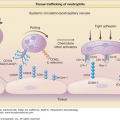
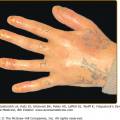
Graft‐versus-Host Disease: Introduction
|
Epidemiology
Approximately 50,000 hematopoietic stem cell transplantation (HCT) procedures are performed worldwide each year for an expanding array of hematologic malignancies and marrow failure syndromes, metabolic disorders, and immunodeficiencies. HCT may utilize autologous, syngeneic, or allogeneic donor hematopoietic stem cell (HC). During autologous transplantation the patient’s own HC are returned to the patient following preparative chemotherapy. Syngeneic transplantation is the transfer of HC between identical twins. Allogeneic HCT (allo-HCT) is the transfer of HC from a related (nonidentical) or unrelated donor to a recipient. Graft-versus-host disease (GVHD) is the primary cause of nonrelapse-related morbidity and mortality in allo-HCT and also rarely occurs following transplantation of solid organs or transfusion of blood products.
Transplantation regimens have advanced rapidly since the first successful allo-HCT was performed in 1968.1 Peripheral blood, rather than bone marrow, is now the primary source of donor HC at many transplant centers, and reduced intensity (nonmyeloablative) conditioning has permitted older patients and others who would not tolerate myeloablative chemotherapy a chance for cure with HCT. More recently, umbilical cord blood has gained prominence as a stem cell source in both pediatric and adult HCT. Donor leukocyte infusions (DLI), the administration of additional donor HC to the recipient weeks or even months after HCT, are also frequently utilized to augment graft-versus-malignancy effect.
These evolving trends in HCT, in conjunction with other known donor/recipient risk variables (Box 28-1), contribute to a wide range of reported GVHD incidence. Nevertheless, the degree of HLA-mismatch between donor and recipient remains the single most important predictor of GVHD.2 Acute GVHD develops in approximately 40% of fully matched sibling donor HCT, whereas 80% of mismatched unrelated HCT result in acute GVHD.3,4 Risk estimates of chronic GVHD also vary widely and confounding factors such as improving early posttransplant survival may be influencing the apparent trend in increasing chronic GVHD incidence.5 The most significant additional risk factor for chronic GVHD is a history of antecedent acute GVHD.5
HLA-incompatibility Patient age (elderly > adult > pediatric) Female donor (especially multiparous)/male recipient Stem cell source (peripheral blood > bone marrow > cord blood) T-cell replete graft Unrelated donor Donor leukocyte infusion Interruption or rapid tapering of immunosuppression |
Skin involvement is often the first indicator of acute GVHD (81%), followed by gastrointestinal (54%) and liver disease (50%).6 Similarly, the majority of patients who develop chronic GVHD manifest skin symptoms at some point in their disease course. The risk of chronic skin involvement is increased by the use of peripheral blood HCT (PB-HCT) compared with bone marrow HCT (BM-HCT). At one center, approximately 90% of patients who developed chronic GVHD following PB-HCT manifested skin symptoms.7 In most published reports, the incidence of sclerotic versus nonsclerotic chronic skin manifestations has not been differentiated. Although sclerotic involvement is less common than “lichenoid” GVHD and tends to occur later post-HCT, sclerotic features, particularly deep-seated fascial changes, may have an insidious onset, and “lichenoid” involvement is not a prerequisite to the development of sclerotic features. In one series of 196 patients post-HCT, only 7 (3.6%) developed sclerotic manifestations (mean 2.0 years after HCT).8 In a review of 133 patients who survived at least 4 months after allo-HCT, the 5-year cumulative incidence of sclerotic GVHD was 10.5% (15.5% among patients with chronic GVHD). In this series, only 21% manifested “lichenoid” changes prior to the onset of skin sclerosis.9 In a referral setting for patients with primarily refractory disease at the NIH, 81/110 (74%) consecutive patients had skin involvement, 58/110 (53%) of whom had sclerotic manifestations.10
Etiology and Pathogenesis
In 1966, Billingham proposed three basic requirements for GVHD: (1) immunocompetent transplanted cells, (2) host antigens recognizable by the transplanted cells and lacking in the donor, and (3) a host incapable of mounting an immune response to the transplanted cells.11 The immunocompetent cells are now known to be T-cells, which target human leukocyte antigens (HLAs) expressed on host tissues. GVHD still develops in 40% of recipients of HLA-identical grafts, however. In this setting, GVHD is due to mismatch of key minor histocompatibility antigens (e.g., HY, HA-3).12 Tissue damage from the recipient’s underlying disease, infection, and pretransplant conditioning also plays a key role in induction of the inflammatory response through pro-inflammatory cytokine production and antigen-presenting cell (APC) activation.13,14
