Fig. 8.1
Description of skin immune cells. LCs Langerhans cells, T RM resident memory T cells, dermal DCs dermal dendritic cells, ILCs innate lymphoid cells, γδT cells gamma delta T cells, NK cells natural killer cells
Atopic dermatitis (AD) is a pruritic, chronic, retractable inflammatory skin disease that is induced by the complex interaction between susceptibility genes encoding skin barrier components and host immune responses [4–10]. Type 2 helper T (Th2) cells and related cytokines such as IL-4, IL-5, and IL-13 have been reported to play important roles in its pathogenesis [10–15]. Cutaneous DCs acquire protein antigens and drive the differentiation/proliferation of a distinct Th cell subset including Th2 cells and provoke antigen-specific T cell responses to external pathogens [16]. In addition to Th2 cells, several recent papers have indicated that type 2 ILCs (ILC2s) are enriched in the lesional skin of AD patients and might play a critical role in driving the Th2-type immune response in AD [17–20].
This review will define the functions of cutaneous DCs and ILCs in the pathogenesis of AD, with a focus on studies performed by Japanese researchers, and will discuss the potential implications of these functions in the context of skin immune responses in AD.
8.2 The Role of Cutaneous DCs in the Pathogenesis of AD
8.2.1 DC Subsets in the Skin
Several subsets of DCs have been identified in the skin of both mice and humans. LCs comprise a skin DC subset that resides in the epidermis. Both human and murine LCs constitutively express a lectin receptor, namely, langerin/CD207, which is capable of binding sugar moieties commonly found on a variety of microorganisms [21]. They also express the E-cadherin and epithelial cell adhesion molecules (EpCAMs), which anchor LCs to keratinocytes [22, 23], and CD205 that is implicated in antigen capture and antigen processing [24, 25]. Human LCs highly express CD1a, a member of the group 1 CD1 protein, which is capable of presenting microbial lipid antigens to T cells [26] (Table 8.1).
Table 8.1
Dendritic cell population in the skin of human and mice
Localization | Cell type | Cellular markers | |
|---|---|---|---|
Human | Epidermis | LC | CD45, MHC class II, CD1a, CD207 (langerin), E-cadherin, EpCAM |
IDEC | CD1a, CD1b, CD1c, CD11c, FcεRI, CD23, HLA-DR, CD11b, CD206, CD36 | ||
Dermis | CD1c+ dDC | CD1c, CD1a+/−, CD45, CD11b, CD11c, MHC class II | |
CD14+ dDC | CD1c, CD45, CD11b, CD11c, CD14, MHC class II, CD209 (DC-SIGN) | ||
CD141+ dDC | CD1c, CD45, CD11c, CD141 | ||
pDC | CD303 (CLEC4C), CD304 (neuropilin), CD123 (IL-3R) | ||
Tip-DC | CD11c, TNF-α, iNOS | ||
Mouse | Epidermis | LC | CD45, CD11b, CD11c, CD24, MHC class II, CD205, CD207, E-cadherin, EpCAM |
Dermis | Langerin− dDC | CD45, CD11bhi, CD11c, CD24, MHC class II, CD205, SIRP-1α | |
Langerin+ dDC | CD45, CD11bdim, CD11c, CD24, CD103, MHC class II, CD207 | ||
pDC | B220, Siglec-H, PDCA-1 |
DCs in the dermis consist of dermal-resident DCs and migratory LCs traveling to the lymphatic vessels [27]. Recent studies have shown that murine dermal-resident DCs are further classified into two populations including langerin-positive dDCs (langerin+ dDCs) and langerin-negative dDCs (langerin− dDCs) [28–30]. The langerin+ dDC population represents 10–20% of the total dDC pool. In contrast to LCs, langerin+ dDCs express integrin αEβ7 (also called CD103) [31], lack the adhesion molecules E-cadherin and EpCAM, and express low levels of the integrin CD11b. Langerin+ dDCs express the same high levels of CD24 as LCs but do not express CX3CR1, F4/80, or signal-regulatory protein alpha (SIRPα); they also express low levels of CD11b and EpCAM [32] (Table 8.1). Langerin− dDCs represent the majority (up to 70%) of the dDC pool and express high levels of integrin CD11b and several macrophage markers, such as F4/80, CX3CR1, and SIRPα [32]. In human, dDCs are classified into three subsets (i.e., CD1a + dDCs, CD14+ dDCs, and CD141+ dDCs), depending on the expression pattern of surface molecules, although their functional differences remain unclear [33] (Table 8.1).
