MIM 144200). EPPK is probably the commonest PPK,9 and is an autosomal dominant trait due to defects in keratin 9, the one keratin specific to palmoplantar skin.42 It presents in childhood as diffuse PPK with a sharp, livid transition to normal skin at the edge of palms and soles (Fig. 50-2C; see also eFig. 50-2.3). Severe cases may be accompanied by blistering, although in adults the epidermal weakness is more commonly manifested by fissuring. Histology of the stratum spinosum shows vacuolated keratinocytes with keratin filament aggregates at electron microscopic level, accompanied by orthohyperkeratosis of the stratum corneum. Defects in keratin 1, the presumed partner of keratin 9 in palmoplantar skin, also cause diffuse transgredient keratoderma with epidermolytic hyperkeratosis at other cutaneous sites, but extrapalmoplantar involvement may be subtle (see eFig. 50-2.4). A specific defect of the 1B domain of keratin 1 causes tubular tonofilament structures to form in some pedigrees.43 Keratoderma may also be a feature of epidermolysis bullosa of the severe Dowling–Meara type (MIM 131760; 44) due to mutations in the basal layer keratins 5 and 14 (see Chapter 62). The mechanism(s) by which keratin gene defects may give rise to the palmoplantar hyperkeratosis are discussed above.
Nonepidermolytic diffuse PPK restricted to palmoplantar skin is heterogeneous. The term Unna–Thost syndrome (MIM 600962) should probably be discarded, as even the original Thost family in fact had epidermolytic PPK.45 In one pedigree a diffuse nonepidermolytic phenotype was due to a defect in the variable region of the keratin 1 gene,46 and in another was mapped to a locus including the type II keratin gene cluster.47 Another locus proximal to the type II keratin gene cluster has been identified.48
Focal PPK (see Fig. 50-2 and eFig. 50-2.5) may complicate punctate PPK on the feet, or may occur in isolation; in the latter situation it may be difficult to distinguish from physiological callosities, and indeed the tendency to these may have a genetic basis.49 Early onset focal keratoderma of whatever cause leads to disability (“hereditary painful callosities”), and patients may be so severely affected as to become wheelchair bound. Autosomal dominant pedigrees of focal PPK are commonly associated with nail dystrophy and mucosal features as part of the pachyonychia spectrum, but even in the absence of nail dystrophy may be due to mutation in keratin 16.50 In the majority of cases of focal PPK in isolation, genetic causes have not been identified, but in three pedigrees it has been ascribed to mutations in keratin 6c.49 Painful focal PPK is also found in tyrosinemia type II (see below and Chapter 131).
Striate PPK (SPPK; MIM 148700) is a distinct variant of focal PPK in which there is prominent linear involvement of the volar surfaces of the digits (see Fig. 50-2B) and less distinctly of corresponding areas of the soles. Histology of autosomal dominant inherited striate PPK shows subtle widening of the intercellular spaces (see Fig. 50-5C). 51,52 Most cases are due to defects in gene encoding desmosomal plaque proteins of which desmoglein 1 (DSG1) is most commonly implicated, but mutations in desmoplakin 1 are also reported.53,54 Keratoderma due to defects in these genes is believed to be due to haploinsufficiency, i.e., reduced amounts of the relevant structural protein expressed in the desmosomal plaque result in mechanical weakness which manifest at points of greatest stress. However, as with keratins, demosomal components are implicated in intracellular signaling and growth regulation. Striate PPK may also be seen with some mutations in keratin 1,55 and indeed striate accentuation is a nonspecific component of milder diffuse keratodermas. Most cases of autosomal dominant SPPK are simple, but defects in desmoplakin also give rise to syndromes of cardiomyopathy and wooly hair (see below).
Figure 50-5
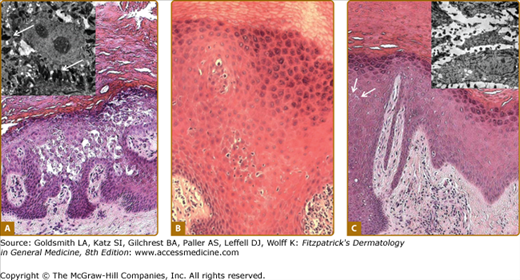
Histology of keratoderma. A. Vacuolation of spinous layer keratinocytes in epidermolytic PPK due to keratin 9 mutations; inset, tonofilament aggregates (arrows), and cytolysis on electron microscopy. B. Eosinophilic cytoplasm but without overt epidermolysis and inflammatory infiltrate in pachyonychia congenita due to keratin 17 mutation. C. Subtly increased cell separation (arrows) in striate PPK due to desmoglein 1 mutation; inset of electron microscopic appearances shows overtly increased cell separation.
