The Human Genome in Dermatology
In the 30 years since the first human gene, placental lactogen, was cloned in 1977, huge investments in time, money, and effort have gone into disclosing the innermost workings of the human genome. The Human Genome Project, which began in 1990, has led to sequence information on more than 3 billion base pairs (bp) of DNA, with identification of most of the estimated 25,000 genes in the entire human genome.1 Although a few relatively small gaps remain, the near completion of the entire sequence of the human genome is having a huge impact on both the clinical practice of genetics and the strategies used to identify disease-associated genes. Laborious positional cloning approaches and traditional functional studies are gradually being transformed by the emergence of new genomic and proteomic databases.2 Some of the exciting challenges that clinicians and geneticists now face are determining the function of these genes, defining disease associations and, relevant to patients, correlating genotype with phenotype. Nevertheless, many discoveries are already influencing how clinical genetics is practiced throughout the world, particularly for patients and families with rare, monogenic inherited disorders. The key benefits of dissection of the genome thus far have been the documentation of new information about disease causation, improving the accuracy of diagnosis and genetic counseling, and making DNA-based prenatal testing feasible.3 Indeed, the genetic basis of more than 2,000 inherited single gene disorders has now been determined, of which about 25% have a skin phenotype. Therefore, these discoveries have direct relevance to dermatologists and their patients. Recently, studies in rare inherited skin disorders have also led to new insight into the pathophysiology of more common complex trait skin disorders.4 This new information is expected to have significant implications for the development of new therapies and management strategies for patients. Therefore, for the dermatologist understanding the basic language and principles of clinical and molecular genetics has become a vital part of day-to-day practice. The aim of this chapter is to provide an overview of key terminology in genetics that is clinically relevant to the dermatologist.
The Human Genome
Normal human beings have a large complex genome packaged in the form of 46 chromosomes. These consist of 22 pairs of autosomes, numbered in descending order of size from the largest (chromosome 1) to the smallest (chromosome 22), in addition to two sex chromosomes, X and Y. Females possess two copies of the X chromosome, whereas males carry one X and one Y chromosome. The haploid genome consists of about 3.3 billion bp of DNA. Of this, only about 1.5% corresponds to protein-encoding exons of genes. Apart from genes and regulatory sequences, perhaps as much as 97% of the genome is of unknown function, often referred to as “junk” DNA. However, caution should be exercised in labeling the noncoding genome as “junk,” because other unknown functions may reside in these regions. Much of the noncoding DNA is in the form of repetitive sequences, pseudogenes (“dead” copies of genes lost in recent evolution), and transposable elements of uncertain relevance. Although initial estimates for the number of human genes was in the order of 100,000, current predictions, based on the essentially complete genome sequence, are in the range of 20,000 to 25,000.1 Surprisingly, therefore, the human genome is comparable in size and complexity to primitive organisms such as the fruit fly. However, it is thought that the generation of multiple protein isoforms from a single gene via alternate splicing of exons, each with a discrete function, is what contributes to increased complexity in higher organisms, including humans. In addition to protein-encoding genes, there are also many genes encoding untranslated RNA molecules, including transfer RNA, ribosomal RNA, and, as recently described, microRNA genes. MicroRNA is thought to be involved in the control of a large number of other genes through the RNA inhibition pathway. Very recently, it has emerged that tracts of the genome are transcribed at low levels in the form of exotic new RNA species, including natural antisense RNA and long interspersed noncoding RNA. These transcripts are emerging as key regulatory molecules. Thus, a much greater proportion of the genome is actively transcribed than was previously recognized and this trend is likely to continue in the current “postgenome” era of human genetics.
The draft sequence of the human genome was completed in 2003. Subsequently, small gaps have been filled, and the sequence has now been extensively annotated in terms of genes, repetitive elements, regulatory sequences, polymorphisms, and many other features recognizable by in silico data mining methods informed, wherever possible, by functional analysis. This annotation process will continue for some time as more features are uncovered. The human genome data, and that for an increasing number of other species, is freely available on Web sites (Table 8-1). Some regions of the genome, particularly near the centromeres, consist of long stretches of highly repetitive sequences that are difficult or impossible to clone and/or sequence. These heterochromatic regions of the genome are unlikely to be sequenced and are thought to be structural in nature, mediating the chromosomal architecture required for cell division, rather than contributing to heritable characteristics.
