Summary and Key Features
- •
Gene arrays can simultaneously analyze the effect of a given bioactive compound on altering the expression of over 5000 skin-specific genes.
- •
Bioactive compounds that inhibit the expression of inflammatory genes often simultaneously stimulate the expression of antiaging genes such as collagen.
- •
Sirtuins play a role in regulating cell aging.
- •
The recent development of high-throughput RNA sequencing can determine the entire transcriptome of a given cell simultaneously and under any condition.
- •
RNA sequencing provides a detailed and complete analysis of all 20,500 human genes that are differentially regulated in normal and diseased tissue.
Introduction
The number of cosmeceutical products on the market that claim a variety of beneficial effects on skin structure and function is growing rapidly with new product introductions occurring almost daily. Products that claim effectiveness in inhibiting skin inflammation, stimulating collagen and elastin production, blocking activity of matrix metalloproteinases, and slowing the aging process are widely available and most advertise that “scientific research” is behind their development. In reality, few ingredients in cosmetic products have been shown, by rigorous laboratory analysis, to have specific antiaging effects. Noteworthy exceptions are retinoic acid and its derivatives, vitamin C, and Matrixyl (palmitoyl-L-lysyl-L-threonyl-L-threonyl-L-lysyl-L-serine), three compounds for which credible scientific data exist to support antiaging claims. The development of truly efficacious cosmeceuticals involves:
- 1
The use of a rigorous cell- and molecular biology–based screening program to identify active compounds with the desired biologic activity (e.g., collagen I, III, or VII gene stimulation)
- 2
The application of this screening program to determine that the identified active ingredient does not also produce undesirable biologic effects on skin cells (e.g., stimulate matrix metalloproteinase-1 gene expression)
- 3
The development of topical formulations that can be shown by skin percutaneous absorption analysis to deliver sufficient amounts of the active ingredient across the stratum corneum and down to the target cells to achieve the required biologic effect
- 4
The use of controlled clinical studies with a sufficient number of patients to generate statistically significant data on product efficacy
Since the first step in developing an effective cosmeceutical product is to demonstrate that the putative active ingredient not only produces the desired biologic action but also does not have any deleterious effect on skin structure or function, it would be advantageous to have access to a biologic screening tool that could accomplish both needs simultaneously. Such a screening method would allow one to predict a compound’s efficacy prior to undertaking any laborious formulation development and before conducting expensive clinical studies. The use of gene array technology and the more robust method of RNA sequencing fulfills these requirements.
Basic Principles of Gene Array Analysis
All cells in the body continuously produce a specific set of proteins that define the structure and function of that particular cell type. For example, liver cells produce unique hormone receptors for glucagon and insulin, while kidney cells produce proteins for the vasopressin receptor and for those involved in ion transport. These proteins are coded for by genes that produce unique mRNAs, and thus each cell type expresses a unique “footprint” of these mRNAs. Under certain conditions, such as ultraviolet (UV) radiation, hormone influence, and aging, this profile of mRNA expression changes as do the proteins coded for by these “messengers.” Thus, for example, in young skin dermal fibroblasts express mRNA for the proteins collagen I, III, and VII, whereas in aged skin the fibroblasts produce less mRNA for the collagens but more mRNA for the enzyme matrix metalloproteinase-1 (MMP-1), which destroys collagen. With the advent of modern molecular biology gene arrays it is now possible to isolate a pool of mRNA from cells expressing different phenotypes (e.g., young and old human fibroblasts) and, from an analysis of these mRNAs, determine which genes are being expressed or repressed in different cell types or in cells exposed to different conditions.
Gene arrays are filters or glass slides to which are bound small pieces of known and unknown (EST; expressed sequence tag) human genes. Typical nylon gene array filters may contain over 5000 different gene sequences on a single filter, and some arrays have been designed with specific tissues or diseases in mind. For example, a gene filter has been designed to which over 4000 skin-specific genes have been bound, allowing one to assess the effects of biologic modifiers such as hormones, cytokines, and UV radiation on the expression of genes important in skin.
