Granulomatous cutaneous T-cell lymphomas (CTCL) and lymphomatoid granulomatosis are considered granulomatous lymphoproliferative disorders. The most common types of granulomatous CTCL are granulomatous mycosis fungoides and granulomatous slack skin. Lymphomatoid granulomatosis is a rare Epstein-Barr virus driven lymphoproliferative disorder. This article reviews the etiopathogenesis, clinical presentation, systemic associations, and management of both granulomatous slack skin syndrome and lymphomatoid granulomatosis.
Key points
- •
Granulomatous cutaneous T-cell lymphomas (CTCL) are rare and present a diagnostic challenge.
- •
Granulomatous mycosis fungoides and granulomatous slack skin, the 2 most common types of granulomatous CTCL, display overlapping histologic findings, but differ clinically by circumscribed areas of pendulous lax skin seen in granulomatous slack skin.
- •
Recent studies have suggested that the prognosis of granulomatous mycosis fungoides is worse than that of classic mycosis fungoides.
- •
Lymphomatoid granulomatosis is a rare Epstein-Barr virus driven lymphoproliferative disease.
- •
Therapeutic options include oral corticosteroids, rituximab, interferon-α, and combined chemoimmunotherapy, and are based on the histologic grading of the lesion.
Granulomatous cutaneous T-cell lymphoma
Introduction
Granulomatous cutaneous T-cell lymphoma (CTCL) is a rare entity. Approximately 2% of all cutaneous lymphomas involve granulomatous infiltrates. The most common types of granulomatous CTCL are granulomatous mycosis fungoides (GMF) and granulomatous slack skin (GSS) ( Table 1 ).
| Clinical and Therapeutic Data from a Series of 19 Patients with Granulomatous Cutaneous T-Cell Lymphoma | ||||
|---|---|---|---|---|
| Patient No. | Stage | Therapy | Treatment Response | Outcome/Follow-Up (years) |
| Granulomatous Mycosis Fungoides | ||||
| 1 | IB | IFN, PUVA | PR | AWD/8 |
| 2 | III | IFN, chemo | SD | DOD/1 |
| 3 | IA | CS, MC, RT, IFN | CR | ACR/20 |
| 4 | IB | CS, MC | PR | AWD/6 |
| 5 | IIB | RT, chemo | PR | DOD/2 |
| 6 | IB | PUVA, RT | PD | DOD/1 |
| 7 | IA | Imiq, IFN, PUVA, RT | CR | ACR/6 |
| 8 | IA | Chemo | SD | AWD/1 |
| 9 | IVA | Chemo | CR | AWD/4 |
| 10 | IIA | Pred | PD | AWD/5 |
| 11 | IA | PUVA, IFN | PR | AWD/4 |
| 12 | IA | PUVA, RT, IFN | PD | DOD/9 |
| 13 | IA | CS, PUVA, IFN, ret | SD | AWD/7 |
| 14 | IB | PUVA, IFN, RT, chemo | PD | DOD/1 |
| 15 | IA | CS, IFN, ret, RT | PD | DOD/5 |
| Granulomatous Slack Skin | ||||
| 16 | IA | PUVA, ret, IFN | PR | AWD/10 |
| 17 | IA | NA | NA | AWD/16 |
| 18 | IA | Excision, CS, MC, PUVA, IFN | PD | AWD/15 |
| 19 | IA | Excision, carmustine | PR | AWD/28 |
GMF is an entity distinct from GSS. GMF is a rare histopathologic variant of mycosis fungoides (MF) characterized by a prominent granulomatous infiltrate. The term GMF was coined by Ackerman and Flaxman in 1970. GSS is recognized by the World Health Organization and European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas as 1 of 3 subtypes of mycosis fungoides. GMF and GSS display overlapping histologic findings, but differ clinically by circumscribed areas of pendulous lax skin seen in GSS.
Etiopathogenesis
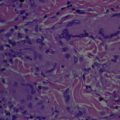
The pathogenesis of granulomatous CTCLs is unknown. Histologically, GMF and GSS show overlapping findings and cannot be discriminated by histologic examination alone. Typically, GMF has an atypical lichenoid CD4 + CD8 − lymphocytic infiltrate with interstitial histiocytes and/or perivascular granulomas with concomitant eosinophils and multinucleated giant cells. Sarcoidal granulomas are also commonly encountered. Tuberculoid, periadnexal, and granuloma annulare–like patterns are occasionally seen.
Classic histologic features of GSS include dense diffuse dermal infiltrate of atypical, irregular, and convoluted lymphocytes that may extend to the subcutaneous tissue. In the dermis, diffuse multinucleate giant cells and numerous histiocytes, which exhibit prominent elastophagocytosis and lymphophagocytosis, are observed. The multinucleated giant cells show 20 to 30 nuclei in the periphery of the cytoplasm. Loss of elastic fibers usually correlates with the extent of the granulomatous infiltrate, and is a universal finding in GSS. In the past, elastolysis involving the full thickness of the dermis was thought to be pathognomonic for GSS. However, a more recent study showed loss of elastic fibers in both patients with GSS and patients with GMF.
Clinical Presentation
The clinical presentation and skin manifestations of GMF are similar to those of classic MF, and patients may present with patches, plaques, tumors, erythroderma, poikilodermatous patches, and granuloma annulare–like lesions. GMF may coexist with classic MF lesions. Unlike GSS, patients with GMF present without pendulous skin folds, and extracutaneous spread is common.