Following activation of host APCs, T-cell activation and differentiation drives the response in acute GVHD. This appears to be primarily a Th1-driven process with massive release of interferon-γ, interleukin-2 (IL2), and TNF-α.14 Genetic polymorphisms in tumor necrosis factor (TNF)-α, interleukin-10, interferon-γ, and transforming growth factor (TGF)-β have been linked to increased risk and severity of GVHD.15–17 Although many therapies for acute GVHD target IL-2 or its receptor (CD25), these approaches (calcineurin inhibitors, daclizumab) may have the unintended consequence of adversely impacting the CD4+CD25+ regulatory T-cell population.14,18 Decreased T-regulatory cells are associated with severity of acute GVHD and poor response to GVHD treatment.19 The final effector phase of acute GVHD is characterized by cell damage via cytotoxic T-cells, natural killer cells, and soluble inflammatory mediators, including TNF-α, interferon γ, and interleukin-1.14
In comparison to acute GHVD, the pathophysiology of chronic GVHD is less well understood. Features of alloimmunity and autoimmunity and the broad spectrum of disease manifestations implicate multiple immunological pathways beyond T-cell alloreactivity. In fact, in contrast to acute GVHD, T-cell depletion of the graft does not necessarily reduce the incidence of chronic disease.20 Murine models of GVHD have demonstrated both Th1 and Th2 responses, depending on the setting; however, these models typically demonstrate specific aspects of GVHD, but do not recapitulate the full breadth of immunological and clinical abnormalities seen in human disease.21
The role of B-cell function in chronic GVHD has garnered renewed interest following the success of the anti-CD20 antibody rituximab in chronic GVHD.22 Autoantibody formation (e.g., antinuclear antibody, anti-ds DNA antibody) is a frequent finding in chronic GVHD, although the antibodies lack the specificity of typical autoimmune disease. B-cells may play several key roles in facilitating the T-cell response in chronic GVHD. Acting as APCs, B-cells prime T-cells to respond to minor histocompatibility antigens (mHA), and high-titers of antibodies directed against mHA are associated with cGVHD.23,24 Similarly, soluble levels of B-cell activating factor of the TNF family (BAFF), a cytokine which inhibits apoptosis of B-cells and promotes differentiation into plasma cells, correlate with cGVHD activity. 25,26
The mechanisms responsible for chronic GVHD-induced fibrosis in the skin and elsewhere (e.g., bronchiolitis obliterans) remains uncertain. A two-phase model has been proposed in which the innate pathway is activated through toll-like receptors, leading to an alloreactive T-cell response. This is followed by a fibrotic phase driven by platelet-derived growth factor (PDGF) and PDGF receptor (PDGFR), which in turn activates TGF-β.21 TGF-β is a potent profibrotic cytokine, capable of stimulating collagen production, abrogating metalloproteinase activity, and sensitizing fibroblasts to a constitutive-activated state via autocrine signaling.27 Furthermore, stimulatory antibodies directed against PDGFR have been identified by one group in patients with chronic GVHD as well as patients with systemic sclerosis.28,29 This has led to significant interest in imatinib mesylate, a multikinase inhibitor with potent activity against PDGFR signaling (and other receptors), for the treatment of GVHD-related fibrosis.30,31 However, to date, detection of PDGFR antibodies in sclerotic skin disease has not been replicated by other groups,32 and administration of imatinib prior to the onset of GVHD does not appear to eliminate the risk of developing skin sclerosis.33 The mechanism of action of imatinib therefore, remains unclear, and other mechanisms, including T-cell inhibition34 and inhibition of fibrosis via “nonclassic” pathways downstream of TGF-β, such as cellular Abelson (c-Abl) may be relevant.27
Clinical Findings
Accurate diagnosis of acute GVHD requires clinicopathologic correlation. Because the skin eruption (and histology) may be nonspecific at the time of first presentation, a careful history is invaluable. Key donor/recipient characteristics include degree of HLA-match, use of related versus unrelated donor, and T-cell depletion of the graft. Reduced-intensity conditioning may delay the onset of acute GVHD symptoms beyond the 100-day period.35 The timing of neutrophil engraftment, new medication exposures, and evidence of other organ involvement (e.g., elevated total bilirubin, diarrhea) provide additional data for clinicopathologic correlation.