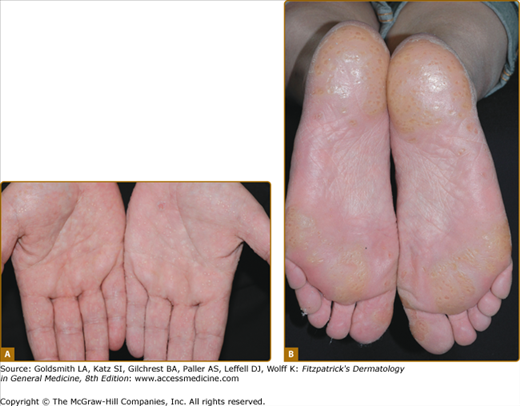
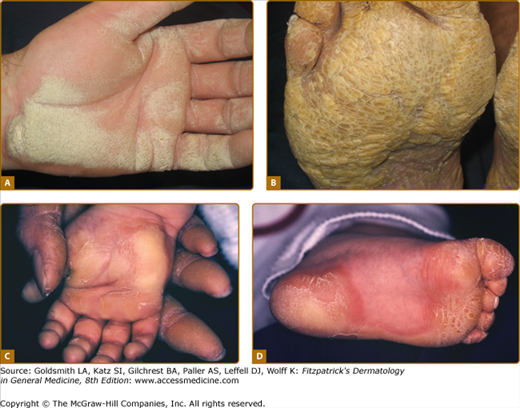
Loricrin keratoderma (syn. Camisa syndrome; MIM 640117) was described as a variant of Vohwinkel syndrome (VS).56 It is characterized by a similar “honeycomb” patterned and transgredient keratoderma (Fig. 50-3A; see also eFig. 50-3.1), leading to circumferential digital involvement, cicatrizing bands, and sometimes autoamputation. However, there is also a mild generalized ichthyosis, and collodion babies or generalized desquamation at birth are reported.57,58 Compared with true VS, due to mutations in Cx26, the edge of the keratoderma at the wrists is diffuse, and deafness is not a feature. Multiple mutations are reported of which the most frequent mutation, 730insG, has been found in families from the United Kingdom, Japan, Germany, and Italy.24,57,59–64 In all cases they are single nucleotide insertions resulting in a shift of the reading frame and a termination codon delayed by 22 amino acids. In the elongated carboxyl-terminal domain many of the glycine residues are replaced by arginine, drastically altering the properties of the loricrin polypeptide by producing nuclear recognition signals (see above).
Figure 50-3
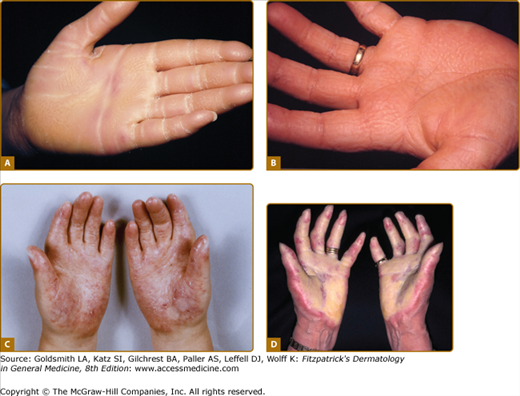
Rare forms of transgredient PPK. A. Patterned keratoderma in a case of loricrin PPK; note circumferential keratoderma and early constricting bands. B. Confluent papules in Vohwinkel PPK with impaired hearing due to connexin 26 mutations; honeycomb lesions and circumferential PPK with constricting bands also occur in this syndrome. C. Huriez syndrome (sclerotylosis), which carries a high risk of squamous cell carcinoma. (Used with permission from Dr. Cameron Kennedy, Bristol Royal Infirmary, United Kingdom.) D. Mal de Meleda due to mutations in ARS component B, with a waxy hyperkeratosis, sclerodactyly, and constricting bands.