Genetic and Genomic Databases
Given the size and complexity of the human genome and other genomes now available, analysis of these enormous datasets in any kind of meaningful way is heavily reliant on computers. Even storage and retrieval of the sequence data associated with mammalian genome require considerable computer power and memory, and even the assembly of the raw sequence of any mammalian genome would have been unfeasible without computers. Many Web browsers for accessing genome data are available and the most useful of these are listed in Table 8-1. Each of these interfaces, which are the ones which the authors find most useful and user-friendly, contains a wide variety of tools for analysis and searching of sequences according to keyword, gene name, protein name, and homology to DNA or protein sequence data.
The main source of historical, clinical, molecular, and biochemical data relating to human genetic diseases is the Online Mendelian Inheritance in Man (OMIM) (see Table 8-1). All recognized genetic diseases and nonpathogenic heritable traits, including common diseases with a genetic component, as well as all known genes and proteins, are listed and reviewed by OMIM number with links to PubMed.
Chromosome and Gene Structure
Human chromosomes share common structural features (Fig. 8-1). All consist of two chromosomal arms, designated as “p” and “q.” If the arms are of unequal length, the short arm is always designated as the “p” arm. Chromosomal maps to seek abnormalities are based on the stained, banded appearance of condensed chromosomes during metaphase of mitosis. During interphase, the uncondensed chromosomes are not discernible by normal microscopy techniques. Genes can now be located with absolute precision in terms of the range of bp that they span within the DNA sequence for a given chromosome. The bands are numbered from the centromere outwards using a system that has evolved as increasingly discriminating chromosome stains, as well as higher resolution light microscopes, became available. A typical cytogenetic chromosome band is 17q21.2, within which the type I keratin genes reside (see Fig. 8-1).
Figure 8-1
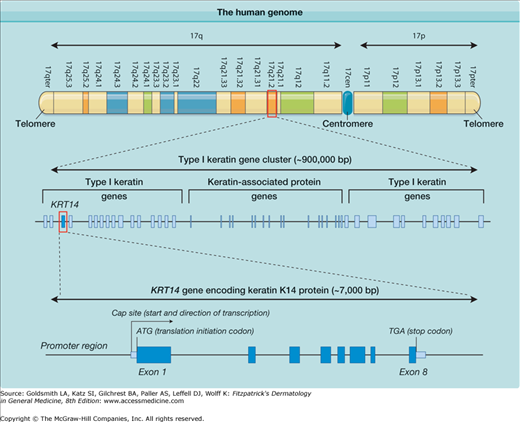
Illustration of the complexity of the human genome. At the top, the short (p) and long (q) arms of human chromosome 17 are depicted with their cytogenetic chromosome bands. One of these band regions, 17q21.2, is then highlighted to show that it is made up of approximately 900,000 base pairs (bp) and contains several genes, including 27 functional type I keratin genes. Part of this region is then further amplified to show one keratin gene, KRT14, encoding keratin 14, which is composed of eight exons.
The ends of the chromosomal arms are known as telomeres, and these consist of multiple tandem repeats of short DNA sequences. In germ cells and certain other cellular contexts, additional repeats are added to telomeres by a protein–RNA enzyme complex known as telomerase. During each round of cell division in somatic cells, one of the telomere repeats is trimmed off as a consequence of the DNA replication mechanism. By measuring the length of telomeres, the “age” of somatic cells, in terms of the number of times they have divided during the lifetime of the organism, can be determined. Once the telomere length falls below a certain threshold, the cell undergoes senescence. Thus, telomeres contribute to an important biological clock function that removes somatic cells that have gone through too many rounds of replication and are at a high risk of accumulating mutations that could lead to tumorigenesis or other functional aberration.5
The chromosome arms are separated by the centromere, which is a large stretch of highly repetitious DNA sequence. The centromere has important functions in terms of the movement and interactions of chromosomes. The centromeres of sister chromatids are where the double chromosomes align and attach during the prophase and anaphase stages of mitosis (and meiosis). The centromeres of sister chromatids are also the site of kinetochore formation. The latter is a multiprotein complex to which microtubules attach, allowing mitotic spindle formation, which ultimately results in pulling apart of the chromatids during anaphase of the cell division cycle.