Features of acute GVHD following recent blood transfusion should raise concern for transfusion-associated GVHD (TA-GVHD). TA-GVHD is an often fatal sequelae of administration of cellular blood products to immunocompromised HCT recipients, and therefore all blood products in these patients are now irradiated. TA-GVHD may also occur following transfusion of unirradiated blood products to children with congenital immunodeficiency, including Wiskott—Aldrich and ataxia-telangiectasia, as well as in the immunocompetent setting. In the latter scenario, the diagnosis may be easily missed. TA-GVHD in the immunocompetent setting follows transfusion of an unirradiated blood product that contains donor lymphocytes that are homozygous for the HLA haplotype of the recipient. A history of blood product transfusion from a relative or genetically similar population is an important feature. For example, in Japan, the estimated risk of randomly receiving blood from a homozygous donor is 1 in 874.36 In this form of TA-GVHD, the donor lymphocytes in the blood product are not recognized as foreign, leading to a GVHD reaction similar to classic acute GVHD. Beginning 10 days after transfusion, fever and skin rash (histologically consistent with GVHD) develops, followed by liver dysfunction and diarrhea. Death from pancytopenia usually occurs within several weeks.37
As with acute disease, a new diagnosis of chronic GVHD is best made based on history, cutaneous examination, and histology. A previous history of acute GVHD is the single greatest risk factor for chronic disease. Because acute symptoms may develop after 100 days posttransplant and chronic symptoms may develop before then, the revised classification of acute and chronic GVHD symptoms includes additional subtypes of GVHD with overlapping features or timing of acute and chronic symptoms (Fig. 28-1).38 Recent tapering of immunosuppressant medication or DLI given to augment the graft-versus-malignancy response are two common triggers of skin activity. DLI, in particular may present with an acute GVHD skin eruption consistent with acute GVHD rather than the papulosquamous eruption of chronic disease (Fig. 28-2D). Cutaneous or systemic infection may also induce a flare of skin GVHD, as will drug exanthems, which can result in a diagnostic challenge given the clinical and histologic similarities between viral exanthem, drug eruption, and GVHD.39 Important clues to sclerotic and fascial disease includes a history of edema of an extremity, muscle cramping, decreased flexibility, and complaints of skin tightness, particularly at the waistband and brassiere-line.10 Although GVHD in other organ systems may not necessarily flare in synchrony with skin involvement, the presence of other organ system involvement is helpful when the cutaneous features are nondiagnostic. Common GVHD symptoms include oral and ocular sicca and oral pain, particularly with spicy foods. Also common, but less specific, are symptoms of fatigue, poor appetite, and weakness. Dysphagia may indicate the presence of esophageal strictures or webbing. Bronchiolitis obliterans manifests as dry cough, wheezing, and dyspnea, but requires pulmonary function tests and computerized tomography (CT) scans to rule out infection and other etiologies. Finally, it is important to remember that despite the phenotypic variability in chronic GVHD of the skin, not every skin manifestation in a patient after HCT is due to GVHD, so a careful dermatologic history to detect other possible diagnoses is prudent.