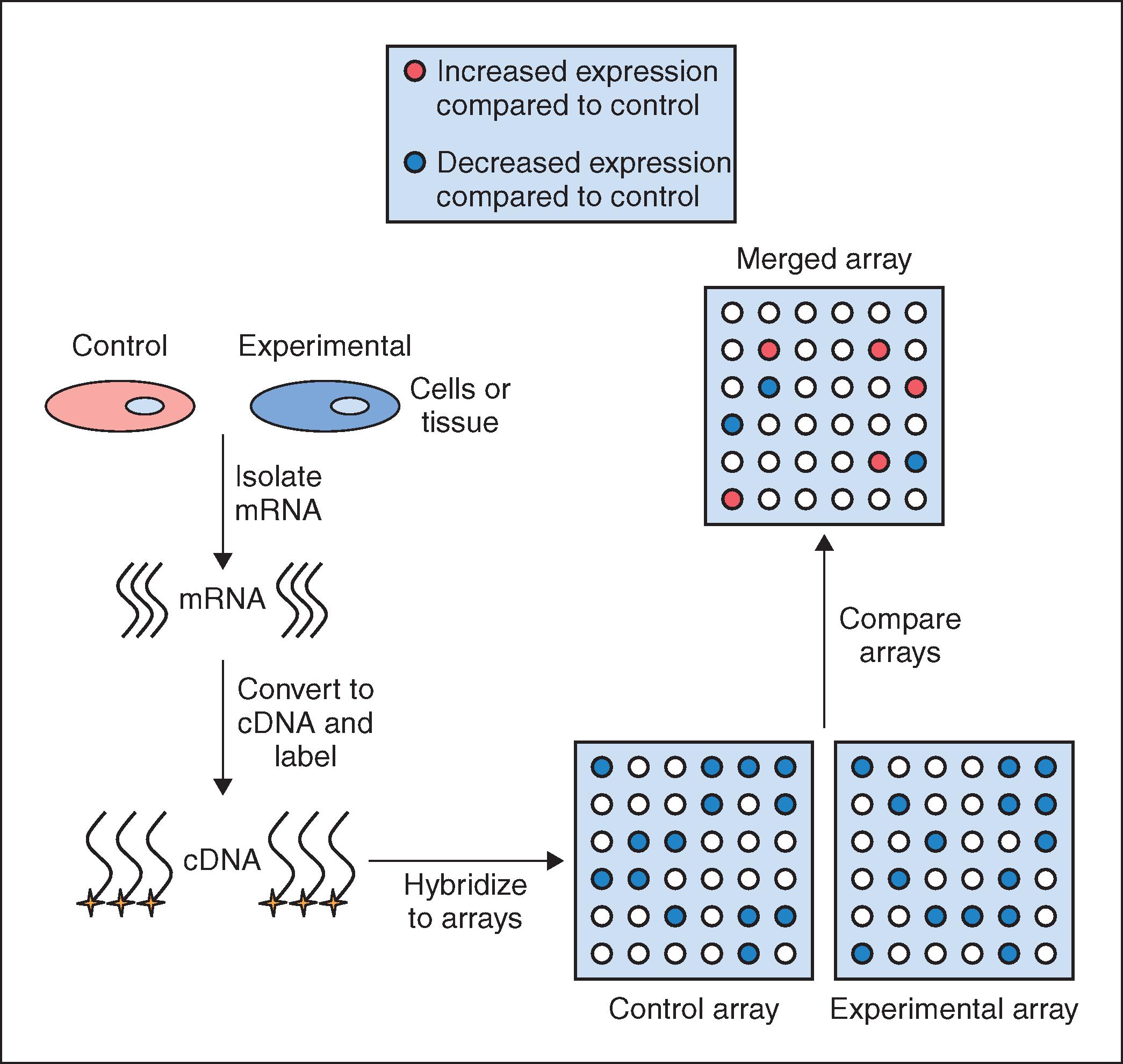
The sequence of steps involved in a gene array analysis is shown in Fig. 18.1 . The first step involves isolating mRNA from cells that represent the control group and from cells exposed to some experimental condition, such as UV radiation (experimental group). The mRNA preparation from each group is then reverse transcribed into complementary DNA (cDNA), which is more stable and hybridizes better to DNA than mRNA. This cDNA is then labeled with either a radioisotope or a fluorescent tag so that each unique cDNA can be detected and identified at the conclusion of the experiment. Once the cDNAs have been tagged, they are incubated with the gene array filter (e.g., the skin-specific array) so that hybridization between a given cDNA and its complementary DNA on the array can occur. Once hybridization is complete, any unbound cDNA is washed away and the hybridized cDNA is detected and quantified. Since the location and identity of each gene on the filter is known, by comparing the quantified spots on the array produced from the control group to those spots that are produced in the experimental array, one can determine whether a particular gene in the experimental group is upregulated or downregulated relative to the control group. Given the complexity of gene arrays, computer software is used to aid in the analysis of the large amount of data that is obtained. The software produces an overlay image of both gene array filters, calculates the difference in expression level for each gene between the control and experimental groups, and then converts the relative expression data into a color image. Typically, a gene that is upregulated in the experimental group relative to the control group is shown as a green spot on the computer-generated image while genes that are downregulated are shown in red. An example of the use of this technology in the identification of a novel antiaging and antiinflammatory active is discussed next.
Application of Gene Arrays to the Identification and Characterization of Antiaging and Antiinflammatory Bioactive Molecules
