11 Genetics and prenatal diagnosis
Synopsis









Human genome
Historical perspective
George Mendel first described the nature of inheritance in 1865 and thereby established the scientific field of genetics and opened the path for genetic discovery (Fig. 11.1). Mendel studied the inheritance pattern in pea plants and found that certain traits show up in offspring without any blending of parent characteristics.1 For instance, the colors of the cross-pollinated plants, either white or purple, were not blended into intermediates in the offspring. However his work did not receive much attention until republished by others, notably Hugo de Vries, Carl Correns, and later by Bateson, who republished Mendel’s work in 1901. Bateson was the first to use the term “genetics,” and was also the main contributor to the rediscovery of Mendel’s research. Several conclusions could be drawn from the works of Mendel and Bateson to form the principles of mendelian or unifactorial inheritance. Genes, or factors according to Mendel’s terminology, come in pairs, one from each parent. Furthermore, genes can occupy different alleles, some of which are dominant while others are recessive (the principle of dominance). The principle of segregation, or Mendel’s first law, stated that alleles segregate from each other, one into each gamete, in the process of meiosis. The principle of independent assortment (Mendel’s second law) states that the segregation of different allelic pairs is independent.
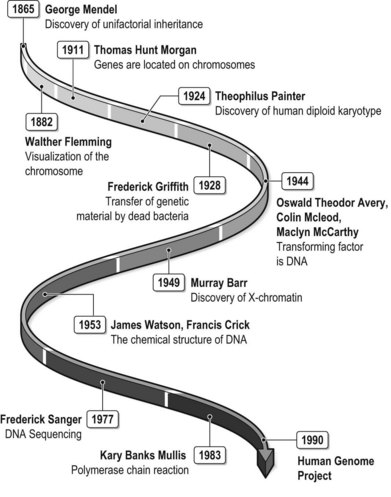
The establishment of the principles of Mendel was followed by the search for the molecules responsible for the mechanisms of inheritance. Flemming first visualized the human chromosome in 1882. Thomas Hunt Morgan become the first to suggest that genes are located on chromosomes and his student, Sturtevant, used genetic linkage to demonstrate that genes are linearly arranged in chromosomes.2,3
In 1924 Painter described the human diploid karyotype with 46 chromosomes, as well as the mechanism of XY sex chromosomes.4 Barr and Bertrand described the X-chromatin, or Barr body, in 1949. The fluorescent F body, or Y chromosome, was discovered 20 years after the publication of Barr’s paper.5,6
Miescher took the first steps towards the discovery of the DNA molecule in the late 19th century. He was able to isolate phosphate-rich chemicals that he called nucleins from white blood cells. Another important contribution was that of Griffith, who discovered that dead bacteria could transfer genetic material to transform other bacteria. The transforming factor was later discovered to be DNA by Avery et al.7
Watson and Crick discovered the chemical structure of the DNA molecule in 1953.8 They used X-ray crystallography to determine the ladder-like double-stranded and coiled helix structure, composed of a phosphate deoxyribose sugar backbone and inward-pointing nucleotide bases. Their discovery was awarded the Nobel Prize in Medicine in 1962. The understanding of the structure of the molecule that carries genetic information laid the foundation for the development of new methodologies to investigate gene structure and function. In 1977 Sanger et al. invented DNA sequencing, a method to read the nucleotide sequence of DNA.9 In 1983 the group of Kary Banks Mullis described polymerase chain reaction (PCR), which enabled the isolation and amplification of specific stretches of DNA.10 These methodologies formed the basis for the sequencing of the entire human genome within the framework of the Human Genome Project.