Figure 28-1
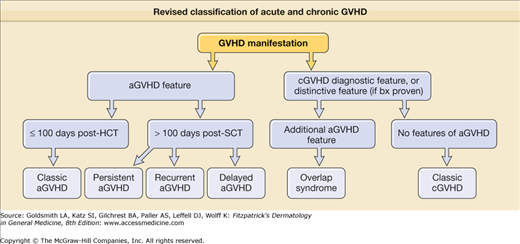
Revised classification of acute and chronic GVHD. (Adapted from Filipovich AH et al: National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 11(12):945-956, 2005.)
Figure 28-2
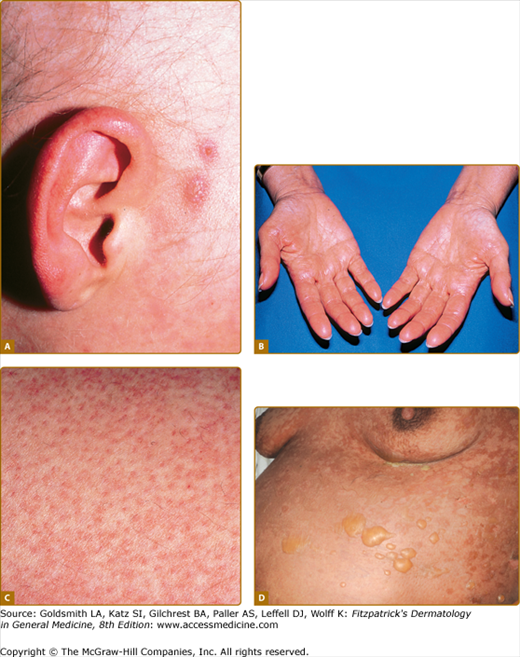
Spectrum of acute graft-versus-host skin manifestations. Acute cutaneous graft-versus-host reaction. Erythematous macules involving the ears (A), palms (B), and soles are characteristic of early cutaneous involvement. C. Follicular graft-versus-host disease. Perifollicular invovlement is an early manifestation of skin involvement. D. GVHD-associated necrolysis. Acute GVHD with bullae formation and skin sloughing following donor leukocyte for relapsed acute lymphoblastic leukemia 10 months following allogeneic HCT.
Acute GVHD initially presents with erythematous-dusky macules and papules of the volar and plantar surfaces and ears that may rapidly become a diffuse morbilliform exanthema (Fig. 28-2A and 28-2B; Box 28-2). Very early involvement may manifest as erythema limited to hair follicles (Fig. 28-2C). Pruritus is variable and is not useful to distinguish acute GVHD from other causes. Erythroderma may develop, and, in severe cases, spontaneous bullae with skin sloughing resembling toxic epidermal necrolysis. Widespread erythrodermic involvement, particularly the presence of skin sloughing portends a very poor prognosis. In contrast to chronic disease, postinflammatory pigmentary changes following acute GVHD are uncommon.
SKIN
|
GASTROINTESTINAL
|
LIVER
|
In appreciation of the tremendous variability in clinical presentation of chronic skin GVHD, it is no longer useful to dichotomize chronic GVHD of the skin into either “lichenoid” or “sclerodermoid” categories. In the transplant community, the term “lichenoid” has been utilized to denote any involvement of the skin in which erythema or scaling is present; however, “lichenoid” is a histologic pattern, not a clinical one, and, therefore, usage of it is best reserved to pathologic description. Futhermore, although chronic GVHD may resemble lichen planus (Fig. 28-3), other patterns are frequently observed, such as poikiloderma (Fig. 28-4) and skin lesions resembling lupus erythematosus, keratosis pilaris, or psoriasis.40 Postinflammatory hyperpigmentation is common following the resolution of epidermal involvement, particularly in darkly pigmented individuals, and may persist for many months after the skin disease becomes quiescent.