Huriez syndrome (PPK with scleroatrophy, sclerotylosis; MIM 181600), an autosomal dominant disorder, is characterized by diffuse PPK that affects mainly palmar skin (Fig. 50-3C).65–70 The underlying gene defect is unknown but linkage to chromosome 4q23 has been reported.71 Sclerodactyly, brachydactyly, and cutaneous erythema and/or atrophy are typical, as are various nail changes; hypohidrosis is also reported. A deficiency of Langerhans cells in affected skin has been identified.72 The condition predisposes to squamous cell carcinomas of affected skin, occurring in the third to fourth decade, which are unusually prone to metastasis.67,68 Causal treatment is not available, but benefit from oral retinoids is recorded.67
Mal de Meleda (MIM 248300) is a rare autosomal recessive disorder originally described in communities of the Mediterranean littoral. The transgredient (i.e., it extends over the lateral margin of the palms and proximally over the wrists) hyperkeratosis has a waxy ivory to yellow appearance and is typically inflamed or macerated, with a livid margin.73 Dermatophyte superinfection, to which many keratodermas are prone, may mimic these appearances. Knuckle pads, similar waxy lesions on acral sites, angular cheilitis, and circumferential sclerosing and cicatrizing lesions of the digits are typical (Fig. 50-3D; see also eFig. 50-3.2B); not surprisingly nails are often dystrophic. The typical Mediterranean disorder is due to mutations in the ARSB gene encoding SLURP-1, an acetylcholine receptor analog.31,74,75
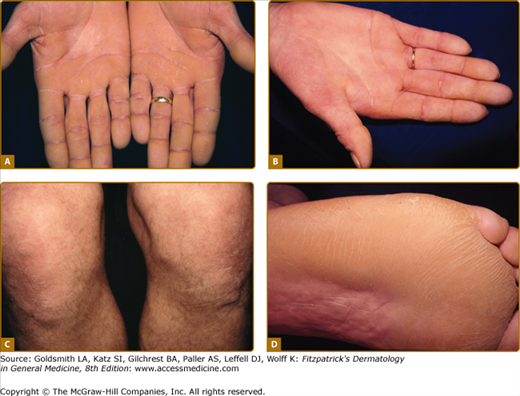
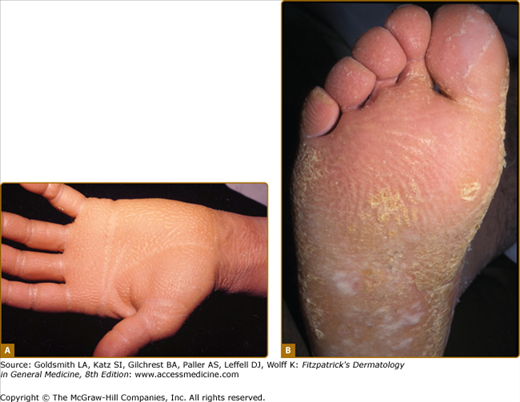
Recessive transgredient and/or mutilating disorders with phenotypes resembling Mal de Meleda are reported,76,77,78,79,80 but appear to be genetically distinct. A very rare autosomal recessive disorder (KLICK syndrome) has been described in which a cicatrizing PPK is associated with ichthyosis and a bizarre striate hyperkeratosis of flexures.81,82 This has recently been shown to be due to defects in POMP which encodes a protein which is a chaperone for proteasome maturation.36 Similarly, pedigrees of autosomal dominant transgredient PPKs have been described. The term Greither syndrome has been used,2,83,84 but may not be a single entity.85 Some autosomal dominant transgredient keratodermas within this spectrum are due to mutations in keratin 1 (Fig. 50-4A),86 but other such pedigrees, for example, Sybert syndrome87 appear not to be keratin disorders.