The majority of chromosomal DNA contains genes interspersed with noncoding stretches of DNA of varying sizes. The density of genes varies widely across the chromosomes so that there are gene-dense regions or, alternately, large areas almost devoid of functional genes. An example of a comparatively gene-rich region of particular relevance to inherited skin diseases is the type I keratin gene cluster on chromosome band 17q21.2 (see Fig. 8-1). This diagram also gives an idea of the sizes in bp of DNA of a typical chromosome and a typical gene located within it. This gene cluster spans about 900,000 bp of DNA and contains 27 functional type I keratin genes, several genes encoding keratin-associated proteins, and a number of pseudogenes (not shown). Because chromosome 17 is one of the smaller chromosomes, Fig. 8-1 starts to give some idea of the overall complexity and organization of the genome.
Protein-encoding genes normally consist of several exons, which collectively code for the amino acid sequence of the protein (or open reading frame). These are separated by noncoding introns. In human genes, few exons are much greater than 1,000 bp in size, and introns vary from less than 100 bp to more than 1 million bp. A typical exon might be 100 to 300 bp in size. The KRT14 gene encoding keratin 14 (K14) protein is one of the genes in which mutations lead to epidermolysis bullosa (EB) simplex (see Chapter 62) and is illustrated in Fig. 8-1. KRT14 is contained within about 7,000 bp of DNA and consists of eight modestly sized exons interspersed by seven small introns. Although all genes are present in all human cells that contain a nucleus, not every gene is expressed in all cells of tissues. For example, the KRT14 gene is only active in basal keratinocytes of the epidermis and other stratified epithelial tissues and is essentially silent in all other tissues. When a protein-encoding gene is expressed, the RNA polymerase II enzyme transcribes the coding strand of the gene, starting from the cap site and continuing to the end of the final exon, where various signals lead to termination of transcription. The initial RNA transcript, known as heteronuclear RNA, contains intronic as well as exonic sequences. This primary transcript undergoes splicing to remove the introns, resulting in the messenger RNA (mRNA) molecule.6 In addition, the bases at the 5′ end (start) of the mRNA are chemically modified (capping) and a large number of adenosine bases are added at the 3′ end, known as the poly-A tail. These posttranscriptional modifications stabilize the mRNA and facilitate its transport within the cell. The mature mRNA undergoes a test round of translation which, if successful, leads to the transport of the mRNA to the cytoplasm, where it undergoes multiple rounds of translation by the ribosomes, leading to accumulation of the encoded protein. If the mRNA contains a nonsense mutation, otherwise known as a premature termination codon mutation, the test round of translation fails, and the cell degrades this mRNA via the nonsense-mediated mRNA pathway.7 This is a mechanism that the cell has evolved to remove aberrant transcripts, and it may also contribute to gene regulation, particularly when very low levels of a particular protein are required within a given cell.
Splicing out of introns is a complex process. The genes of prokaryotes, such as bacteria, do not contain introns, and so mRNA splicing is a process that is specific to higher organisms. In some more primitive eukaryotes, RNA molecules contain catalytic sequences known as ribozymes, which mediate the self-splicing out of introns without any requirement for additional factors. In mammals, splicing involves a large number of protein and RNA factors encoded by several genes. This allows another level of control over gene expression and also facilitates alternative splicing of exons, so that a single gene can encode several functionally distinct variants of a protein. These isoforms are often differentially expressed in different tissues. In terms of the gene sequences important for splicing, a few bp at the beginning and at the end of an intron, known as the 5′ splice site (or splice donor site) and the 3′ splice site (or splice acceptor site) are crucial. A few other bp within the intron, such as the branch point site located 18–100 bp away from the 3′ end, are also critical. Mutations affecting any of the invariant residues of these splice sites lead to aberrant splicing and either complete loss of protein expression or generation of a highly abnormal protein.