Basic molecular biology
DNA
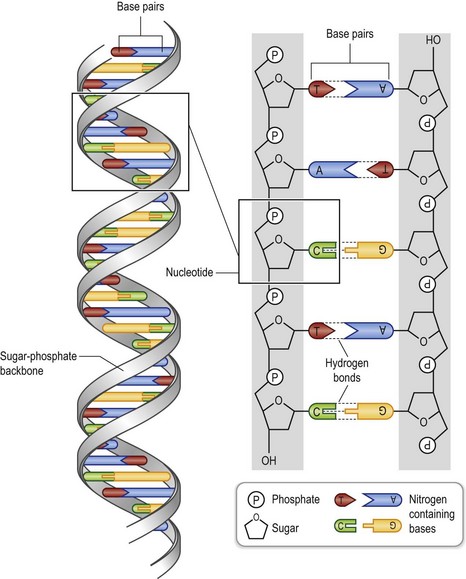
Genes are made of the molecule deoxyribonucleic acid, or DNA (Fig. 11.2). The DNA molecule is a long polymer of units called deoxyribonucleotides, or nucleotides. Each nucleotide is in turn composed of the phosporylated sugar phospodeoxyribose, and one of the four nucleotide bases adenine (A), guanine (G), cytosine (C), and thymine (T). Two such polymers form a two-stranded double helix with the sugar phosphates as hydrophilic backbones on the outside, and the hydrophilic bases on the inside. The bases of one strand bind to the bases of the other, so that A pairs with T and G pairs with C. As a consequence, the DNA chain is complementary and the sequence of one strand generates the sequence of the other. At one end the DNA chain starts with a sugar phosphorylated at its 5′ carbon and at the other end the chain ends with a 3′ hydroxylated carbon. The two chains are in antiparallel orientation with the 3′ end of one chains binding to the 5′ end of the complementary chain.
Organization of genomic DNA
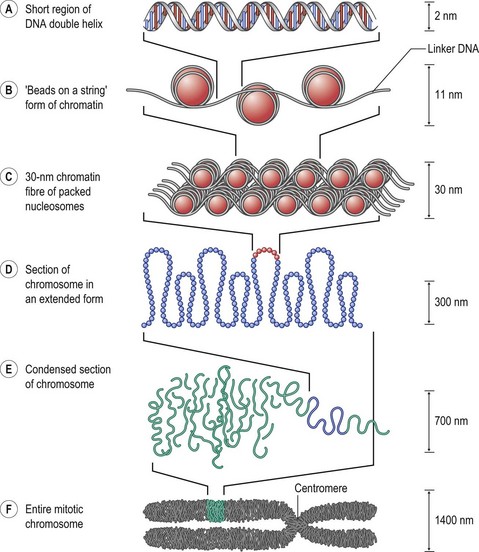
One DNA molecule makes up one chromosome and each human chromosome contains about 1.3 × 108 basepairs. Human germ cells are haploid, which means they have one set of 23 different chromosomes, and somatic cells are diploid and contain 23 pairs, or 46 chromosomes. The autosomes are the 22 pairs that are not either of the two different sex chromosomes, X or Y. The 46 DNA molecules that make up the genome must be packed into the confined space of the somatic cell nucleus. This is accomplished with the help of DNA-binding proteins, or histones, and the consequent formation of chromatin (Fig. 11.3). Two of each histones 2A, 2B, 3, and 4 form a protein complex called the nucleosome. The DNA molecule winds around the nucleosomes and each nucleosome is separated by a piece of linker DNA. In this unfolded state this structure is viewed as “beads on a string” in the electron microscope. The nucleosomes are brought together by another histone, H1, to form the 30-nm chromatin fiber. Nonhistone proteins further condense the chromatin in a process called supercoiling. The condensed chromatin visible under the light microscope is called heterochromatin and the less compacted chromatin is called euchromatin. The chromosomes are most condensed just before mitosis. At this stage the cell is about to divide, the DNA has been replicated, and each chromosome is therefore composed of two chromatids. Each chromosme has a long (q) and a short (p) arm separated from each other by the centromere.