As results from microarray technology have become more reliable and reproducible, it has become possible to determine the effects of candidate bioactive molecules on skin cells with increased confidence. Furthermore, the use of microarrays has expanded from conducting basic research studies for candidate compound identification to screening tissue samples in a clinical setting to determine an individual’s susceptibility to certain diseases such as cancer. Due to the vast amount of data obtained from one particular experiment (e.g., 4000 genes of interest on one DNA filter), it is advantageous to only select a highly restricted set of critical genes of interest for investigation. For example, if antiinflammatory activity is desired, one may investigate the regulation of genes such as cyclooxygenase-2 (COX-2, a PGE 2 -producing gene), interleukin 1α (IL-1α), IL-6, IL-8, and tumor necrosis factor-alpha (TNF-α). Alternatively, if an antiaging bioactive was desired, one would focus on the expression of extracellular matrix genes such as collagens, elastin, and proteoglycans combined with the inhibition of matrix degrading proteases such as collagenase and the gelatinases. Microarray technology also offers the opportunity to identify new beneficial effects that would not have been discovered otherwise using typical single-gene experiments.
We have used gene array technology to identify unique compounds that have antiaging and/or antiinflammatory effects in multiple skin cell types. In one study we assessed the ability of a novel nitrone spin-trap compound to modulate the expression of aging-related genes in older human dermal fibroblasts. The fibroblasts were grown in the presence (experimental) or absence (control) of the spin-trap nitrone compound CX-412 for 48 hours, at which time the mRNA from each cell culture group was isolated, converted to a cDNA, labeled with radioactive nucleotides, and hybridized to IntegriDerm DermArray gene filters. These microarray filters contain over 4400 unique cDNAs specifically chosen due to their expression in skin cells and relevance to dermatologic research. Genes that are commonly expressed in skin are spotted in duplicate at different sites on the gene filters to provide an estimate of reproducibility of the hybridization reaction. The hybridization images are imported into a computer program that normalizes the data set and provides a color-coded picture of which expressed genes in the CX-412–treated fibroblasts were upregulated (coded in green) or downregulated (coded in red) relative to the untreated fibroblast cultures ( Fig. 18.2C ). Figs. 18.2A and B show the actual filter images of the hybridized radioactive cDNAs that were upregulated or downregulated by the CX-412 compound.
