25 Flap pathophysiology and pharmacology






Introduction
Flaps, both pedicled and free, are routinely used to reconstruct defects resulting from injury, excision of tumors, ulceration, or congenital malformation.1–3 The clinical problem is that flap failure as a result of ischemic necrosis occurs in both pedicle and free flaps. In free flaps alone, ischemic necrosis occurs in 5–10% of patients, even in experienced hands.4–10 Flap necrosis can be partial or total.11–13 Flap failure is time-consuming and costly because it requires repeated surgery and prolonged hospitalization. In the US, the additional operating room cost ranges from about $40 000 to $68 000 for each total free flap failure, and the additional surgeon reimbursement ranges from $5000 to $35 000 for each surgery.14,15 In addition, repeated surgery increases the incidence of donor site deformity and/or morbidity, with devastating effects on the patient. Therefore, we need to understand the pathophysiology of ischemic necrosis in flap surgery because this information may lead us to develop effective pharmacological therapies to prevent or salvage flap failure.
Pathophysiology of flap failure
Vasospasm and thrombosis in pathogenesis of pedicle and free flap failure
Clinically and experimentally, ischemic necrosis occurs mainly in the distal portion of both pedicle and free flaps. The general consensus is that vasospasm and thrombosis due to surgical trauma and insufficient distal vascularity are the main pathogenic factors in flap failure,3, but little is known about the pathogenic mechanism. However, there are published reviews on the role of vasoactive neurohumoral substances in the local regulation of peripheral vascular tone in normal and diseased states.16–21 These articles may provide insight into the pathogenesis of vasospasm and thrombosis in flap surgery, as illustrated in Figure 25.1. Briefly, endothelium-derived relaxing factors (EDRFs) such as prostacyclin (PGI2) and nitric oxide (NO) cause relaxation of vascular smooth muscle and inhibit platelet aggregation. On the other hand, endothelium-derived contracting factors (EDCFs) such as thromboxane A2 (TXA2), and endothelin-1 (ET-1) raise vascular tone. Under physiological conditions, a balance of vascular effects between EDCFs and EDRFs maintains adequate tissue perfusion. However, an imbalance can occur as a result of surgical trauma, as shown in Figure 25.1. Specifically, traumatized sympathetic nerve endings release norepinephrine (NE), causing vasoconstriction and platelet aggregation. The NE released by the sympathetic nerve endings, leukotrienes, serotonin (5HT2) and TXA2 released by the platelets, and the ET-1 released by the traumatized vascular endothelial cells can cause vasoconstriction and intravascular platelet aggregation, especially in the small arteries in the distal portion of the flap where the perfusion pressure is low and the concentration of these vasoconstrictive substances is high due to the downstream effect. Hemoglobin from hemolyzed red blood cells (e.g., hematoma) is also a potent vasoconstrictor. The histamine released by the mast cells changes the membrane permeability, resulting in edema formation. Furthermore, the synthesis and release of EDRFs such as PGI2 and NO from the traumatized vascular endothelium are depressed. In addition, the rate of endothelial degradation of NE and 5HT2 by catechol-O-methyl transferase and monoamine oxidase, respectively, is reduced in situations of impaired endothelial function. The end result is that there are high local levels of vasoconstrictive and prothrombotic neurohumoral substances in surgical trauma and these substances exacerbate vasospasm and promote thrombosis in flap surgery.
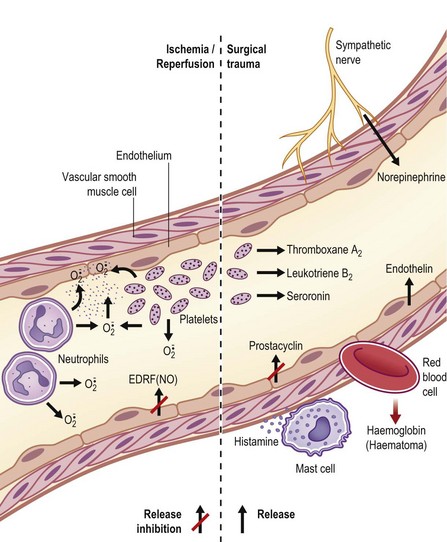
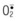 ) are produced by platelets and neutrophils. These free radicals can damage blood vessels.