Gene expression
In eukaryotes, mRNA transcripts undergo several modifications. The 5′ end is methylated to form the 7-methyl guanosine cap, while the 3′ end is altered by the above-mentioned polyadenylation to form the poly-A tail. Both the capping and the addition of the poly-A tail increase the stability of mRNA molecules towards degradation. Before the mRNA molecule can be used as a template for protein synthesis it undergoes the process of splicing, in which the parts of the mRNA sequence that correspond to introns, or noncoding regions, are removed. In alternative splicing, exons are rearranged in different ways to create different splice variants, or isoforms, of the protein in question. This constitutes an important regulatory mechanism by which cells can synthesize different variants of a peptide at different time points. However, alternative splicing to produce aberrant proteins is also highly implicated in disease. In syndromic craniosynostosis, mutations in the gene coding for FGFR2 may produce splice sites that lead to the synthesis of aberrant receptor protein.11
Gene regulation
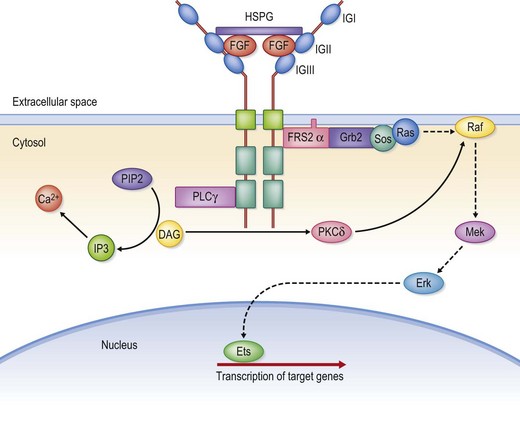
The control of gene expression is an extraordinarily complex process. The entire genome, with all its genes, is present in every somatic cell. However, the phenotype and behavior of cells during development, tissue repair, and homeostasis are directed by a differential and highly selective pattern of gene expression. The moment-to-moment alterations in gene expression are mainly regulated at the level of gene transcription. Cells respond and adapt with changes in gene expression to a wide range of stimuli from the external microenvironment, such as soluble factors, cell–cell contacts, or cell–matrix contacts. The effect of these stimuli on gene expression is in turn mediated by intracellular signal transduction mechanisms. Upon extracellular stimulation, complex cascades of intracellular enzymatic reactions are activated which ultimately result in the activation of a specific set of transcription factors (Fig. 11.4). Transcription factors bind to the promoter sequence and together with the RNA polymerase they form the preinitiation complex. Other transcription factors bind to so-called enhancer sequences, located upstream or sometimes downstream of the gene. The transcription factors encoded by homeobox-containing genes are particularly important for the developmental process.12 A homeobox is a DNA sequence of about 180 basepairs that encodes for a so-called homeodomain, an amino acid sequence that binds DNA. Complicated molecular pathways involving several different growth factors regulate the timely and coordinated expression of genes during embryogenesis. Two growth factor families with particular importance in this regard are the transforming growth factor-β (TGF-β) superfamily and the fibroblast growth factors (FGF).13 Bone morphogenic proteins (BMP) are members of the TGF-β superfamily and modulate transcription of homeobox-containg genes, such as MSX1 and MSX2. MSX1 and MSX2 have both been implicated in the formation of orofacial clefts.14 Mutations in FGFR genes that lead to an increase in receptor signaling are the principal mechanisms behind FGFR-related craniosynostosis syndromes.15 The role of these factors in craniofacial development is elaborated below.
Epigenetic mechanisms
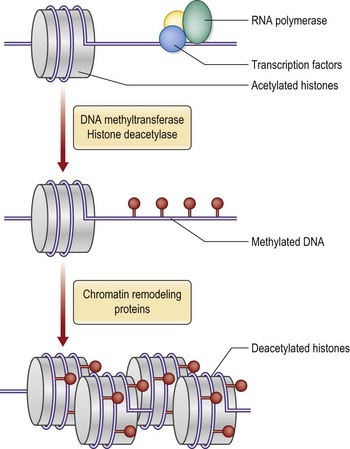
The three-dimensional architecture of chromatin regulates gene expression.16 Unfolding of the chromatin structure exposes the DNA sequence to transcription. Protein-encoding genes are as a consequence expressed in an environment of euchromatin and, conversely, the highly condensed heterochromatin inhibits transcription. The physical three-dimensional arrangement of chromatin is in turn regulated by different biochemical epigenetic mechanisms that may be transferred through cell division. The histones may be modified by methylations or acetylations. The pattern of methylation and acetylation of the histones in turn directs the binding of other chromatin-binding proteins that regulate gene transcription. Another mechanism is methylation of CpG dinucleotides of the DNA molecule by enzymes called DNA methyl transferases. DNA methylation generally silences gene transcription and the pattern of DNA methylations acts in concert with histone modifications to determine chromatin structure and regulate transcription (Fig. 11.5).