In inflamed skin, two additional subsets of DCs can be found: a DC population derived from blood monocytes (monocyte-derived DCs) [19] and plasmacytoid DCs (pDCs) [2, 34] (Table 8.1, Fig. 8.2). Another subset of the identified DC subpopulation referred to as TNF-α and inducible NO synthase (iNOS)-producing DCs (Tip-DCs) has been reported to be critical against bacterial infections [35]. In AD patients, inflammatory dendritic epidermal cells (IDECs) appear in the epidermis (Table 8.1, Fig. 8.2).
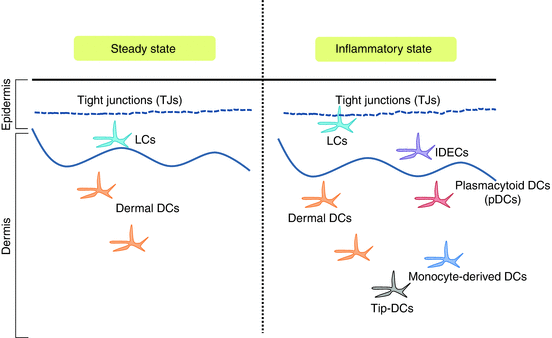
Fig. 8.2
Cutaneous dendritic cells in the steady state and in the inflammatory state. TJs tight junctions, LCs Langerhans cells, dermal DCs dermal dendritic cells, IDECs inflammatory dendritic epidermal cells, Tip-DCs tumor necrosis factor and inducible nitric oxide synthase-producing dendritic cells, pDCs plasmacytoid dendritic cells. In steady state, LCs reside in the epidermis beneath the TJ barrier. Dermal DCs reside in the dermis. In inflammatory condition, such as AD, LCs elongate their dendrite through TJs, capturing external antigens. IDECs recruit into the epidermis. Tip-DCs, pDCs, and monocyte-derived DCs also recruit into the dermis from the circulation
8.2.2 Cutaneous DCs in the Pathogenesis of AD
Since AD is a Th2-type immune response to protein antigens, and cutaneous DCs can induce Th2 cell differentiation, it has been suggested that cutaneous DCs initiate AD [36]. It remains unclear, however, which cutaneous DC subset mediates epicutaneous sensitization to protein antigens.
Kubo et al. showed that protein antigens of large molecular size are localized above the size-selective barrier, the tight junction (TJ), and that activated LCs extend their dendrites through the TJ to take up antigens [37]. Therefore, it can be hypothesized that LCs, not dermal DCs, initiate epicutaneous sensitization with protein antigens in the development of AD.
Nakajima et al. and Ouchi et al. showed that, in a murine AD model, which is induced by epicutaneous sensitization with protein antigen, depletion of LCs led to significantly attenuated induction of IgE/IgG1 upon epicutaneous sensitization with protein antigens [38, 39]. Moreover, Nakajima et al. showed that thymic stromal lymphopoietin (TSLP) receptor (TSLPR) expression on LCs is enhanced upon protein antigen exposure to the skin and that mice lacking TSLPR on LCs exhibited significantly attenuated Th2 cell differentiation in the AD model [38].
TSLP is a cytokine that is produced mainly by non-hematopoietic cells such as fibroblasts and epithelial cells, including epidermal keratinocytes. It has been reported that TSLP plays an important role in the induction of Th2 responses through the activation of DCs via the expression of OX40L [40]. TSLP stimulation also causes LCs to express OX40L [38]. In addition, cutaneous DCs can elicit a Th2 response in a mechanical injury model, in which TSLP is produced by keratinocytes [41]. These results indicate that TSLP produced by keratinocytes acts on LCs, which induces Th2-type immune responses in the murine AD model [38] (Fig. 8.3). In keeping with this, it has been reported that TSLP is highly expressed in the lesional skin of AD patients and that it activates human LCs in vitro [42–44], suggesting that LCs induce Th2-type immune response in human AD as well.