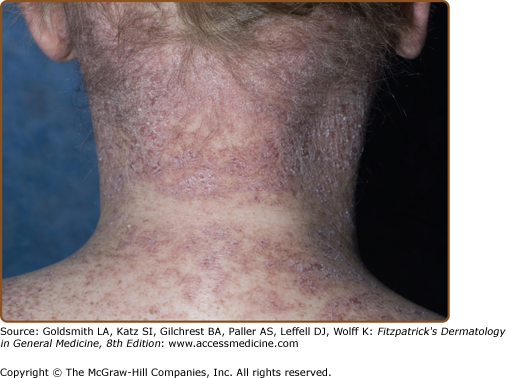
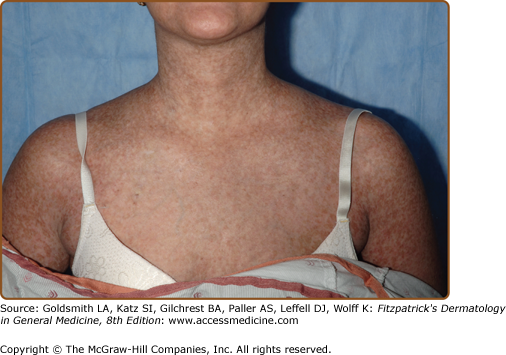
The fibrotic changes of chronic GVHD are also remarkably variable, and the term “sclerodermoid” is an inadequate descriptor of the varied sclerotic tissue abnormalities in the dermis, subcutaneous tissue, and fascia (Fig. 28-5). As in systemic sclerosis, an edematous phase may herald the onset of skin fibrosis, but fingers and toes are usually spared and the typical acral to proximal progression characteristic of systemic sclerosis is not seen in chronic GVHD. In constrast to systemic sclerosis, facial involvement is rarely involved in sclerotic-type GVHD. As mentioned above, fibrosis may occur primarily in the upper dermis, through the full-thickness of dermis, or in the subcutaneous fat and fascia. Early superficial fibrotic involvement resembles lichen sclerosus, often manifesting as porcelain-white atrophic plaques on the upper back (Fig. 28-5A). A common pattern of GVHD-associated fibrosis involves patchy sclerotic plaques with hypo- and hyperpigmentation mimicking morphea. Sclerosis of this type may exhibit an isomorphic response, localizing to the sites of minor skin trauma, particularly the waistband area, or may develop at sites of previous scar formation (Fig. 28-5B).41 Diffuse dermal involvement may result in a “pipe-stem” appearance of the lower extremities with marked reduction in limb volume and overlying shiny hidebound skin with loss of hair resembling scleroderma (Fig. 28-5C). Deeper involvement of the subcutaneous fat results in irregular hyperpigmented sclerotic plaques with intervening areas of edematous skin closely resembling deep morphea/morphea profunda.42 Bullae may develop at sites of fibrosis, particularly on the lower legs, as a result of dermal edema, as has been described in bullous morphea profunda.43 Patchy hyperpigmentation (“leopard spots”) may be visible prior to the diagnosis of dermal sclerotic involvement.44
Figure 28-5
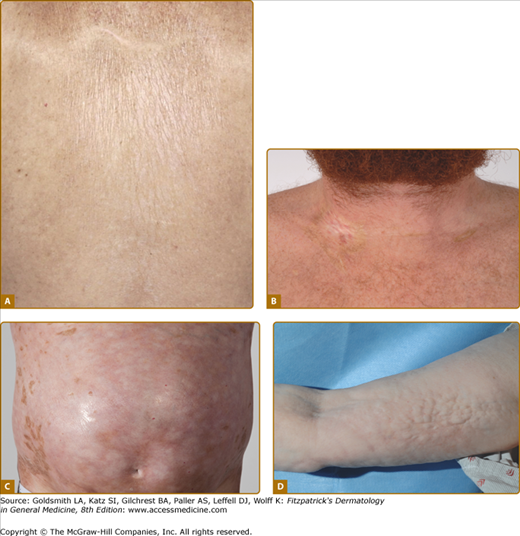
Clinical spectrum of sclerotic GVHD skin manifestations. A. Guttate white plaques on the upper back resembling lichen sclerosus. B. Morphea-like sclerotic plaques at sites of previous indwelling line placement near the clavicle (isotopic response). C. Diffuse dermal sclerosis resembling scleroderma on the anterior torso with patchy hyperpigmentation. D. Subcutaneous fibrosis of chronic GVHD. There is prominent rippling with a firm nodular texture extending along the medial arm resembing eosinophilic fasciitis. There is associated decreased range of motion at the elbow.