The epigenetic mechanisms give rise to so-called epigenetic phenomena.17 X-inactivation is the phenomenon by which one of the X chromosomes in females is randomly inactivated. The inactivated X chromosome can be visualized as a highly condensed chromatin structure, the Barr body. By inactivating one of the two X chromosomes in the somatic cells of females, the transcription of genes on the X chromosome, and consequent protein synthesis, is kept at the same level in males and females. During embryonic life paternal and maternal X chromosomes are inactivated in a random fashion. Therefore in any tissue there will be an equal number of cells with the active X chromosomes from either parent. This explains why most X-linked diseases are not expressed in females. Nonrandom X inactivation is an epigenetic abnormality that may lead to the occurrence of X-linked disease in females.
Inheritance
Mendelian patterns of inheritance
Autosomal inheritance
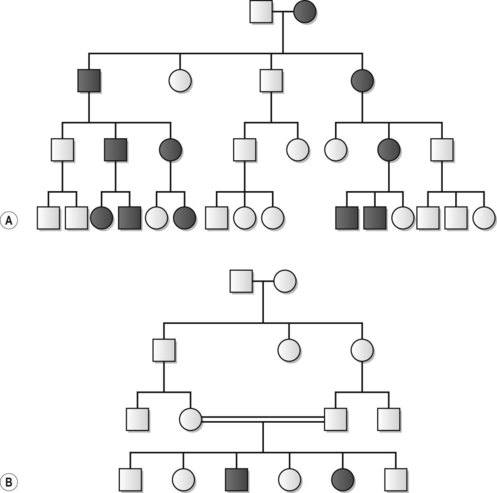
Autosomal inheritance refers to the transmission of traits directed by genes on any of the 22 pairs of autosomes. Autosomal inheritance may be either dominant or recessive (Fig. 11.6). Dominant traits are expressed in heterozygous states, while recessive traits are only expressed in individuals homozygous for that particular genetic locus. Thus, in autosomal-dominant disease the risk for the offspring to an affected individual is 50%. In autosomal-recessive disease, two parents, both heterozygous for the disease-causing gene, will have a 25% risk of having an affected child. Importantly, autosomal-dominant disease may arise as a result of inheritance, or from de novo gene mutation. A and B blood group antigens are both autosomal-dominant, but may be simultaneously expressed, a phenomenon referred to as codominance. Phenotypes determined by inherited or de novo mutated genes may be expressed to a varying degree. Thus, the same disease, caused by mutations in the same gene, may present with anything from very mild to severe manifestations. The explanation for this variable expressivity may be caused by different mutations in the same gene, other modifying genetic and molecular mechanisms, or environmental factors. Nonpenetrance refers to the situation when individuals heterozygous for an autosomal-dominant trait do not express the corresponding phenotype. This explains why autosomal-dominant disease may skip generations to reappear in coming generations.
Sex-linked inheritance
The genes on the X and Y chromosomes are inherited according to a pattern of sex-linked inheritance. X-linked dominant traits may be expressed in both males and females, but will never be transmitted from affected father to son (Fig. 11.7). Daughters with affected fathers will on the other hand have a 100% risk of inheriting the trait. Affected mothers will have a 50% risk of transmitting an X-linked dominant trait to both daughters and sons. X-linked recessive traits are much more common and are almost exclusively expressed in males, as the two copies of the X chromosome in females, one from each parent, will usually ascertain heterozygocity. Examples of X-linked recessive disease are Duchenne’s muscular dystrophy, Becker’s muscular dystrophy, and hemophilias A and B. Y-linked inheritance affects only males, exemplified by inheritance of the H-Y histocompatibility antigen.