The mRNA also contains two untranslated regions (UTR): (1) the 5′UTR upstream of the initiating ATG codon and (2) the 3′UTR downstream of the terminator (or stop codon, which can be TGA, TAA, or TAG). The 5′ UTR can and often does possess introns, whereas the 3′UTR of more than 99% of mammalian genes does not contain introns. The nonsense-mediated mRNA decay pathway identifies mutant transcripts by means of assessing where the termination codon occurs in relation to introns. The natural stop codon is always followed immediately by the 3′UTR, which, in turn, does not normally possess any introns. If a stop codon occurs in an mRNA upstream of a site where an intron has been excised, this message is targeted for nonsense-mediated decay. The only genes that contain introns within their 3′UTR sequences are expressed at extremely low levels. This is one of the ways in which the cell can determine how much protein is made from a particular gene.
Gene complexity is widely variable and not necessarily related to the size of the protein encoded. Some genes consist of only a single small exon, such as those encoding the connexin family of gap junction proteins. Such single exon genes are rapid and inexpensive to analyze routinely. In contrast, the type VII collagen gene, COL7A1, in which mutations lead to the dystrophic forms of EB (see Chapter 62), has 118 exons, meaning that 118 different parts of the gene need to be isolated and analyzed for molecular diagnosis of each dystrophic EB patient. The filaggrin gene (FLG) on chromosome 1, recently shown to be the causative gene for ichthyosis vulgaris (see Chapter 49) and a susceptibility gene for atopic dermatitis (see Chapter 14), has only three exons. However, the third exon of FLG is made up of repeats of a 1,000-bp sequence and varies in size from 12,000 to 14,000 bp among different individuals in the population. This unusual gene structure makes routine sequencing of genes such as COL7A1 or FLG difficult, time consuming, and expensive.
Gene Expression
Each specific gene is generally only actively transcribed in a subset of cells or tissues within the body. Gene expression is largely determined by the promoter elements of the gene. In general, the most important region of the promoter is the stretch of sequence immediately upstream of the cap site. This proximal promoter region contains consensus binding sites for a variety of transcription factors, some of which are general in nature and required for all gene expression, others are specific to particular tissue or cell lineage, and some are absolutely specific for a given cell type and/or stage of development or differentiation. The size of the promoter can vary widely according to gene family or between the individual genes themselves. For example, the keratin genes are tightly spaced within two gene clusters on chromosomes 12q and 17q, but these are exquisitely tissue specific in two different ways. First, these genes are only expressed in epithelial cells, and therefore their promoters must possess regulatory sequences that determine epithelial expression. Therefore, these regulatory elements are specific for cells of ectodermal origin. Second, these genes are expressed in very specific subsets of epithelial cells, and so there must be a second level of control that specifies which epithelial cell layers express specific keratin genes. This is best illustrated in the hair follicle, where there are many different epithelial cell layers, each with a specific pattern of keratin gene expression (see Chapter 86).8
Transcription factors are proteins that either bind to DNA directly or indirectly by associating with other DNA-binding proteins. Binding of these factors to the promoter region of a gene leads to activation of the transcription machinery and transcription of the gene by RNA polymerase II. The transcription factor proteins are encoded by genes that are in turn controlled by promoters that are regulated by other transcription factors encoded by other genes. Thus, there are several tiers of control over gene expression in a given cell type, and the intricacies of this can be difficult to fully unravel experimentally. Nevertheless, by isolation of promoter sequences from genes of interest and placing these in front of reporter genes that can be assayed biochemically, such as firefly luciferase that can be assayed by light emission, the activity of promoters can be reproduced in cultured cells that normally express the gene. Combining such a reporter gene system with site-directed mutagenesis to make deletions or alter small numbers of bp within the promoter can help define the extent of the promoter and the important sequences within it that are required for gene expression. A variety of biochemical techniques, such as DNA footprinting, ribonuclease protection, electrophoretic mobility shift assays, or chromatin immunoprecipitation, can be used to determine which transcription factors bind to a particular promoter and help delineate the specific promoter sequences bound. Expression of reporter genes under the control of a cloned promoter in transgenic mice also helps shed light on the important sequences that are required to recapitulate the endogenous expression of the gene under study. Keratin promoters are unusual in that, generally, a small fragment of only 2,000 to 3,000 bp upstream of the gene can confer most of the tissue specificity. For this reason, keratin promoters are widely used to drive exogenous transgene expression in the various specific cellular compartments of the epidermis and its appendages for experiments to determine gene, cell, or tissue function.9