Primary involvement of the subcutaneous fat and fascia results in a diffuse firm, rippled pattern to the skin resembling eosinophilic fasciitis (Fig. 28-5D).45 Features of overlying epidermal GVHD involvement and pigmentary changes may be absent. Fascial involvement is often most visible on the medial arms and thighs and be accentuated by abduction and supination of the arm. Prominent “grooving” demarcating fascial bundles and along the path of superficial vessels may be observed. Careful palpation of the skin is helpful in detecting deep-seated irregularities in skin texture and differentiation from cellulite. Dermal fibriosis or fascial involvement without overlying dermal thickening may lead to progressive loss of joint range of motion and contracture formation.
Nail involvement in chronic GVHD typically results in longitudinal ridging and thin, easily broken nails. Partial or complete anonychia and dorsal pterygium formation may occur. Other unusual skin sequelae of chronic GVHD include milia formation, porokeratosis, often on the buttock area,46 angioma formation at sites of skin sclerosis,47 nipple hyperkeratosis,48 vitiligo,49 and alopecia, either diffuse or focal areas of alopecia areata.50
Different manifestations of sclerotic and and nonsclerotic skin disease may be present in the same individual, making accurate quantification of disease activity challenging. The chronic GVHD NIH Consensus Development Project provided more precise terminology for organ system involvement and defined features specific for diagnosis of chronic GVHD in the setting of HCT (Box 28-3). Diagnostic cutaneous features of GVHD include poikiloderma, lichen-planus-like lesions, and sclerotic skin changes.38
SKIN AND MUCOSAL INVOLVEMENT |
Skin |
Alopecia |
Angiomatous papules |
Bullae |
| Erythema |
Hypo- or hyperpigmentation |
Ichthyosis-like |
Keratosis-pilaris-like |
| Lichen planus-likea |
Lichen sclerosus-likea |
Maculopapular |
| Morphea-likea |
Poikilodermaa |
Scleroderma-likea |
Sweat impairment |
Ulceration |
Nails |
Brittleness |
| Longitudinal ridging or splitting |
Onycholysis |
Pterygium unguis |
Subcutaneous tissue |
Fasciitisa |
Panniculitis |
Oral mucosa |
Erythema |
Gingivitis |
Hyperkeratotic plaquesa |
Lichen planus-likea |
Mucocele |
Mucosal atrophy |
Mucositis |
Pseudomembrane |
Restriction of oral opening from sclerosisa |
Ulcer |
Xerostomia |
Genital mucosa |
Lichen planus-likea |
Vulvar erosions/fissures |
Vaginal scarring/stenosisa |
| OTHER ORGAN SYSTEM INVOLVEMENT |
Cardiovascular |
Pericardial effusion |
Cardiac conduction abnormality |
Cardiomyopathy |
Ophthalmologic |
Blepharitis |
Cicatricial conjunctivitis |
Confluent punctuate keratopathy |
Keratoconjunctivitis sicca |
Photophobia |
Gastrointestinal |
Esophageal weba |
Esophageal stricture/stenosisa |
Exocrine pancreatic insufficiency |
Hematopoeitic |
Eosinophilia |
Hypo-/hypergammaglobulinemia |
Lymphopenia |
Thrombocytopenia |
Hepatic |
Elevated total bilirubin |
Elevated alkaline phosphatase |