) are produced by platelets and neutrophils. These free radicals can damage blood vessels.In reperfusion of ischemic blood vessels, superoxide radicals (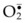 ) are produced by platelets, neutrophils, and endothelial cells and these free radicals can damage vascular walls during reperfusion (Fig. 25.1).
) are produced by platelets, neutrophils, and endothelial cells and these free radicals can damage vascular walls during reperfusion (Fig. 25.1).
Xanthine dehydrogenase/xanthine oxidase enzyme system in pathogenesis of ischemia–reperfusion injury in free flap surgery
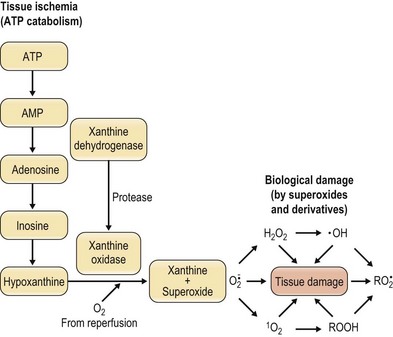
In free flap surgery, skin and muscle are subjected to warm (room-temperature) global ischemia under vascular clamp control during transfer from donor site to recipient site prior to reanastomosis. Human muscle and skin can withstand 2–2.5 hours and 6–8 hours of warm global ischemia, respectively.22–26 Excessive ischemic insult can result in ischemia–reperfusion injury caused by energy depletion and formation of oxygen-derived free radicals, as shown in Figure 25.2. Specifically, during prolonged ischemia, adenosine triphosphate (ATP) in skin and muscle is catabolized stepwise to hypoxanthine, with concomitant increase in cytosolic Ca2+.27 At the same time, a cytosolic protease is activated by intracellular Ca2+ and it converts xanthine dehydrogenase to xanthine oxidase.28,29 During reperfusion, the xanthine oxidase generates superoxide (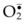 ) by univalent reduction of molecular oxygen in the presence of hypoxanthine.30 The unstable
) by univalent reduction of molecular oxygen in the presence of hypoxanthine.30 The unstable 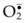 forms H2O2 spontaneously by dismutation. Furthermore, the unstable
forms H2O2 spontaneously by dismutation. Furthermore, the unstable 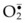 also interacts with H2O2 in the presence of a transition metal (e.g., iron) to form the most potent cytotoxic hydroxyl radical (OH•) through the Haber–Weiss (Fenton) reaction,31 as shown in Figure 25.2. There is evidence to suggest that this hypoxanthine/xanthine oxidase system is a main source of oxyradicals in ischemic rat skin and muscle.32,33 Furthermore, inhibition of xanthine oxidase activity by allopurinol and depletion of xanthine oxidase with a tungsten diet have been shown to attenuate the microvascular damage in rat skeletal muscle subjected to 2 hours of ischemia and 30 minutes of reperfusion.34
also interacts with H2O2 in the presence of a transition metal (e.g., iron) to form the most potent cytotoxic hydroxyl radical (OH•) through the Haber–Weiss (Fenton) reaction,31 as shown in Figure 25.2. There is evidence to suggest that this hypoxanthine/xanthine oxidase system is a main source of oxyradicals in ischemic rat skin and muscle.32,33 Furthermore, inhibition of xanthine oxidase activity by allopurinol and depletion of xanthine oxidase with a tungsten diet have been shown to attenuate the microvascular damage in rat skeletal muscle subjected to 2 hours of ischemia and 30 minutes of reperfusion.34
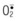 , superoxide; H2O2, hydrogen peroxide; •OH, hydroxyl radical, 1O2, singlet oxygen;
, superoxide; H2O2, hydrogen peroxide; •OH, hydroxyl radical, 1O2, singlet oxygen; 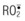 , peroxy radicals; ROOH, hydroperoxide.