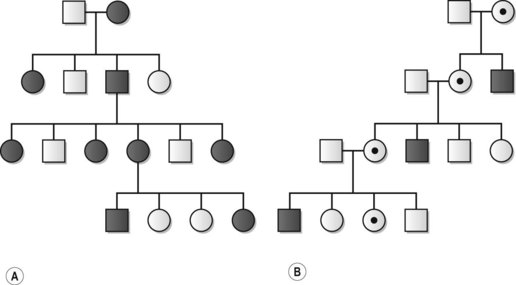
Nonmendelian patterns of inheritance
Uniparental disomy
In uniparental disomy the individual has inherited both chromosomes, or part of a chromosome, from a single parent. Thus, the corresponding genomic sequence from the other parent will be missing. Normally this does not affect phenotype; however, if combined with imprinting, as described above, disease may occur. Illustrative examples are the syndromes of Prader–Willi and Angelman.18 About 75% of patients with Prader–Willi syndrome have deletion in the proximal portion of the long arm of chromosome 15, and in the majority of cases this occurs on the paternally derived chromosome. Patients with Angelman syndrome have the same deletion, but on the maternal chromosome. The deleted segments contain the disease-causing genes and the disease develops due to imprinting of these genes on the chromosome-derived form the other parent.
Genetic causes for disease
Inherited chromosomal abnormalities
Errors in disjunction of chromosomes during meiosis or mitosis may cause an abnormal number of chromosomes, or a numerical abnormality. The most common abnormality seen in human disease is aneuploidy, or a number of chromosomes that is not a multiple of the haploid set of chromosomes. Aneuploidy in humans is most often due to an abnormal number of sex chromosomes. Supernumerary X chromosomes may lead to increased dosage of gene products from the X chromosome. However, if all but one of the X chromosomes are inactivated, the effect on phenotype may be very slight. Monosomy for X chromosome, or 45X, is the most common of all numerical abnormalities. However, 45X usually leads to abortion in the first trimester. Autosomal numerical abnormalities are usually in the form of trisomy. The most common of all chromosomal conditions is Down syndrome, caused by trisomy 21. Certain chromosomal sites are prone to various rearrangements that produce structural chromosomal abnormalities. Segments of chromosomes may be deleted or duplicated. CATCH 22 (see below) is a syndrome that may include abnormalities of the palate and is caused by deletion of locus 22q11.19 Other types of structural abnormalities are translocations, where there is an exchange of segments between chromosomes, and inversions, referring to a situation when a chromosomal segment is detached, inverted, and reinserted.
The gene as a focus for therapy
The development of the various methodologies for gene transfection opened up for the prospect of therapeutic modification of gene expression. The first phase I clinical gene therapy trials for adenosine deaminase deficiency and malignant melanoma were reported in 1990.20,21 In gene therapy the introduction of DNA or RNA into cells is utilized for curing disease or altering gene expression to achieve transient advantage of tissues in processes such as tissue repair and regeneration. The expression of individual genes can be modified or abnormal genes can be corrected. The delivered DNA or RNA may be categorized as gene inhibitors, gene vaccines, or gene substitutes. Gene inhibitors, such as siRNA, inhibit the expression of defective genes. Gene vaccines encode for specific pathogens that activate cell-mediated and humoral immune responses. Gene substitutes are complete genes that substitute for absolute or relative deficiency of a specific gene product. There are currently about 1500 approved clinical trial protocols worldwide. In all, 64.5% of all ongoing trials concern cancer disease, 8.7% are for cardiovascular disease, and 7.9% concern monogenetic disease.22
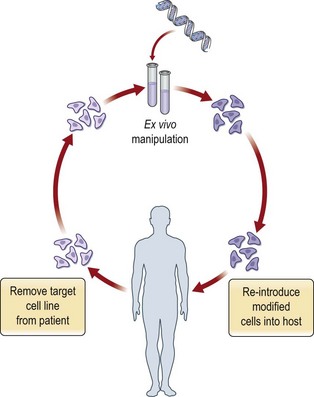