Elevated transaminases |
Musculoskeletal |
Arthralgia |
Arthritis |
Edema |
Myalgia |
Myositis/polymyositis |
Neurologic |
Peripheral neuropathy |
Pulmonary |
Bronchiolitis obliterans +/− organizing pneumoniaa |
Pleural effusion |
Renal |
Nephrotic syndrome |
Rheumatologic |
Autoantibodies |
Myasthenia gravis |
Acute GVHD is primarily a disorder of the skin, GI tract, and liver (Box 28-2), typically presenting with skin rash, new onset elevation of total bilirubin, and/or voluminous diarrhea. By contrast, chronic GVHD is remarkably diverse in its breadth of organ system manifestations (Box 28-3). The most frequently affected sites are skin and nails, oral mucosa, eyes, liver, lungs, and marrow (usually thrombocytopenia).5 Esophageal webs/strictures, vagino-vulvar disease, myositis, nephrotic syndrome, and pericarditis are less frequent sequelae of chronic disease.
Mucosal disease is second only to skin involvement in frequency in chronic GVHD. Mucoceles are common, as are erosions, lichen-planus-like changes with Wickham’s striae, and sicca symptoms. Dryness and violaceous erythema of the lips are common. Genital involvement significantly impairs sexual function and quality of life and may be overlooked if a specific examination and directed questions regarding genital symptoms are not undertaken. Involvement of the penis may induce phimosis. Vulvo-vaginal involvement presents as erythema, erosions/fissures, vestibulitis, vaginal stenosis, labial resorption, or complete agglutination of the introitus leading to hematocolpos (Fig. 28-6).51
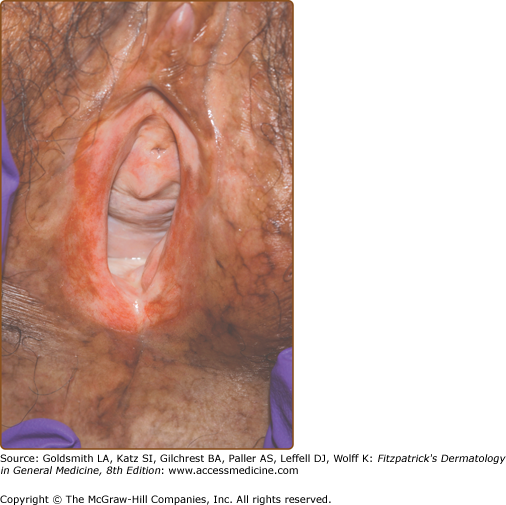
The histological grading scale for acute GVHD is shown in Table 28-1. The hallmark feature of acute GVHD is the presence of necrotic keratinocytes accompanied by a dermal lymphocytic infiltrate (usually sparse) and basal vacuolar alteration (Fig. 28-7). Early GVHD involvement with follicular erythema correlates with involvement limited to the hair follicle. Subepidermal cleft formation (Grade III) is indicative of more severe involvement, whereas complete separation of epidermis from dermis (Grade IV) correlates with clinical findings resembling toxic epidermal necrolysis. Grade IV involvement may be impossible to differentiate histologically from drug-induced toxic epidermal necrolysis and requires careful clinical correlation. The presence of eosinophils has been used in the past to argue against a diagnosis of GVHD; however, the presence of scattered eosinophils may lead to a false diagnosis of drug eruption52 and, unless very large numbers of eosinophils are present, this feature cannot be used as a reliable indicator of a drug hypersensitivity reaction.53 Engraftment syndrome is a poorly understood phenomenon at the time of neutrophil engraftment following autologous-HCT or allo-HCT characterized by a nonspecific erythematous skin eruption, fever, and pulmonary edema.54 Histologically, it may not be possible to distinguish engraftment rash from early (Grade I) acute GVHD.