Some promoter or enhancer sequences act over very long distances. In some cases, sequences located millions of bp distant, with several other genes in the intervening region, somehow influence expression of a target gene. In some genetic diseases, mutations affecting such long-range promoter elements are now emerging. These types of mutations appear to be rare, but since they occur so far away from the target gene and are therefore very difficult to find, this class of mutation may, in fact, be more common than is immediately obvious. In general, relatively few disease-causing mutations have been shown to involve promoters, but this class of defect is probably greatly underrepresented because the sequences that are important for promoter activity are poorly characterized. Prediction of transcription factor binding sites by computer analysis is an area for further study. Although these undoubtedly exist, there are relatively few examples so far of pathogenic defects in microRNA or other noncoding regulatory RNA species.
Finding Disease Genes
In establishing the molecular basis of an inherited skin disease, there are two key steps. First, the gene linked to a particular disorder must be identified, and second, pathogenic mutations within that gene should be determined. Diseases can be matched to genes either by genetic linkage analysis or by a candidate gene approach.10 Genetic linkage involves studying pedigrees of affected and unaffected individuals and isolating which bits of the genome are specifically associated with the disease phenotype. The goal is to identify a region of the genome that all the affected individuals and none of the unaffected individuals have in common; this region is likely to harbor the gene for the disorder, as well as perhaps other nonpathogenic neighboring genes that have been inherited by linkage disequilibrium. Traditionally, genome-wide linkage strategies make use of variably sized microsatellite markers scattered throughout the genome, although for recessive diseases involving consanguineous pedigrees, a more rapid approach may be to carry out homozygosity mapping using single nucleotide polymorphism (SNP) chip arrays. By contrast, the candidate gene approach involves first looking for a clue to the likely gene by finding a specific disease abnormality, perhaps in the expression (or lack thereof) of a particular protein or RNA, or from an ultrastructural or biochemical difference between the diseased and control tissues. Nevertheless, the genetic linkage and candidate gene approaches are not mutually exclusive and are often used in combination. For example, to identify the gene responsible for the autosomal recessive disorder, lipoid proteinosis (see Chapter 137), genetic linkage using microsatellites was first used to establish a region of linkage on 1q21 that contained 68 genes.11 The putative gene for this disorder, ECM1 encoding extracellular matrix protein 1, was then identified by a candidate gene approach that searched for reduced gene expression (lack of fibroblast complementary DNA) in all these genes. A reduction in ECM1 gene expression in lipoid proteinosis compared with control provided the clue to the candidate gene because there were no differences in any of the other patterns of gene expression. Ultrastructural and immunohistochemical analyses can also provide clues to underlying gene pathology. For example, loss of hemidesmosomal inner plaques noted on transmission electron microscopy and a complete absence of skin immunostaining for the 230-kDA bullous pemphigoid antigen (BP230) at the dermal–epidermal junction, led to the discovery of loss-of-function mutations in the dystonin (DST) gene, which codes for BP230, in a new form of autosomal recessive epidermolysis bullosa simplex.12
Having identified a putative gene for an inherited disorder, the next stage is to find the pathogenic mutation(s). This can be done by sequencing the entire gene, a feat which is becoming easier as technologic advances make automated nucleotide sequencing faster, cheaper, and more accessible. However, the large size of some genes may make comprehensive sequencing impractical, and therefore initial screening approaches to identify the region of a gene that contains the mutation may be a necessary first step. There are many mutation detection techniques available to scan for sequence changes in cellular RNA or genomic DNA, and these include denaturing gradient gel electrophoresis, chemical cleavage of mismatch, single stranded conformation polymorphism, heteroduplex analysis, conformation sensitive gel electrophoresis, denaturing high-performance liquid chromatography and the protein truncation test.13
The most critical factor that determines the success of any gene screening protocol is the sensitivity of the detection technique. In addition, when choosing a mutation screening strategy using genomic DNA, the size of the gene and its number of exons must be taken into account. The sensitivities of these methods vary greatly, depending on the size of template screened. For example, single-stranded conformation polymorphism has a sensitivity of >95% for fragments of 155 bp, but this is reduced to only 3% for 600 bp. Once optimized, denaturing gradient gel electrophoresis has a sensitivity of about 99% for fragments of up to 500 bp, and conformation sensitive gel electrophoresis is expected to have a sensitivity of 80% to 90% for fragments of up to 600 bp. Chemical cleavage of mismatch, on the other hand, has a sensitivity of 95% to 100% for fragments >1.5 kilobases (kb) in size and is ideal for screening compact genes where more than one exon can be amplified together using genomic DNA as the template. All these techniques detect sequence changes such as truncating and missense mutations as well as polymorphisms; however, the protein truncation test screens only for truncating mutations and is predicted to have a sensitivity of >95% and can be used for RNA or DNA fragments in excess of 3 kb.