Gene therapy often refers to stable transformation, where the introduced gene is integrated into the target cell genome to cause a permanent modification of gene expression. Inherited monogenic disorders are the best candidates for this kind of therapy. Examples are hemophilia A and B, cystic fibrosis, and Huntington’s disease. Stable transformation has however so far only been achievable in therapy directed at the modification of germ cells, which is widely practiced in agriculture and which is used to generate clones of genetically modified laboratory animals. Germ cell gene therapy raises complex moral and ethical issues and has so far not been practiced in humans. Transient transformation is more appropriate to enhance tissue function during a limited therapeutic period in conditions such as wound healing and cancer. Regardless of the therapeutic aim, gene therapy relies on methodology to deliver genes effectively to cells and tissues. Gene therapy may be combined with cell therapy, where transfection is performed ex vivo followed by transplantation of the transformed cells to the patient (Fig. 11.8). The ex vivo cell therapy approach caries several advantages such as more efficient transfection, reduced risk for undesired local and systemic reactions to vectors, and improved possibility of targeting specific cell types. Despite the establishment of various methods for gene delivery and transgene expression in humans, there has been limited success with genetic therapy in clinical praxis thus far. Significant challenges directly related to gene transfer technology include achievement of sufficient transgene levels, appropriate duration of transgene expression, and increased therapeutic specificity. In addition, treatment of multifactorial, polygenetic disease requires further identification of appropriate target genes.
Gene delivery
Gene delivery is achieved through physical, chemical, and biological approaches.23 All these approaches can be used in vivo or ex vivo. Transgene delivery in the form of naked DNA is often less efficient mainly due to rapid degradation by nucleases. Therefore, various specialized technologies have been developed to increase transfection efficiency. The ideal gene delivery approach should: (1) allow for delivery of appropriate transgene size; (2) evade immunologic/toxic reactions or other detrimental effects; (3) allow for target cell specificity; (4) produce efficient delivery; and (5) provide gene modification of appropriate duration.
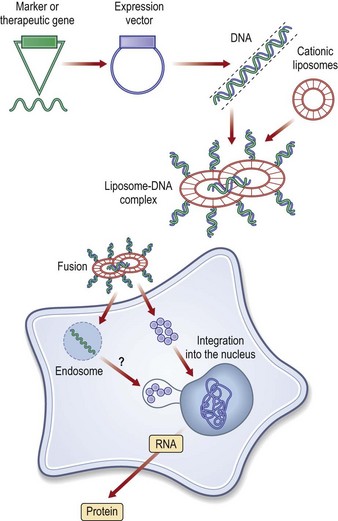
There are nonbiological and biological methods for gene delivery. The nonbiological methods may in turn be divided into needle injection of naked DNA, physical and chemical methods.24 Physical approaches, such as microseeding, electroporation, ballistic, ultrasound or hydrodynamic delivery, employ a physical force to deliver the transgene into the cell through the plasma membrane. Chemical approaches use synthetic or naturally occurring chemical compounds as transgene carriers. The predominating type of nonbiological method is chemical, through delivery by cationic liposomes. The liposomes are synthetic vectors composed of positively charged cationic lipids and co-lipids, which may be noncharged phospholipids or cholesterol. These liposomes are mixed with transgene-containing plasmids to form particles called lipoplexes. The DNA bound in these particles is resistant against degradation by nucleases. Cellular uptake is presumed to occur through endocytosis and the DNA exits the intracellular vesicles before degradation in lysosomal compartments (Fig. 11.9).