Histologic Grading Scheme for Acute Cutaneous Graft-Versus-Host Reaction | |
|---|---|
Grade | Description |
0 | Normal skin or changes not referable to graft-versus-host disease |
1 | Basal vacuolization of the dermalepidermal junction |
2 | Basal vacuolization, necrotic epidermal cells, lymphocytes in the dermis and/or epidermis |
3 | Subepidermal cleft formation plus grade 2 changes |
4 | Separation of epidermis from dermis plus grade 2 changes |
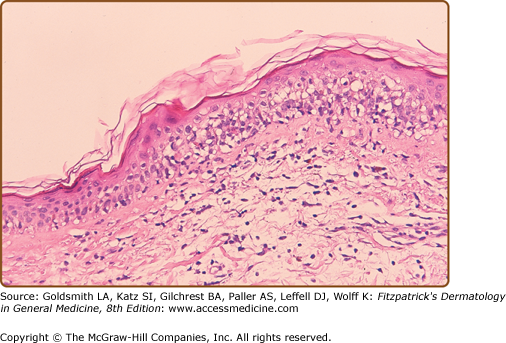
Epidermal changes in chronic GVHD may be indistinguishable from those of acute disease (Fig. 28-8A). Acanthosis and wedge-shaped hypergranulosis may be seen. Sclerotic involvement of the upper dermis may resemble lichen sclerosus, with atrophy, hyperkeratosis, follicular plugging, and pale, homogenized appearance of the upper dermis collagen (Fig. 28-8B).45 If epidermal changes of GVHD are not present, dermal fibrosis with thickened collagen bundles and loss of periadnexal fat involvement may be indistinguishable from morphea/scleroderma. Subcutaneous and fascial involvement accordingly demonstrates changes in the fat septae and fascia, including thickening, edema, and fibrosis. Variable lymphocytes, histiocytes, and eosinophils may be seen.45
Figure 28-8
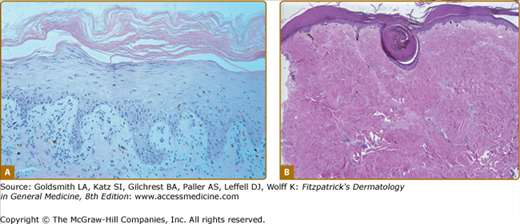
Histologic features of epidermal and sclerotic-type chronic cutaneous graft-versus-host disase. A. Histopathologic features of a lichen planus-like reaction. Acanthosis, hypergranulosis, hyperkeratosis, and pointed rete ridges are present. The inflammatory infiltrate is less dense than that usually seen in idiopathic lichen planus. B. Sclerotic-type GVHD. There is mild, compact hyperkeratosis or the epidermis with keratin plugging. There is hyalinization of the collagen throughout the dermis with loss of appendegeal structures.
Histology of involvement of the oral mucosa reflects similar interface changes as those seen in epidermal GVHD, but without associated acanthosis.55 Lymphocytic infiltration of the salivary glands resembles changes seen in Sjögren’s syndrome.
Laboratory Tests
Diagnosis of acute GVHD skin involvement is based on histopathologic correlation, particularly exclusion of drugs and infectious causes. The presence of a normal leukocyte count is indicative of engraftment but no specific laboratory testing is diagnostic. Liver function testing and total bilirubin levels and quantification of diarrhea volume are used in conjunction with skin disease to stage the disease (Table 28-2).
Stage | Skin | Liver | Gut |
|---|---|---|---|
1 | Rash <25% BSA | Bilirubin 2 mg/dL to <3 mg/dL | Diarrhea 500–1,000 mL/day or persistent nausea |
2 | Rash 25%–50% BSA | Bilirubin 3–6 mg/dL | Diarrhea 1,000–1,500 mL/day |
3 | Rash >50% BSA | Bilirubin 6–15 mg/dL |