, peroxy radicals; ROOH, hydroperoxide.There is also evidence to indicate that hypoxanthine/xanthine oxidase system is not a main source of oxyradicals in pig and human skin and skeletal muscle. Specifically, control pig and human skin samples were found to contain minimal xanthine oxidase activity, almost 40 times less than that of the rat, and xanthine oxidase activity did not rise during the first 8 hours of ischemia.35 In pigs, intravenous allopurinol given 60 minutes before ischemia did not protect cutaneous or musculocutaneous flaps from necrosis when subsequently subjected to 8 hours of warm ischemia and 5 days of reperfusion.36 Similarly, it was reported that xanthine oxidase activity in pig and human skeletal muscle was minute (<0.5 mU/g wet weight) compared with that of the rat.37 In addition, 5 days of competitive xanthine oxidase inhibitor treatment (allopurinol 25 mg/kg i.v., b.i.d.) starting 2 days before ischemia or 3 days of noncompetitive xanthine oxidase inhibitor treatment (oxypurinol, 25 mg/kg, i.v., b.i.d.) starting 15 minutes before reperfusion did not attenuate pig latissimus dorsi muscle necrosis when subjected to 5 hours of ischemia and 48 hours of reperfusion.37
Neutrophilic nicotinamide adenine diphosphate (NADPH) and myeloperoxidase (MPO) enzyme system in pathogenesis of ischemia/reperfusion injury in free flap surgery
There is accumulated evidence to indicate that neutrophils may play an important role in ischemia–reperfusion injury in free flap surgery. Specifically, it is well known that activated neutrophils produce large amounts of 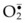 via NADPH oxidase, and these
via NADPH oxidase, and these 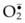 dismutates yield high concentration of H2O2 and OH•, causing tissue damage.38 MPO, which is unique and abundant in neutrophils, catalyzes the conversion of H2O2 to hypochlorous acid (HOCl), a potent cytotoxic oxidizing agent (H2O2 + Cl− + H+ → HOCl + H2O).39,40 Furthermore, it was reported that treatment with monoclonal antibodies against neutrophil–endothelium adhesion molecules attenuated ischemia–reperfusion-induced skin necrosis in rabbit ears,41 rat epigastric island skin flaps,42 and skin and muscle in pig latissimus dorsi musculocutaneous flaps.43 Treatment with monoclonal antibody against neutrophil–endothelium adhesion molecules also attenuated arteriolar vasoconstriction induced by ischemia–reperfusion injury.44 Finally, neutrophil depletion (~95%) with mechlorethamine significantly reduced necrosis in pig latissimus dorsi muscle flaps subjected to 5 hours of warm ischemia and 48 hours of reperfusion.37 Also, neutrophil depletion attenuated vascular injury in dog gracilis muscle subjected to 4 hours of warm ischemia and 1 hour of reperfusion.45
dismutates yield high concentration of H2O2 and OH•, causing tissue damage.38 MPO, which is unique and abundant in neutrophils, catalyzes the conversion of H2O2 to hypochlorous acid (HOCl), a potent cytotoxic oxidizing agent (H2O2 + Cl− + H+ → HOCl + H2O).39,40 Furthermore, it was reported that treatment with monoclonal antibodies against neutrophil–endothelium adhesion molecules attenuated ischemia–reperfusion-induced skin necrosis in rabbit ears,41 rat epigastric island skin flaps,42 and skin and muscle in pig latissimus dorsi musculocutaneous flaps.43 Treatment with monoclonal antibody against neutrophil–endothelium adhesion molecules also attenuated arteriolar vasoconstriction induced by ischemia–reperfusion injury.44 Finally, neutrophil depletion (~95%) with mechlorethamine significantly reduced necrosis in pig latissimus dorsi muscle flaps subjected to 5 hours of warm ischemia and 48 hours of reperfusion.37 Also, neutrophil depletion attenuated vascular injury in dog gracilis muscle subjected to 4 hours of warm ischemia and 1 hour of reperfusion.45
On the other hand, there are arguments against the important role of neutrophils in the pathogenesis of animal or human myocardial ischemia–reperfusion injury.46–48 Specifically, ischemia–reperfusion injury could be induced in neutrophil-free systems such as in cultured animal cardiomyocytes49 and human atrial strips50,51 and in isolated perfused animal hearts.52–54 In clinical studies, free oxyradical scavengers were not effective in protecting myocardium from ischemia–reperfusion injury.55,56 Furthermore, although monoclonal antibody to intercellular adhesion molecule-1 and anti-CD18 antibodies were effective in protecting myocardium against ischemia–reperfusion in laboratory animals,57,58 clinical trials with these agents yielded negative results.59–61 More recently, it was reported that ischemia–reperfusion injury was also induced in human rectus abdominis muscle strips cultured in neutrophil-free buffer.62 There is the possibility of species difference in the role of neutrophils in the pathogenesis of ischemia–reperfusion injury. Therefore, it is important to clarify the causal role of neutrophils in ischemia–reperfusion injury in human skin and skeletal muscle.