Figure 50-4
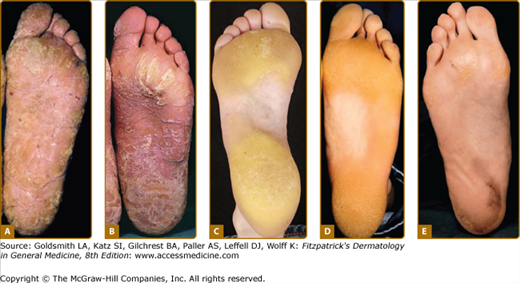
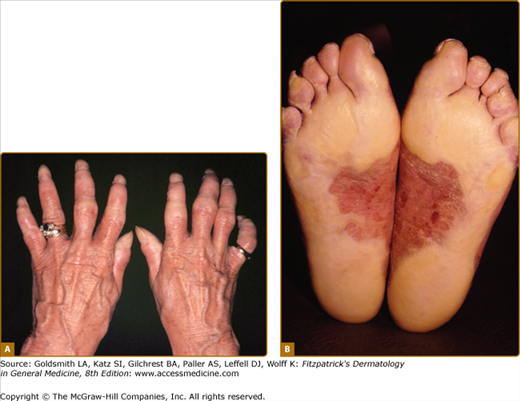
The pattern of plantar PPK is often not diagnostic. A. Diffuse fissured PPK due to mutation in keratin 1 but with only localized epidermolytic hyperkeratosis of elbows, knees and flexures. B. Similar changes in Papillon-Lefèvre syndrome due to mutation in Cathepsin C. (Used with permission from Barts and the London NHS Trust, United Kingdom.) C. Howel–Evans syndrome (PPK with familial esophageal carcinoma) showing subconfluent keratoderma sparing the instep. D. Similar changes due to an insertion mutation in keratin 1. E. Focal PPK with impaired hearing due to A7445G mutation in mitochondrial DNA.
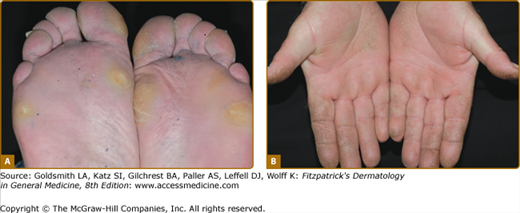
Pachyonychia congenita (PC) is a spectrum of dominantly inherited disorders associated with focal PPK. In addition to painful focal PPK and hypertrophic dystrophy of the distal nail, there are numerous reported associations88 (see eFig. 50-4.1). Two main syndromes are recognised clinically,89 in which various associated features can cause significant additional morbidity and disability. PC-1 (Jadassohn–Lewandowsky; MIM 167210) is associated with follicular keratoses, oral leukokeratosis and hoarseness. Severe oral lesions can resemble mucosal candidiasis90 and laryngeal involvement may produce infantile respiratory obstruction.91 The rarer PC-2 (Jackson–Lawler; MIM 167210) is distinguished by multiple pilosebaceous cysts, teeth present at birth, and hair changes such as protuberant eyebrows. Extensive and infected flexural, vulval or scrotal cysts can present as hidradenitis suppurativa.92 PC-1 is typically associated with mutations in KRT6A or KRT16 and PC-2, with mutations in KRT6B or KRT17, reflecting the expression patterns of the relevant keratins3: for example K16 is a major secondary keratin in orogenital epithelia. Severity varies within and between families, and incomplete phenotypes such as PPK without nail dystrophy or steatocystoma multiplex without PPK may be caused by mutations in the same genes. However, detailed analysis of patients with KRT16 mutations suggests that mutations which predict a more disruptive effect on K16 protein structure produce a more severe phenotype.93 A recent large study casts doubt on the consistency of the association of clinical syndrome and mutated keratin94 and it has been proposed instead that PC be designated according to the affected keratin—PC-6a, PC-6b, etc. and PC-U (unknown).95 Genotypic classification has the aim of individualising targeted molecular therapy, already promising in this disorder.8
eFigure 50-4.1
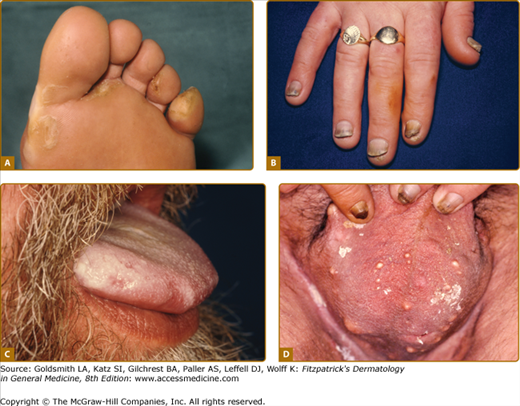
Features of pachyonychia congentia. A. Blistering of the feet can mimic epidermolysis bullosa. B. Finger-nail dystrophy is on dominant hand especially in manual workers. C. Oral leukokeratosis of tongue is found with keratin 6a and 16 mutations. D. Multiple cysts are seen with keratin 6b and 17 mutations although such marked scrotal involvement is unusual.