GSS is an extremely rare type of CTCL. GSS usually presents initially as asymptomatic, erythematous papules and plaques that progress, with elastic tissue loss, to bulky pendulous skin folds with a predilection for the axillae and groin of Caucasian men in their third to fifth decades of life. Unlike granulomatous MF, extracutaneous spread is rare. Most patients have a very slow progressive course.
Systemic Associations
Patients with GMF and GSS are at risk for the development of second lymphoid neoplasias. Hodgkin lymphoma is the most common second neoplasia in patients with granulomatous CTCL and occurs in approximately 50% of cases. Granulomatous CTCL may also be associated with MF, CD30 + anaplastic large-cell lymphoma, follicular lymphoma, chronic lymphocytic leukemia, diffuse large B-cell lymphoma, and lymphomatoid papulosis.
Patients with CD8 + granulomatous CTCL appear to have an associated immunodeficiency. In a retrospective study of 4 patients with CD8 + granulomatous CTCL, nodal involvement was not seen but lung granulomas were found in all cases. All 4 patients had a history of immunodeficiency, either primary or iatrogenic, in addition to slowly progressive disease. Evaluation for an underlying immunodeficiency may be considered in patients with CD8 + granulomatous CTCL.
Evaluation and Management
Granulomatous CTCLs are a diagnostic challenge. Predominant granuloma formation may obscure the atypical lymphoid infiltrate, delaying diagnosis and treatment. Epidermotropism may or may not be present. Detection of a monoclonal T-cell receptor (TCR) gene rearrangement can be a useful diagnostic adjunct in granulomatous CTCLs. However, a monoclonal T-cell population may also be found in reactive granulomatous disorders. In a recent study, rearrangement of TCRγ genes had sensitivity of 94% and specificity of 96% for granulomatous CTCL over reactive granulomatous disorders. Diagnosis of GSS should be made on clinical findings and be restricted to patients presenting with lax skin folds.
Granulomatous CTCLs show a therapy-resistant, slowly progressive course. Treatment options for patients with GMF are similar to those recommended for patients with classic MF. To date, there is no standard treatment for GSS. Multiple treatment modalities, both individually and in combination, have been tried with some success. Partial remissions have been reported with psoralen in combination with ultraviolet A light (PUVA), radiotherapy, (poly)chemotherapy, systemic steroids, azathioprine, immunomodulating drugs such as interferon-α and interferon-γ, surgery, and some combination therapies. Rapid recurrences of GSS after surgical excision have been reported.
Clinical Results and Outcomes in the Literature
In a study of 15 patients with GMF and 4 with GSS, the disease showed a slowly progressive course. The most common treatment modalities were PUVA and interferon-α in addition to radiotherapy. Extracutaneous disease was observed in one-third of patients with GMF, and 6 of 15 patients with GMF died of the disease. By contrast, all patients with GSS were alive after a median follow-up of 17 years. GSS was therapy resistant, and complete remission was not achieved in any patient.
In a recent retrospective case-control study of 27 patients with GMF and age-matched and stage-matched patients with classic MF, patients with GMF had poorer response to skin-directed therapies and progressed more frequently. Compared with patients with classic MF, patients with GMF achieved fewer partial or complete responses with topical (57% vs 83%) or ultraviolet light (62% vs 90%) therapy. Patients with GMF also required a longer time to achieve best response (median 35 vs 9 months) compared with patients with classic MF. The 5- and 10-year progression-free survival rates were significantly lower in the GMF group (59% and 33%) compared with the classic MF group (84% and 56%). However, this significant difference did not translate into worse overall survival. The 5- and 10-year overall survival rates were similar in patients with GMF (86% and 72%) and those with classic MF (85% and 85%) ( Table 2 ).
| Treatments | N = 25 | |
|---|---|---|
| No. of Patients | No. of Patients Achieving PR or CR | |
| Skin directed | ||
| Topical therapy a | 23 | 13 (57%) |
| PUVA or NB-UVB | 13 | 8 (62%) |
| TSEBT | 4 | 4 (100%) |
| Systemic | ||
| Oral bexarotene | 9 | 5 (56%) |
| Interferon | 1 | 1 (100%) |
| Romidepsin/vorinostat | 5 | 1 (20%) |
| Alemtuzumab/zanolimumab | 3 | 1 (33%) |
| Denileukin diftitox | 2 | 1 (50%) |
| Pralatrexate | 4 | 2 (50%) |
| Methotrexate | 4 | 3 (75%) |
| Chemotherapy b | 9 | 7 (78%) |
| Allogeneic SCT | 2 | 2 (50%) |
| No treatment | 1 | 1 (100%) |
| Treatments (median) | ||
| Topical | 2 | |
| Systemic | 1 | |
| Total | 4 | |
| Systemic therapy | ||
| Yes | 16 (67%) | |
| No | 8 (33%) | |
| Time to best response, mo | ||
| Median | 35 | |
| Mean | 56 | |
| Best response | ||
| CR | 9 (38%) | |
| PR | 13 (54%) | |
| Stable disease | 2 (8%) | |
a Topical corticosteroids, nitrogen mustard, and bexarotene.
b Cladribine, liposomal doxorubicin, gemcitabine, vinorelbine, and combination chemotherapy with cyclophosphamide, doxorubicin, vincristine, prednisone (CHOP) or CHOP-based.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