Intracellular Ca2+ overload in pathogenesis of ischemia–reperfusion injury in free flap failure
Recently, experimental evidence indicates that intracellular Ca2+ overload plays a key role in causing cell death during myocardial reperfusion.63 The pathogenic mechanism is summarized in Figure 25.3. Specifically, in sustained ischemia, mitochondrial ATP synthesis ceases and glycolysis ensues, resulting in a net breakdown of ATP and an accumulation of lactate and intracellular H+,64 causing intracellular acidosis. This build-up of intracellular H+ activates the Na+/H+ exchange isoform-1 (NHE-1) antiporter, resulting in extrusion of H+ and accumulation of intracellular Na+ to restore intracellular pH. There is a further increase in intracellular Na+ accumulation because Na+ extrusion is limited by inactivation of the energy-dependent Na+-K+-ATPase pump.65,66 Elevation of intracellular Na+ concentration causes an increase in intracellular Ca2+ by activation of the Na+/Ca2+ exchanger causing Ca2+ influx.65,67–70 If these events continue, the cystolic Ca2+ will be overloaded, and significant uptake of Ca2+ from the cytosol to the mitochondria will occur, resulting in mitochondrial Ca2+ overload71 which causes depolarization of mitochondria and impairs ATP synthesis, resulting in cell necrosis52 (Fig. 25.3). However, the NHE-1 may be inhibited72 if the extracellular acidosis is more pronounced than the intracellular acidosis after a prolonged period of ischemia.73 At reperfusion, the rapid washout of the extracellular H+ reactivates the NHE-1, resulting in further extrusion of intracellular H+, and further accumulation of intracellular Na+, causing further cystolic Ca2+ overload through Na+/Ca2+ exchange.74,75 Again, cytosolic Ca2+ overload causes mitochondrial Ca2+ overload, impairing ATP synthesis and resulting in cell death.52,71 Recently, there is evidence to suggest that mitochondrial Ca2+ overload plays an important role in ischemia–reperfusion in skeletal muscle and this will be discussed later, along with the therapeutic treatment aimed at preventing mitochondrial Ca2+ overload.
Pathogenesis of no-reflow phenomenon in free flap surgery
Rabbit island epigastric skin free flaps were used to study the pathogenesis of the no-reflow phenomenon in free flap surgery.76 It was observed that ischemia induced swelling of the endothelial and parenchymal cell, narrowing of the capillary lumen, intravascular aggregation of blood cells, and leakage of intravascular fluid into the interstitial space to form edema. This pathology increased with the increase in length of ischemic time from 1 to 8 hours and the obstruction of blood flow reached a point of irreversibility after 12 hours of ischemia, leading to no reflow and ultimate death of the flap. Three pathogenic mechanisms have been suggested to play a central role in the development of no-reflow phenomenon in the skeletal muscle of laboratory animals: (1) oxygen-derived free radicals causing damage in the endothelial and parenchymal cells; (2) this cell membrane damage allowing Ca2+ influx, resulting in intracellular overload; and (3) change in arachidonic acid metabolism resulting in synthesis of less vasodilating and antithrombotic PGI2 by the endothelium and increased synthesis of vasoconstricting and thrombotic TXA2 by platelets.77
Surgical manipulation for augmentation of pedicle flap viability
Flap design in augmentation of pedicle flap viability
One of the misleading principles in plastic surgery is that the viable length of a skin flap depends on the width of the pedicle. Milton was the first to disprove this principle.1,78–80 Using a random-pattern skin flap model in the pig, it was demonstrated that the ultimate surviving length of a pedicle flap is determined by the balance between perfusion pressure and vascular resistance. Increasing the width of pedicle flaps merely adds additional vessels of the same type and the same perfusion pressure and thus cannot increase the length of flap viability (Fig. 25.4). However, in other locations of the body, increasing the width of the pedicle may increase the chance of including a large artery. Therefore, one of the surgical manipulations to augment flap viability is the conversion of a random-pattern skin flap to an arterialized skin flap by incorporating a direct artery or a larger perforator.