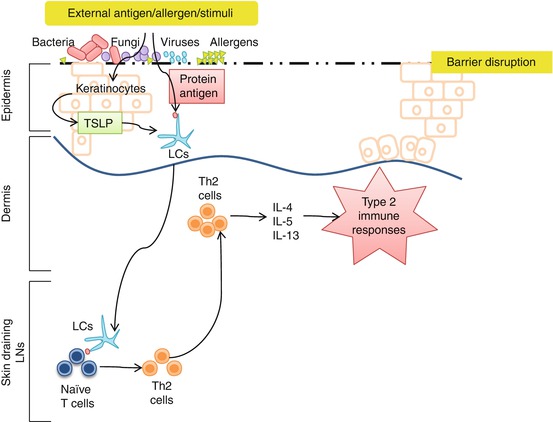
Fig. 8.3
Role of LCs in the pathogenesis of AD. Upon stimulation, keratinocytes produce TSLP. TSLP will activate LCs and induce LCs to uptake external antigens, migrate to the skin-draining lymph nodes, and induce Th2 cell differentiation
In the epidermis of human AD patients, both LCs and IDECs have been detected. Both LCs and IDECs express FcεRI on the surface and have been assumed to enable specific allergen uptake through IgE-dependent antigen capture [45, 46]. Yoshida et al., however, revealed that LCs, not IDECs, extend their dendrites through the TJ with a polarized distribution of langerin but not FcεRI in human AD skin [47]. These results again suggest that LCs mediate Th2-type immune responses in a langerin-dependent manner in the pathogenesis of AD.
8.3 The Role of ILCs in the Pathogenesis of AD
8.3.1 ILCs in the Skin
ILCs are part of a family of innate immune cells that are derived from a common lymphoid progenitor, and their development is partially or completely dependent on the common γ-chain (γc or CD132), IL-7, Notch, and the transcription factor inhibitor of DNA binding 2 [48]. ILCs are predominantly tissue-resident cells that lack antigen-specific receptors, such as T and B cell receptors. Instead, ILCs rapidly respond to various environmental stimuli with cytokine production [49].
The ILC family can be subdivided into three groups based on their requirement for activating cytokines, expression of transcription factors, and production of effector cytokines (Fig. 8.4). Group 1 ILC comprises NK cells and ILC1s; both of them are activated by IL-12 depending on transcription factor T-bet and produce interferon (IFN)-γ. Groups 2 ILC contains ILC2s that are activated by epithelial cell-derived cytokines/chemokines, such as IL-25, IL-33, and TSLP. ILC2s express GATA3 and produce Th2-type cytokines, e.g., IL-4, IL-5, and IL-13 upon activation. Group 3 ILC comprises lymphoid tissue inducer (LTi) cells and ILC3s. ILC3s can be further subdivided based on their expression of natural cytotoxicity receptors. They are activated by IL-1β and IL-23, are dependent on RORγt, and produce IL-17A and/or IL-22 [50]. Intriguingly, all of the ILC-activating cytokines can be produced by skin immune cells, such as keratinocytes, LCs, and dDCs.
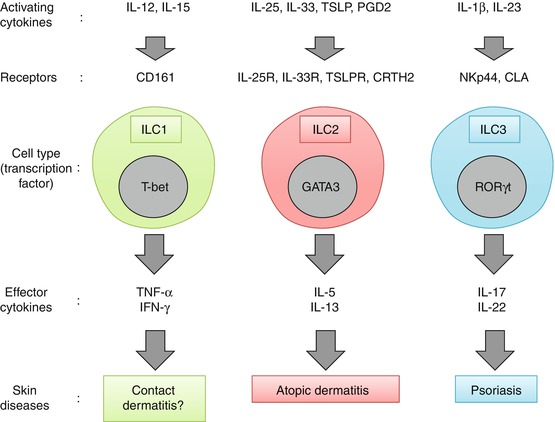
Fig. 8.4
Regulation and function of each subset of innate lymphoid cells (ILCs) in the human skin. Human skin ILC1s express CD161 and can be activated by IL-12 and IL-15 to produce its effector cytokines TNF-α and IFN-γ. Human skin ILC2s can be activated by epithelial cell-derived cytokines/eicosanoid (e.g., IL-25, IL-33, TSLP, and PGD2) and produce its effector cytokines IL-5 and IL-13; they have thus been implicated in AD. Human skin ILC3s produce IL-17 and IL-22 in response to IL-1β and IL-23 and have been implicated in psoriasis
All three ILC subsets have been identified in healthy human adult skin, where ILC3s are the most abundant and ILC2s make up 25% of the total ILC pool [20, 51–54]. In addition to NK cells, ILC2s were the first ILCs to be identified in both healthy and AD skin [52]. Although ILCs are present in both epidermis and dermis, the majority are found in the dermis, where ILC2s constitute 5–10% of CD45+ cells [55].