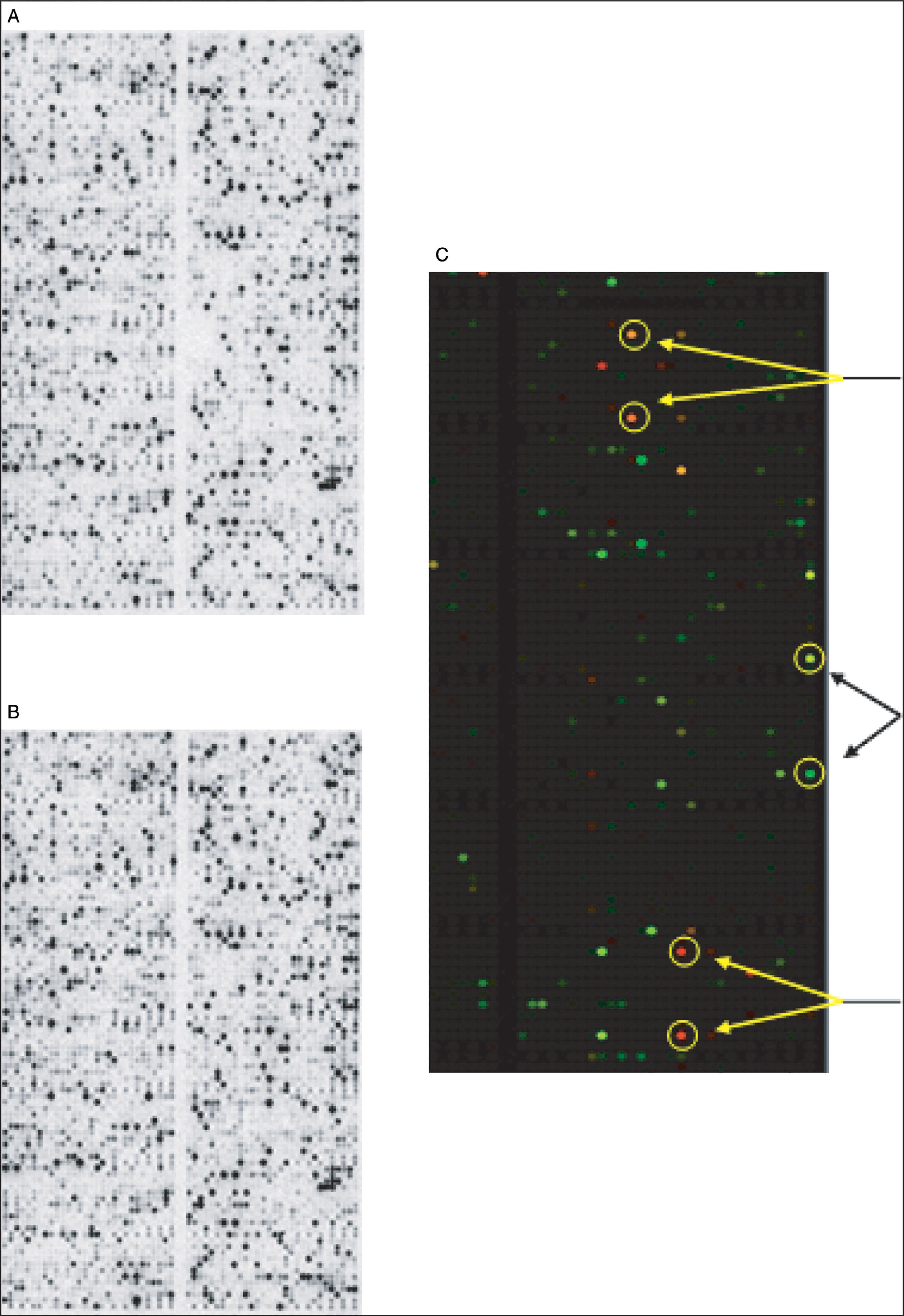
After quantifying the level of all expressed genes in the control and CX-412–treated cells, we found that aged fibroblasts treated with CX-412 shifted their expression patterns from matrix destruction to matrix production ( Box 18.1 ). For example, inhibition of the MMPs collagenase I and 92 kDa gelatinase was noted, while the naturally occurring inhibitors of MMPs, tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2, were upregulated. Furthermore, the expression of collagen types I, II, and III was enhanced in fibroblasts. In addition to age-related genes we also found that certain inflammation-associated genes, including uPA, tPA, PAI-1, IL-1α, and IL-6, were markedly inhibited by the spin-trap compound tested.