Surgical delay in augmentation of pedicle flap viability
Surgical delay and vascular delay are proven techniques for augmenting flap viability in human patients,81–85 and in laboratory animals such as mice, rats, rabbits, and pigs.86–99 It takes two to three stages in surgical delay of pedicle skin flaps in order to augment flap viability. Specifically, a skin flap is mapped out on the donor site and incised on its two longitudinal sides. The flap is then undermined to form a bipedicle flap and is sutured back to the donor site. Two to three weeks after construction of the bipedicle flap, the third side (distal end) is cut in one or two stages at 2–3 days apart. At the end of this stage, a single pedicle flap is completely raised and the distal portion of the flap is moved to the recipient site for wound coverage without skin necrosis.1,100 Studies with pig random-pattern skin flaps showed that surgical delay increased skin flap capillary blood flow between 2 and 7 days of delay.101,102 This increase in capillary blood flow was mainly in the distal random portion of the delayed skin flaps.102
Vascular delay in augmentation of pedicle flap viability
In mice, rats, and rabbits, vascular delay for augmenting blood flow and viability in the distal portion of latissimus dorsi muscle flaps was achieved by dividing distal perforating arteries at 1–2 weeks prior to raising of muscle flaps.94,96,97 This phenomenon was also achieved in transverse rectus abdominis myocutaneous (TRAM) flaps in laboratory animals and human patients. Division of perforators or one or two dominant arteries that supply blood to the rectus abdominis muscle 2–3 weeks before flap surgery significantly augmented viability in TRAM flaps in rats92,93,95,98 and augmented skin and muscle blood flow and viability in TRAM flaps in pigs.92,93 In human patients, ligation of the deep inferior epigastric arteries 2–4 weeks before flap surgery augmented skin blood supply84,103,104 and viability in TRAM flaps.81,82,83,85
Surgical and vascular delay is proven clinically effective in augmenting flap viability, but these surgical procedures are costly and time-consuming. They require at least one additional surgical step done under general anesthesia. Vascular delay by embolization does not require general anesthesia, but it requires local anesthesia and catheterization performed in the interventional radiology department.105 Recently, “recharging” of the TRAM has been described as an alternative technique to augment flap perfusion.106 The evolution of microsurgery saw the development of free flaps. The idea was to improve flap blood flow and viability. TRAM free flaps are a good example as the free TRAM is perfused by the more dominant deep inferior epigastric pedicle rather than the less dominant superior epigastric vessels that supply the pedicled TRAM.106–111 However, free flap surgery is not always available and it is expensive. It requires specialized microsurgical staff and equipment and long operating room time. Furthermore, free flap surgery is not without morbidity and flap ischemic necrosis due to thrombosis and vasospasm occurs in 5–10% of patients.4–10,111 Therefore, there is the need to understand the mechanism of the surgical delay phenomenon through research to identify pharmacological strategies to prevent/treat ischemic necrosis.