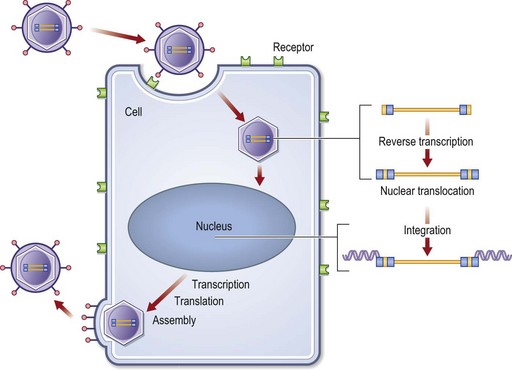
Liposomes have been of limited value for in vivo gene therapy mainly due to a risk of toxic reactions and rapid plasma clearance. Other examples of chemical methods are receptor-mediated and cationic polymers. Viral gene delivery is the predominating form of biological gene delivery.25 Commonly used viruses are adenoviruses, herpes simplex viruses, and retroviruses. Viral gene delivery is based on the natural ability of viruses to infect host cells with genetic material. In order to be used as vectors, viruses first have to be genetically modified. The genes required for viral replication are deleted and replaced by the gene of interest. To compensate for this inability to replicate, the viral vectors are propagated in packaging cell lines that provide the genes encoding for the structural proteins for the viral particle. In general, the viral approaches are associated with higher transfection efficiency and more stable transformations. The main concerns with biological methods are the risks of transmitting disease, inducing insertional mutations, or evoking immunological or toxic reactions. In retroviral vectors, the retroviral single-stranded RNA genome is reverse-transcribed into double-stranded DNA and the transgene is integrated into the genomic DNA of host cells. This offers the possibility of stable transfection, however, with a risk for insertional mutations (Fig. 11.10). The risks of toxicity of viral gene therapy were clearly demonstrated by the tragic death of an 18-year-old male patient treated for ornithine transcarbamylase deficiency.26
Immunosuppression has been used to reduce immunological responses to viral vectors. However, this strategy considerably increases the risks and side-effects of the treatment. Nonviral biological vectors such as bacterial, bacteriophages and virus-like particles may become valuable delivery tools in certain gene therapy niches.27 Today, about two-thirds of clinical gene therapy trials are based on viral vectors, and merely 5–10% deploy lipofection as a mode of gene delivery.22 Features of the various techniques for gene delivery are summarized in Table 11.1. There is a need for more efficient and safer methods for gene delivery, and ongoing research aims at the development of improved technologies for delivery.
Table 11.1 Common techniques for gene delivery
| Gene delivery technique | Advantages | Disadvantages |
|---|---|---|
| Direct injection of naked DNA/plasmid DNA | Simple, local delivery, unlimited gene size, nontoxic and nonimmunogenic | Only direct injection, very low transfection efficiency, transient gene expression only |
| Gene gun | Can deliver large amounts of DNA; technically simple | Nonspecific, physical damage to cell required for DNA uptake, low transfection efficiency, potential foreign-body reaction |
| Microseeding | Can deliver large amounts and different types of DNA | Low transfection efficiency, cellular damage, limited experience |
| Electroporation | Large transgene size, nontoxic | Nonspecific, complex equipment, damage to cell membrane required for DNA uptake |
| Cationic liposomes | Technically simple, can transfect any cell type, large transgene size, local delivery, low immunogenicity | No targeting, toxicicity, low transfection efficiency, transient transfection, rapid plasma clearance in vivo |
| Retrovirus | Tranduces many different cell types, high efficiency of ex vivo transduction, long-term gene expression | Transduces mainly dividing cells, inefficient transduction in vivo, risk of insertional mutagenesis, limited transgene size, difficult to propagate in culture |
| Adenovirus | Transfects virtually all cell types, dividing and nondividing cells, good transfection efficiency in vitro and in vivo, no integration into host genome, large transgene size | Immune response, lack of permanent expression, pre-existing antibodies are common, potential wild-type breakthrough infection |
| Adeno-associated virus | Transduces dividing and nondividing cells, integrates to specific site at chromosome 19, long-term gene expression, low immunogenicity | Difficult to grow to high titers, risk of insertional mutagenesis, limited transgene size, pre-existing antibodies, complex and expensive to manufacture |
| Hepres simplex virus 1 | Transduces wide variety of cell types, neurotropism, large transgene size, long-term expression feasible, low immunogenicity |