Involvement of the genitourinary tract has been described in many different types of epidermolysis bullosa (EB). Pathology may be broadly divided into problems resulting in obstruction, that may in turn lead to hydroureter or hydronephrosis, or disease primarily affecting the renal parenchyma. Left unrecognized and untreated, renal tract disease may lead to chronic renal failure, and consequent problems associated with providing renal replacement therapy. Management of the urogenital tract in EB should therefore focus on detecting symptoms suggestive of obstruction and regular monitoring to detect problems as early as possible.
Genitourinary tract obstruction in epidermolysis bullosa
Some forms of epidermolysis bullosa (EB) are particularly susceptible to stenosis or obstruction of the genitourinary tract. Patients typically present with a reduced urinary flow, pain on voiding, or recurrent infections, depending on the site and severity of obstruction. Ulceration and scarring of the glans penis and labia may occur and are a particular feature in the inversa form of dystrophic EB (DEB). Urethral meatal stenosis is also well recognized in both sexes, particularly in patients with recessive DEB and junctional EB (JEB), in whom it has been described in 3% to 4% of patients. In severe cases, this may lead to bladder distension and subsequently hydroureter and hydronephrosis, with chronic renal failure the ultimate consequence if not treated. Partial labial fusion and reflux of urine into the vagina and uterine cavity has been described in one case of DEB. The urethra is also prone to developing strictures ( Fig. 1 ), and these may frequently recur after interventions such as catheterization, urethral dilatation, or electroresection. If instrumentation of the urogenital tract is necessary, it is probably prudent to use the smallest caliber instruments possible to avoid undue damage, or to avoid surgery if at all possible.
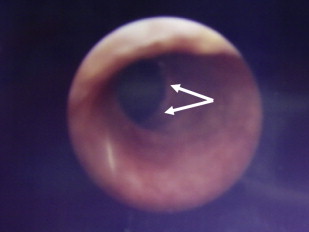
The vesicoureteric junction is another site that is particularly predisposed to stenosis, as well as reflux, particularly in patients with JEB with pyloric atresia. Again, this may result in hydroureter and hydronephrosis. Blistering of the bladder mucosa, and thickening and fibrosis of the bladder wall have also been described in patients with EB. Expression of proteins implicated in different types of EB, such as laminin 332 and α6β4 integrin, in the urogenital epithelium, particularly in the urethra and at the vesicoureteric junction, presumably leads to fragility at these sites, causing mucosal separation and secondary inflammation and scarring of the underlying structures.
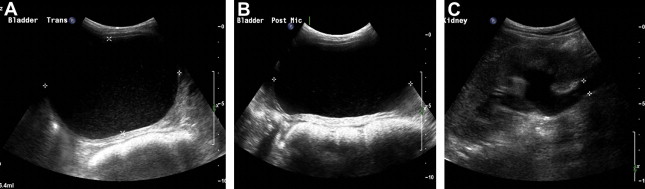
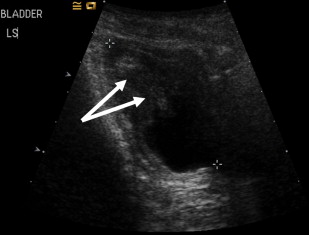
Severe constipation in patients with severe generalized recessive DEB may also result in urinary outflow obstruction with hydroureter and hydronephrosis ( Figs. 2 and 3 ), which may resolve on adequate treatment of the constipation (J.E. Mellerio, personal communication, 2009).
EB-associated renal parenchymal disease
The renal parenchyma itself may be affected in patients with EB, particularly those with the more severe forms, notably severe generalized recessive DEB and JEB with pyloric atresia, although there are scant data regarding the frequency with which this complication occurs. Three main patterns of renal parenchymal disease are observed in EB. First, post-infectious glomerulonephritis usually occurs after streptococcal infections of the skin, which are a relatively frequent problem in EB. Presentation is usually with hematuria, hypertension, and deteriorating renal function. Second, mesangial IgA nephropathy may occur, and is also postulated to be secondary to repeated mucocutaneous infections. Patients usually have hematuria, proteinuria, and hypertension. The third entity seen in EB patients is secondary renal amyloidosis, which usually presents with nephrotic range proteinuria, edema, and hypoalbuminemia. Elevated serum amyloid A protein levels may be observed in patients with severe generalized forms of EB without evidence of renal amyloidosis, and the significance of this is therefore unclear. All 3 forms of renal parenchymal disease may progress to chronic renal insufficiency, even if factors amenable to treatment (eg, streptococcal infection) are addressed.
Because infection and inflammation seem to be implicated in the pathogenesis of all 3 patterns of renal parenchymal disease described in EB patients, efforts to reduce bacterial colonization and infection of the skin may be beneficial in reducing the risks of renal impairment. However, the chronicity of skin ulceration in the more severe forms of EB probably means that this is not a realistic option in addition to the measures that are usually taken to primarily improve wound healing.
Hereditary nephritis has also been described in 2 siblings with pretibial DEB, and congenital focal segmental glomerulosclerosis identified in one child with JEB with pyloric atresia. The latter case had homozygous missense mutations in the β4 integrin gene, and reduced β4 integrin expression in the skin and glomerular podocytes, indicating that this protein may have an important role in normal glomerular function.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