Surgical delay procedure reduces arteriovenous (AV) shunt flow
Reinisch reported good correlation between postoperative fluorescein staining and the eventual skin viability in pig skin flaps. He detected warm skin temperature beyond the fluorescein dye marker in the distal portion of the pig skin flap.112 He also demonstrated 51Cr-labeled red blood cells and technetium and 85Sr-microsphere (15 µm) activity beyond the fluorescein dye staining in acute pig skin flaps.112 Taken together, he hypothesized that, in acute skin flap surgery, distal ischemic necrosis was caused by opening of AV shunt flow as a result of sympathetic denervation. He speculated that shunt flow occurred throughout the skin flap and the flow in the proximal areas is sufficient to supply both the AV and capillary (nutrient) blood flow, but the shunting became lethal in the distal areas of the skin flap where the total blood flow was low. In surgical delay, the bipedicle skin flap provided sufficient blood supply during the early period of sympathetic denervation and opening of AV shunts. Pearl presented data from rat abdominal arterial skin flaps, which supported Reinish’s hypothesis, and also suggested that surgical delay allowed the skin flap to recover from its hyperadrenergic state before converting the bipedicle to single-pedicle skin flaps.113 However, these observations were not supported by other investigators. For example, Prather et al., using various laboratory techniques (fluorescein dye test, xenon clearance, angiograph, 51Cr-tagged red blood cells and thermometry), failed to detect any evidence of vascular perfusion in the nonfluorescein distal portion of acute axial pattern skin flaps in the pig.114 In contrast to Palmer’s findings115 in rat dorsal skin flaps, Cutting et al.116,117 did not observe persistent adrenergic denervation in delayed bipedicle skin flaps. Later, Guba used 15 µm and 50 µm radioactive microspheres to measure capillary (nutrient) and total blood flow, respectively, and to calculate AV shunt flow in bipedicle axial pattern pig skin flaps. Both capillary and AV shunt flow increased between 2 and 7 days of delay in pig bipedicle skin flaps, but this increase in capillary blood flow in delayed bipedicle skin flaps was not the result of a decrease in AV shunt flow.101
Subsequently, using 15 µm and 50 µm radioactive microspheres as well as fluorescein dye, Kerrigan118 found no evidence of AV shunt flow in the distal portion of acute random and arterial pig skin flaps destined to necrose, as indicated by lack of fluorescein penetration. Finally, using a similar radioactive microsphere technique, Pang et al. demonstrated that AV shunts played no important role in the pathogenesis of distal ischemic necrosis in random-pattern skin flaps in the pig and augmenting distal skin viability by surgical delay did not rely on closing of AV shunts, but vasodilation of existing blood vessels.102,119 It is important to point out that there may also be species differences in the extent of AV shunt flow in the skin assessed by the radioactive microsphere technique. Specifically, Pang et al. observed in the pig that the AV shunt flow in the control skin and in the skin of acute and delayed random-pattern skin flaps within 6 hours postoperatively was ~60% of the total blood flow.102 However, Sasaki and Pang120 observed in the rat that the AV shunt flow in abdominal island skin flaps within 6 hours postoperatively was ~10% of the total skin blood flow. More recently, Kreidstein et al.121 observed that the AV shunt flow in isolated perfused human paraumbilical skin was ~1%. Taken together, AV shunt flow does not seem to play an important role in the pathogenesis of distal ischemic necrosis in acute pedicle skin flaps.
Surgical delay procedure depletes vasoconstriction and prothrombotic substances in the skin flap
Local tissue content of vasoconstricting and prothrombotic substances such as NE, TXA2, 5HT and ET-1 are known to be elevated by surgical trauma.3,122–136 These substances are released locally by traumatized blood cells, endothelial cells, and sympathetic nerve endings (Fig. 25.1). These are potent vasoconstrictors in skin vasculature.135,137–144 There is a general consensus that vasospasm and thrombosis play an important role in the pathogenesis of ischemic necrosis in acute skin flaps and the surgical delay procedure reduces local production and also allows time to deplete the vasoconstricting and prothrombic substances before converting the bipedicle flap to single-pedicle flaps. In the past, research in skin flap surgery was focused on the use of vasodilating and antithrombotic drugs to augment skin flap viability. So far, the outcomes of pharmacological therapy for prevention or treatment of pedicle flap ischemic necrosis are disappointing and will be discussed later.