Whichever approach is taken, having identified a difference in the patient’s DNA compared with the control sample, the next stage is to determine how this segregates within a particular family and also whether it is pathogenic or not. Very recently, great advances have been made in DNA sequencing technology, with the emergence of “next generation sequencing” (NGS) technology. Currently, it is quite feasible to carry out whole exome sequencing in an individual using NGS, i.e., sequencing of all the protein-encoding exons in the genome, in a matter of days and for only a few thousand dollars. It is expected that whole genome sequencing, at a cost of $1,000 or less will be a commonplace in 2–3 years. This incredible new technology is set to revolutionize human genetics once more, and in particular, will facilitate identification of mutated genes in small kindreds that are not tractable by genetic linkage methods. These advances will also impact on diagnosis—in the near future it may be faster and cheaper to sequence a patient’s whole genome rather than to do targeted sequencing of specific genes or regions.
Gene Mutations and Polymorphisms
Within the human genome, the genetic code of two healthy individuals may show a number of sequence dissimilarities that have no relevance to disease or phenotypic traits. Such changes within the normal population are referred to as polymorphisms (Fig. 8-2). Indeed, even within the coding region of the genome, clinically irrelevant substitutions of one bp, known as SNPs, are common and occur approximately once every 250 bp.14 Oftentimes, these SNPs do not change the amino acid composition; for example, a C-to-T transition in the third position of a proline codon (CCC to CCT) still encodes for proline, and is referred to as a silent mutation. However, some SNPs do change the nature of the amino acid; for example, a C-to-G transversion at the second position of the same proline codon (CCC to CGC) changes the residue to arginine. It then becomes necessary to determine whether a missense change such as this represents a nonpathogenic polymorphism or a pathogenic mutation. Factors favoring the latter include the sequence segregating only with the disease phenotype in a particular family, the amino acid change occurring within an evolutionarily conserved residue, the substitution affecting the function of the encoded protein (size, charge, conformation, etc.), and the nucleotide switch not being detectable in at least 100 ethnically matched control chromosomes. Nonpathogenic polymorphisms do not always involve single nucleotide substitutions; occasionally, deletions and insertions may also be nonpathogenic.