Upregulated by CX-412
- •
Tissue inhibitor of metalloproteinase 1
- •
Tissue inhibitor of metalloproteinase 2
- •
Type I collagen
- •
Type II collagen
- •
Type III collagen
Downregulated by CX-412
- •
Collagenase 1 (matrix metalloproteinase-1)
- •
72 kDa gelatinase (matrix metalloproteinase-2)
- •
Urokinase-type plasminogen activator
- •
Tissue-type plasminogen activator
- •
Plasminogen activator inhibitor 1
- •
Plasminogen activator inhibitor 2
- •
Chemokine receptor I
- •
Activated leukocyte cell adhesion molecule
- •
Interleukin-1α
- •
Interleukin-6
- •
Interleukin-13
- •
Basic fibroblast growth factor-2
Once the data from gene arrays are analyzed, simpler tests, including ELISAs and newly developed protein antibody arrays, can be used to confirm the gene array results for any given compound and to provide the necessary dose-response data needed to decide on the optimum concentration of the compound to use in topical formulations.
Gene Arrays and the Search for Aging Genes
As discussed previously, gene arrays can be used to identify compounds that can either upregulate (e.g., collagen) or downregulate (e.g., MMPs) gene activity to produce beneficial effects on the structure and function of skin cells. In this regard, a number of naturally occurring compounds, including curcumin, resveratrol, quercetin, luteolin, and epigallocatechin gallate (EGCG), have been shown to reduce the activity of inflammatory genes in the skin and increase the expression of matrix building genes including collagen, elastin, and hyaluronic acid synthase. But beyond the search for natural compounds that can alter the activity of genes that are important for maintaining youthful, structurally strong and resilient skin, there has been considerable interest in trying to find the “antiaging genes,” that is, genes (or even a key “primary” gene) that control the entire aging process, including skin aging. With the findings several years ago that certain species, including mice and monkeys, lived longer when placed on a calorically restricted (CR) diet, considerable excitement and research has been focused on utilizing gene arrays to identify genes that are subject to epigenetic regulation by diet and which may function to prolong life. In one study on monkeys, microarrays were used to identify genes that were expressed differentially in young and old animals and in those fed either normal diets or CR diets. The gene array analysis, performed as described in this chapter, was capable of analyzing over 4500 cDNAs expressed in monkey muscle tissue. The results showed the following:
- 1
Aging was associated with at least a twofold or higher increase in the expression of 300 genes.
- 2
Of these 300 “aging” genes, 24 were involved in inflammation, at least 4 were involved in oxidative stress, and 3 were associated with neuronal cell death.
- 3
The study also found that 149 genes were downregulated in aging animals and, not surprisingly, these included genes involved in energy metabolism (production of cellular energy), cell growth, and structural components.
- 4
When gene array analysis was carried out on age-matched “middle-aged” monkeys that had been either fed normal diets or put on CR diets, 107 genes were found to be upregulated in the CR-treated monkeys, indicating that diet can produce epigenetic changes in gene expression. Many were for structural components including collagen I, VII, and III. Of these induced genes, monkeys showed an inhibition of 93 genes including those involved in energy metabolism.
The question of whether or not caloric restriction can increase life span in primates was seemingly answered in 2009 when a 20-year study published in Science showed that CR-fed monkeys lived longer than their well-fed counterparts. Although these data suggested that caloric restriction is somehow linked to increased life span, another 20-year study on the same strain of monkeys found no correlation between food intake and longevity. Reducing the amount of calories consumed does not prolong life, although it likely reduces the incidence of diseases associated with obesity. An examination of the gene array data from the monkey studies failed to reveal any common group of genes that may be specifically important for the increased life span seen in the CR animals. Thus the search for a unique gene or sets of genes that control aging continues.