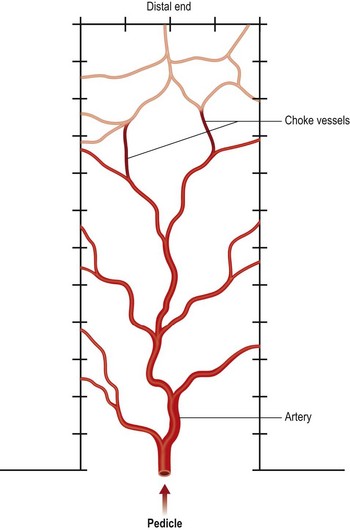
Surgical delay procedure induces vascular territory expansion by opening existing choke arteries
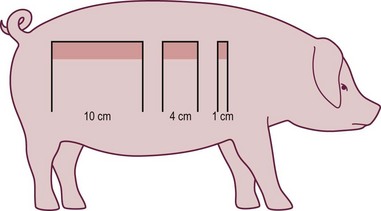
Pang et al. studied the capillary blood flow in delayed random-pattern pig skin flaps. We found that the capillary blood flow increased significantly within 2 days of delay and a maximum increase in skin flap capillary blood flow occurred between 2 and 3 days of delay, and remained unchanged between 4 and 14 days of delay without an increase in density of arteries (arteriogenesis) assessed by histology. This increase in capillary blood flow occurred mainly in the distal portion of the skin flap.102 Similar increase in capillary blood flow was also seen in vascular delay of pig TRAM flaps within 3–4 days of delay and it is unlikely that an increase in arterial density (arteriogenesis) could have occurred during this short period of time. Therefore, these investigators described this phenomenon as vascular territory expansion by recruitment (opening) of existing arteries, as illustrated in Figure 25.5.91 Opening of existing blood vessels was demonstrated by Taylor and colleagues in vascular delay of skin flaps in the guinea pig, rabbit, dog, pig, and human,145–148 and by Yang and Morris in vascular delay of rat skin flaps,149,150 and this phenomenon was labeled by these investigators as angiosome territory expansion by opening of existing choke blood vessels.
Surgical delay procedure induces angiogenesis
Lineaweaver et al. reported that vascular delay increased the viability of the skin paddle of rat TRAM flaps and this protective effect of vascular delay was associated with a significant increase in gene expression of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (FGF) in the skin paddle of the rat TRAM flaps within 12 hours of vascular delay. These investigators speculated that these cytokines induced vasodilation and angiogenesis to augment skin paddle viability in rat TRAM flaps.151 The vascular mechanism of surgical delay is still unclear. However, it was previously reported that the angiogenesis inhibitor endostatin inhibited ischemia-induced microvascular density and viability of mouse dorsal random-pattern skin flaps.152 Therefore, endostatin can be used in the future to investigate the role of angiogenesis and arteriogenesis in surgical delay in skin flap surgery in laboratory animals.
Pharmacological therapy for augmentation of pedicle flap viability
Drug therapy against vasoconstriction and thrombosis in pedicle and free flap surgery
Vasoconstricting and prothrombotic substances such as NE, TXA2, 5HT, and ET-1 are known to be elevated in skin flap surgery as a result of surgical trauma.122,123,125,126,153–160 These are very potent vasoconstrictors.135,161–168 In the past, research in skin flap surgery was focused on the use of vasodilating and antithrombotic drugs to augment skin flap viability. These studies were reviewed in detail up to 1990 by various investigators and will not be discussed here.3,118,169,170 The categories of drugs included: α-adrenoreceptor antagonists; drugs causing depletion of catecholamine in nerve terminals; drugs preventing catecholamine release from the nerve terminal; β-adrenoreceptor agonists; direct vasodilators, calcium channel blockers, hemorrheological drugs; vasodilating eicosanoids and their synthesis inhibitors; anti-inflammatory drugs; drugs inhibiting adherence and accumulation of neutrophils; and free radical scavengers. More recently, research in drug therapy for augmenting flap viability also focused on vasodilation,171–187 antithrombosis,188 and inhibition of neutrophil from adherence and accumulation.189 In general, the results with these vasodilating and antithrombotic drug treatments in augmenting pedicle flap viability were controversial, inconclusive, or very modest at best, compared with surgical delay. Finally, most of these studies were performed in loose-skinned laboratory animals (e.g., rats, rabbits) whose skin vasculature and anatomy are different from those of the human.190 Using more clinically relevant pig pedicle skin flap models, Pang and colleagues tested the following drugs, which were reported to augment rat skin flap viability by other investigators. It was observed that neither skin blood flow nor viability was significantly improved by the following categories of drug: glucocorticoid,191 α-adrenoreceptor antagonists,192 vascular smooth-muscle relaxants,193,194 β-adrenoreceptor agonist,193 TXA2 synthesis inhibitor,195 TXA2 receptor antagonist,195 and vasodilating prostanoids.196 It was observed that 5HT2 receptor antagonists augmented skin flap viability in the pig, but the beneficial effect was modest compared with surgical delay.195,197 In conclusion, the mechanism of surgical or vascular delay is unclear. So far, there is no effective drug therapy which can mimic surgical or vascular delay in augmenting skin flap viability.