Figure 8-2
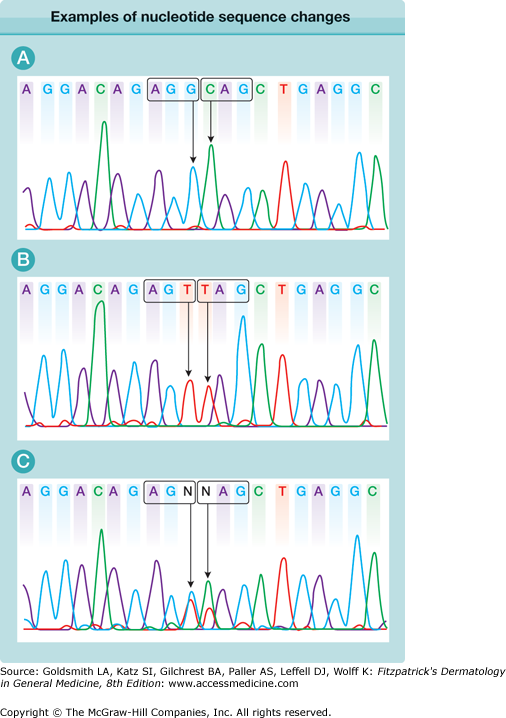
Examples of nucleotide sequence changes resulting in a polymorphism and a nonsense mutation. A. Two adjacent codons are highlighted. The AGG codon encodes arginine and the CAG codon encodes glutamine. B. The sequence shows two homozygous nucleotide substitutions. The AGG codon now reads AGT (i.e., coding for serine rather than arginine). This is a common sequence variant in the normal population and is referred to as a nonpathogenic missense polymorphism. In contrast, the glutamine codon CAG now reads TAG, which is a stop codon. This is an example of a homozygous nonsense mutation. C. This sequence is from one of the parents of the subject sequenced in B and shows heterozygosity for both the missense polymorphism AGG > AGT and the nonsense mutation CAG > TAG, indicating that this individual is a carrier of both sequence changes.
A mutation can be defined as a change in the chemical composition of a gene. A missense mutation changes one amino acid to another. Mutations may also be insertions or deletions of bases, the consequences of which will depend on whether this disrupts the normal reading frame of a gene or not, as well as nonsense mutations, which lead to premature termination of translation (see Fig. 8-2). For example, a single nucleotide deletion within an exon causes a shift in the reading frame, which usually leads to a downstream stop codon, thus giving a truncated protein, or often an unstable mRNA that is readily degraded by the cell. However, a deletion of three nucleotides (or multiples thereof) will not significantly perturb the overall reading frame, and the consequences will depend on the nature of what has been deleted. Nonsense mutations typically, but not exclusively, occur at CpG dinucleotides, where methylation of a cytosine nucleotide often occurs. Inherent chemical instability of this modified cytosine leads to a high rate of mutation to thymine. Where this alters the codon (e.g., from CGA to TGA), it will change an arginine residue to a stop codon. Nonsense mutations usually lead to a reduced or absent expression of the mutant allele at the mRNA and protein levels. In the heterozygous state, this may have no clinical effect [e.g., parents of individuals with Herlitz junctional EB are typically carriers of nonsense mutations in one of the laminin 332 (laminin 5) genes but have no skin fragility themselves; see Chapter 62], but a heterozygous nonsense mutation in the desmoplakin gene, for example, can result in the autosomal dominant skin disorder, striate palmoplantar keratoderma (see Chapter 50). This phenomenon is referred to as haploinsufficiency (i.e., half the normal amount of protein is insufficient for function).
Apart from changes in the coding region that result in frameshift, missense, or nonsense mutations, approximately 15% of all mutations involve alterations in the gene sequence close to the boundaries between the introns and exons, referred to as splice site mutations. This type of mutation may abolish the usual acceptor and donor splice sites that normally splice out the introns during gene transcription. The consequences of splice site mutations are complex; sometimes they lead to skipping of the adjacent exon, and other times they result in the generation of new mRNA transcripts through utilization of cryptic splice sites within the neighboring exon or intron.
Mutations within one gene do not always lead to a single inherited disorder. For example, mutations in the ERCC2 gene may lead to xeroderma pigmentosum (type D), trichothiodystrophy, or cerebrofacioskeletal syndrome, depending on the position and type of mutation. Other transacting factors may further modulate phenotypic expression. This situation is known as allelic heterogeneity. Conversely, some inherited diseases can be caused by mutations in more than one gene (e.g., non-Herlitz junctional EB; see Chapter 62) and can result from mutations in either the COL17A1, LAMA3, LAMB3, or LAMC2 genes. This is known as genetic heterogeneity. In addition, the same mutation in one particular gene may lead to a range of clinical severity in different individuals. This variability in phenotype produced by a given genotype is referred to as the expressivity. If an individual with such a genotype has no phenotypic manifestations, the disorder is said to be nonpenetrant. Variability in expression reflects the complex interplay between the mutation, modifying genes, epigenetic factors, and the environment and demonstrates that interpreting what a specific gene mutation does to an individual involves more than just detecting one bit of mutated DNA in a single gene.