Given the conflicting data from the monkey studies, is there any evidence from gene array studies for a key antiaging gene whose expression can be regulated by external factors such as caloric restriction or by phenolic antioxidants? The answer is yes. Numerous studies have now shown that sirtuin genes play a key role in controlling cell aging and that these proteins, in turn, can be regulated by caloric restriction and by naturally occurring botanical compounds that are known to have antiaging or antiinflammatory effects. There are seven sirtuins produced in mammals, and these are proteins that have either deacetylase activity or ribosyltransferase activity. As a result of this activity, sirtuins can deacetylate histones, thereby causing transcriptional repression of certain genes. In addition, by deacetylating enzymes such as those involved in glycolysis, sirtuins can switch energy utilization away from glucose and toward fatty acid oxidation (advantageous during caloric restriction). Further, by deacetylating the p65 subunit of nuclear factor-kappa B (NF-κB), sirtuins can block inflammatory events. There is now evidence that sirtuins have antiaging effects on cells, delay senescence, and play a role in cancer cell growth. Finally, many studies have shown that certain sirtuins, including SIRT1 and SIRT5, are activated by botanically derived compounds, including resveratrol and quercetin.
In regard to skin aging, sirtuins (e.g., SIRT1 and SIRT6) have been shown to upregulate collagen I gene expression and downregulate MMP-1. Further, since skin aging is caused, in part, by ongoing inflammation that suppresses genes for collagen, hyaluronic acid synthase, and other dermal matrix proteins, the inhibition of inflammatory gene activity by sirtuins can produce antiaging skin benefits. Because sirtuin gene expression is upregulated by natural compounds, including resveratrol, many studies have now used gene array technology to determine what other genes, besides sirtuins, are regulated by resveratrol. These studies have shown that resveratrol downregulates the expression of genes involved in skin inflammation.
As will be discussed in the next section, a newer gene expression analysis method, called RNA sequencing, is providing far more detailed information on the effect of various compounds, drugs, and disease states on gene expression than was possible using gene arrays.
Application of RNA Sequencing to Study Gene Expression
Although gene array analysis using commercially available microarrays is a valuable tool for determining the differential expression of genes, one drawback of the method is that since microarray “chips” or “filters” are typically coated with small sequences of known genes, it is not possible to identify mRNAs that do not have a corresponding DNA sequence bound to the filter. For example, the search for unknown mRNAs that may be specifically transcribed in cancer cells and that may be important for the malignant process would not be identified using commercially available microarrays that contain small sequences of only known expressed genes. Another drawback of microarray analysis is that the background levels are typically high due to cross-hybridization. This makes it difficult to analyze mRNAs whose expressions are very low. Further, microarrays are not designed to identify unique forms of RNA, including microRNAs, noncoding RNAs, or alternatively spliced RNAs. Finally, obtaining quantitative data on mRNA levels is difficult without the use of complicated normalization methods.
If one wants to identify all of the RNAs being transcribed in a particular cell or tissue, a technique that is able to sequence all of these mRNAs simultaneously is needed. Over the past 15 years, methods have been developed that allow for the analysis of the entire transcriptome of a given cell. This new technology is referred to as transcriptomics , since it refers to the complete analysis of every mRNA produced by a cell at any time. The ability to carry out this transcriptome analysis has been made possible by significant advances in high-throughput DNA sequencing. There are several next-generation sequencing (NGS) platforms being used commercially today to sequence all the RNAs (abbreviated as RNA-Seq) produced in a cell. The use of this high-throughput sequencing methodology has led to a rapid increase in the number and types of studies being carried out to study gene expression. To date there have been over 8000 scientific references that have used RNA sequencing to study gene expression in human cells and tissues, and there are several excellent reviews that describe this technology.