Angiogenic cytokine protein or gene therapy for augmentation of pedicle flap viability
Angiogenic cytokines such as VEGF, FGF, and platelet-derived growth factors (PDGF) are known to induce an increase in capillary density (angiogenesis). Recently, flap research has been focused on the use of local angiogenic cytokine protein therapy to augment flap viability. For example, improved viability was observed following local subdermal injection of VEGF165 immediately after elevation of rat dorsal random-pattern skin flaps198,199; local interarterial injection of VEGF165 immediately after raising of rat epigastric island skin flaps200,201; and injection of VEGF165 into the rectus abdominis muscle 20 days before construction of TRAM flap in the rat.202 There is also evidence to indicate that FGF can augment skin flap viability. This has been observed following intradermal injection of FGF 30 minutes before surgery in rat dorsal random-pattern skin flaps203; subdermal injection of FGF immediately after surgery and at 48 hours postoperatively in rat dorsal random-pattern skin flaps204; and subdermal injection of FGF 18 days before construction of arterial skin flaps in mouse ears.205 Last but not least, it was reported that local PDGF treatment mimicked the surgical delay phenomenon in augmenting latissimus dorsi muscle viability in the mouse.206 So far none of these angiogenic cytokines has been tested on tight-skinned animals such as the pig. Since the angiogenic cytokines were effective in augmenting skin flap viability when given several days before or immediately after surgery, it is possible that vasodilation may play an important role this mechanism. Khan et al. used the rat dorsal random-pattern skin flap model to study the mechanism of acute local intradermal VEGF165.207 It was observed that subdermal injection of VEGF165 at the time of surgery effectively attenuated pedicle skin flap ischemic necrosis in a dose-dependent manner, mainly by inducing the synthesis/release of the vasorelaxing factor NO. The mechanism by which VEGF165 augmented skin flap viability appeared to depend on the vasodilator effect of VEGF165 in the early stage (within 6 hours) after surgery, followed by the angiogenic effect (i.e., increase in capillary density) of VEGF165 in the later stage after surgery. It is important to point out that VEGF165 is a potent vasodilator in skin vasculature. Specifically, it has been reported that VEGF165 is seven times more potent than acetylcholine in inducing skin vasodilation in isolated perfused pig island buttock skin flaps.208
The biological half-life of VEGF165 is 30–45 minutes in normoxic and 6–8 hours in hypoxic conditions.209 It is possible that the effectiveness of VEGF165 protein therapy is limited by its short half-life and VEGF165 gene therapy may be the key to providing a steady release of VEGF165 perioperatively.210 Specifically, local intradermal or subcutaneous injection of liposomal or adenoviral vectors encoding the cDNA of VEGF165 (Ad.VEGF165) given at 0.5, 2, 3, 7, or 14 days before surgery was shown to augment skin flap viability in the rat.211–214 Local subcutaneous injection of VEGF165 plasmid DNA 7 days preoperatively also increased skin viability in rat musculocutaneous (TRAM) flaps.215 Of interest is the observation that the number of capillaries and arterioles were significantly increased in the skin of rat musculocutaneous (TRAM) flaps when Ad.VEGF165 was injected subcutaneously 14 days before surgery.216 However, it is important to point out that review of the data in the literature thus far indicates that the efficacy of rat dorsal skin flap viability augmentation was similar between VEGF165 protein and gene therapy, and the skin flap viability was about 15–20% lower than that of surgical delay.207,217 Therefore, more than VEGF165 is required to mimic surgical delay in augmenting flap viability.
Pharmacological therapy for augmentation of free flap viability
Drug therapy for prevention of vasospasm and thrombosis in free flap surgery
Drug therapy is used by some surgeons to prevent or treat anastomotic vasospasm and thrombosis in free flap surgery. These drugs can be classified into three categories and their dosage, efficacy, and treatment guidelines have been reviewed.218–220