Mendelian Disorders
There are approximately 5,000 human single-gene disorders and, although the molecular basis of less than one-half of these has been established, understanding the pattern of inheritance is essential for counseling prospective parents about the risk of having affected children. The four main patterns of inheritance are (1) autosomal dominant, (2) autosomal recessive, (3) X-linked dominant, and (4) X-linked recessive.
For individuals with an autosomal dominant disorder, one parent is affected, unless there has been a de novo mutation in a parental gamete. Males and females are affected in approximately equal numbers, and the disorder can be transmitted from generation to generation; on average, half the offspring will have the condition (Fig. 8-3). It is important to counsel affected individuals that the risk of transmitting the disorder is 50% for each of their children, and that this is not influenced by the number of previously affected or unaffected offspring. Any offspring that are affected will have a 50% risk of transmitting the mutated gene to the next generation, whereas for any unaffected offspring, the risk of the next generation being affected is negligible, providing that the partner does not have the autosomal dominant condition. Some dominant alleles can behave in a partially dominant fashion. The term semidominant is applied when the phenotype in heterozygous individuals is less than that observed for homozygous subjects. For example, ichthyosis vulgaris is a semidominant disorder in which the presence of one or two mutant profilaggrin gene (FLG) alleles can strongly influence the clinical severity of the ichthyosis.
Figure 8-3
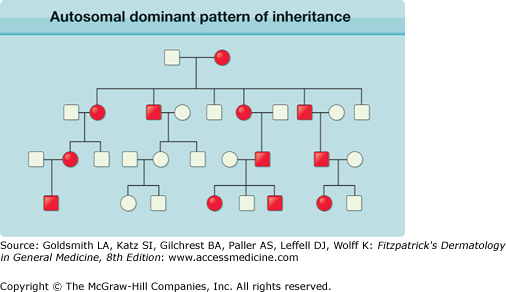
Pedigree illustration of an autosomal dominant pattern of inheritance. Key observations include: the disorder affects both males and females; on average, 50% of the offspring of an affected individual will be affected; affected individuals have one normal copy and one mutated copy of the gene; affected individuals usually have one affected parent, unless the disorder has arisen de novo. Importantly, examples of male-to-male transmission, seen here, distinguish this from X-linked dominant and are therefore the best hallmark of autosomal dominant inheritance. Filled circles indicate affected females; filled squares indicate affected males; unfilled circles/squares represent unaffected individuals.
In autosomal recessive disorders, both parents are carriers of one normal and one mutated allele for the same gene and, typically, they are phenotypically unaffected (Fig. 8-4). If both of the mutated alleles are transmitted to the offspring, this will give rise to an autosomal recessive disorder, the risk of which is 25%. If one mutated and one wild-type allele is inherited by the offspring, the child will be an unaffected carrier, similar to the parents. If both wild-type alleles are transmitted, the child will be genotypically and phenotypically normal with respect to an affected individual. If the mutations from both parents are the same, the individual is referred to as a homozygote, but if different parental mutations within a gene have been inherited, the individual is termed a compound heterozygote. For someone who has an autosomal recessive condition, be it a homozygote or compound heterozygote, all offspring will be carriers of one of the mutated alleles but will be unaffected because of inheritance of a wild-type allele from the other, clinically and genetically unaffected, parent. This assumes that the unaffected parent is not a carrier. Although this is usually the case in nonconsanguineous relationships, it may not hold true in first-cousin marriages or other circumstances where there is a familial interrelationship. For example, if the partner of an individual with an autosomal recessive disorder is also a carrier of the same mutation, albeit clinically unaffected, then there is a 50% chance of the offspring inheriting two mutant alleles and therefore also inheriting the same autosomal recessive disorder. This pattern of inheritance is referred to as pseudodominant.
Figure 8-4