RNA sequencing has many advantages over previously used methods such as gene microarrays and quantitative reverse transcription PCR (qRT-PCR), including the following:
- •
Unlike microarrays, which are designed with known gene fragments, RNA-Seq can detect novel RNAs.
- •
RNA-Seq provides sensitive and accurate quantitation of both low and highly expressed RNA transcripts.
- •
RNA-Seq detects a larger number of differentially expressed genes, particularly those with low expression levels.
- •
RNA-Seq can identify different forms of RNA including alternatively spliced RNAs, noncoding RNAs, and microRNAs.
- •
RNA-Seq reveals the full transcriptome, not just a few selected and known transcripts.
- •
Unlike Sanger sequencing, which can only sequence a single DNA fragment at a time, NGS can sequence millions of DNA fragments simultaneously per run.
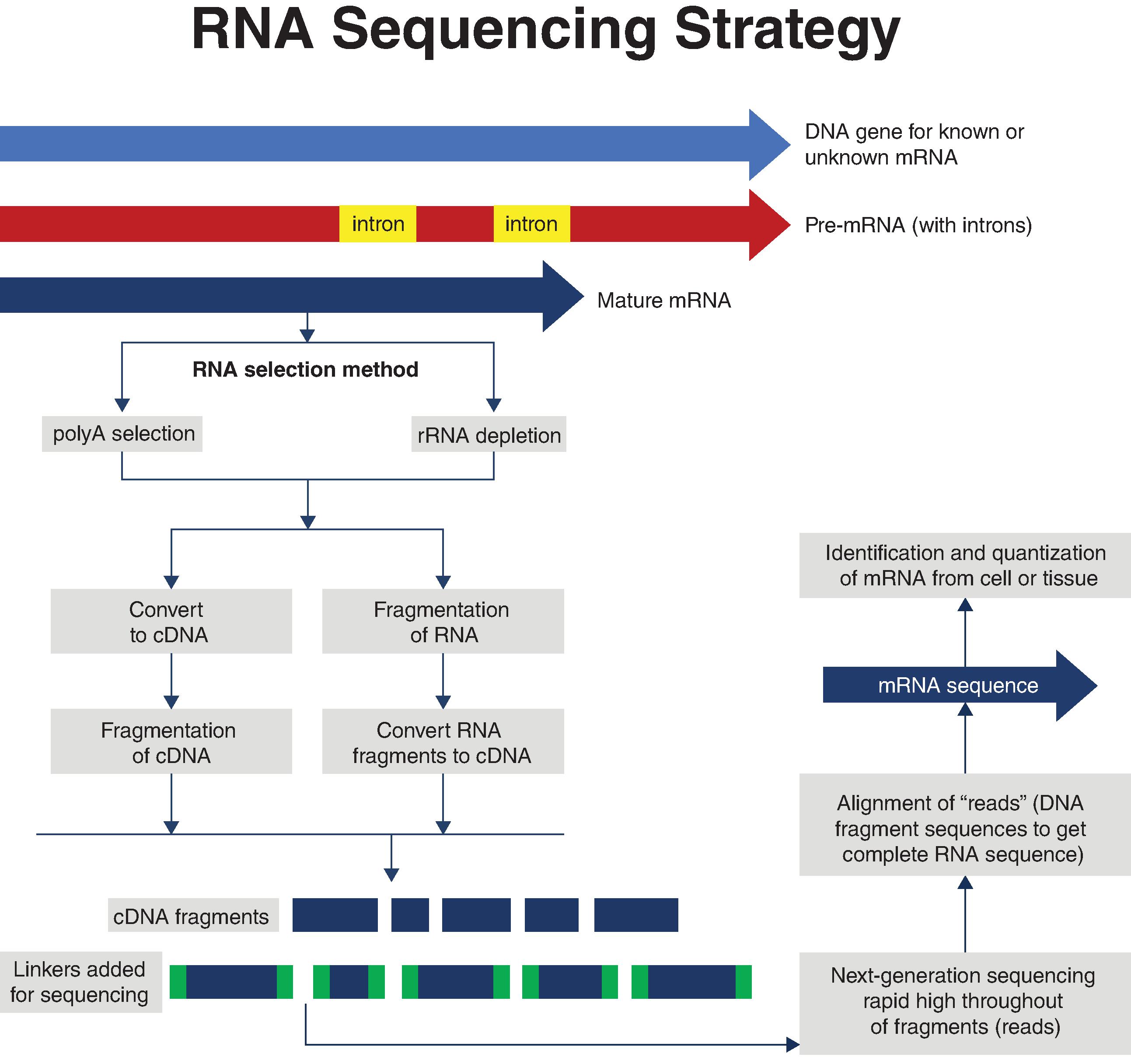
As shown in Fig. 18.3 , the steps involved in RNA sequencing are (1) isolating mRNA, (2) converting RNA to cDNA, (3) fragmenting the cDNA into smaller segments needed for sequencing, (4) sequencing the DNA sequences using NGS methods such as Illumina sequencing, and (5) aligning the DNA sequence “reads” to obtain the entire sequence. This method can be applied to identifying novel unknown RNAs as well as known mRNAs in tissues and even single cells. As was the case for gene array analysis, the first step of isolating RNA from the cells of interest typically involves isolating mRNAs that contain 3′polyadenylated (polyA) tails. However, this isn’t the only option. Since there are some mRNAs (e.g., histone mRNA) and other types of RNA that do not have polyA tails, if only polyadenylated mRNAs are isolated, it is possible that some unknown mRNAs that may be important for cell functioning are not being recovered. To circumvent this, total RNA can be prepared and then processed to remove ribosomal RNA. Once either polyadenylated mRNA or rRNA-depleted RNA is prepared, the RNA can then either be (1) fragmented into smaller segments that are then reverse transcribed to cDNA, or more frequently, (2) reverse transcribed into cDNA that is then fragmented. Regardless of which approach is used, the preparation of small cDNA fragments of between 30 and 400 base pairs is necessary for the subsequent rapid, high-throughput sequencing steps. As shown in Fig. 18.3 , prior to DNA sequencing, adapter “linkers” are added to the cDNA fragments. These adapters allow the cDNA fragments to bind to complementary oligonucleotides that have been bound to the flow cell matrix (or another matrix, depending on the system used) where DNA synthesis and sequencing will occur. The actual steps involved in NGS-based high-throughput RNA sequencing of the bound cDNA fragments vary depending on the particular methodology being used. One of the most widely used methodologies is the reversible dye terminator method used by Illumina. The first step in this method is to prepare thousands of copies of each cDNA fragment that are then bound to the oligonucleotides on the flow cell matrix (“chip”). The thousands of identical copies of each cDNA are referred to as a cluster . The next step involves the use of nucleotides, where each nucleotide has a unique fluorescent dye blocker that only allows DNA polymerase to add one nucleotide at each cycle to the cDNA fragment template bound to the flow cell matrix. The addition of this blocker nucleotide halts the synthesis. After each round of synthesis, the fluorescent dye that was added is recorded, and because each nucleotide has a different dye with a unique fluorescent signature, the computer can tell which nucleotide was added. After the camera records the fluorescent signature of the nucleotide, a chemical de-blocking agent is added to remove the blocking group, and the polymerase reaction then proceeds to add another fluorescently labeled nucleotide, which then stops the reaction again. The newly added fluorescently tagged nucleotide is identified, the blocking dye removed, and the synthesis step reinitiated. This continues until the entire DNA fragment has been sequenced. A sequenced fragment of the cDNA bound to the matrix is known as a read . The sequencing event occurs for millions of clusters at once, and given that each cluster has about 1000 identical copies of a cDNA, the speed and efficiency of this procedure are remarkable. Once all the fragments bound to the flow cell matrix have been sequenced, the fragments must be aligned through their overlapping sequences to obtain the full-length RNA sequence. A more complete review of this type of high-throughput NGS methodology can be found in several reviews.