8.3.2 ILC2s in the Pathogenesis of AD
ILC2s were originally identified in the gut and gut-associated lymphoid tissues and were found to contribute to immunity against parasitic helminth in the absence of an adaptive immune system [56–59]. Subsequent studies showed that ILC2s play key roles in airway hyperreactivity [60] and epithelial tissue repair in the lung [61]. These studies demonstrated that ILC2s were predominantly regulated by the epithelial cell-derived cytokines IL-25 and IL-33 [62] (Fig. 8.5).
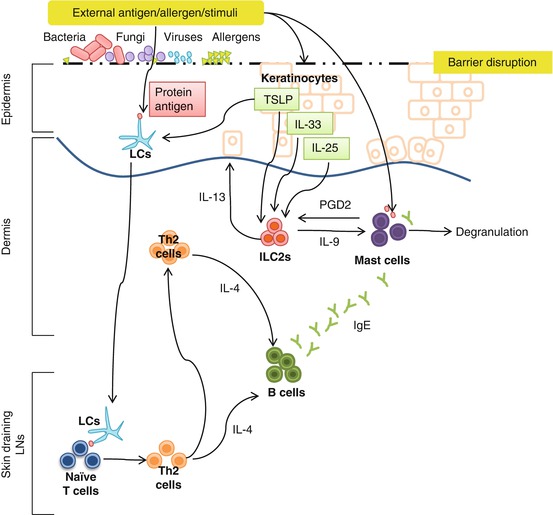
Fig. 8.5
Potential roles of ILC2s in AD. ILC2s are activated by epithelial cell-derived cytokines IL-33, IL-25, and TSLP, all of which are associated with barrier disruption and are upregulated in AD. ILC2 cell-derived IL-9 may increase mast cell numbers in the skin and enhance their responses to IgE-mediated activation in AD. Activated mast cell-derived PGD2 promotes ILC2 migration in the skin
When ILC2s were first identified in the skin, they were also found to be enriched in the lesional skin of AD patients [20, 52]. Type 2 cytokines, including IL-5 and IL-13, have long been suspected to play a key role in the pathogenesis of AD [63–65]. It was also revealed that ILC2s that produce IL-5 and IL-13 were found to be both necessary and sufficient for the development of AD-like disease in a low-dose vitamin D3-induced murine AD model [52, 55].
Mast cell numbers are elevated in chronic AD and likely drive IgE-mediated pathology in the disease [66]. Mast cells can also be directly activated by toxins derived from the skin-resident bacteria, Staphylococcus aureus, which is commonly found in the lesional skin of AD patients [67, 68]. Mast cell hyperplasia during inflammation is mediated by IL-3 and IL-9 [69, 70]. IL-9 can also augment the inflammatory cytokine response by mast cells following IgE-mediated activation [55]. ILC2s are a key source of IL-9 in vivo [71], suggesting that ILC2s potentially affect the severity of AD by regulating the number and function of mast cells in the lesional skin (Fig. 8.5). It has also been shown that prostaglandin D2 (PGD2) promotes the migration of skin ILC2s and induces type 2 cytokine production from skin ILC2s. Moreover, the supernatant of IgE/anti-IgE-activated mast cells induces migration and cytokine production by human skin ILC2s [72]. Since mast cells produce PGD2 by means of activation through FcεRI, they might play a role in migration and cytokine production by ILC2s through the effects of PGD2.
As mentioned earlier, TSLP is a crucial cytokine that causes cutaneous DCs to induce Th2 cells. ILC2s express the receptor for TSLP, and TSLP signaling in ILC2s promotes cytokine production [73]. Activation of cutaneous ILC2s was found to be dependent on TSLP signaling, suggesting that ILC2s were critical for disease pathogenesis [52] (Fig. 8.5). In addition to TSLP, Imai et al. showed that transgenic overexpression of IL-33 under a keratin 14 promoter is sufficient to induce spontaneous AD-like dermatitis in association with the accumulation of ILC2s [74]. This human and mouse evidence indicates that ILC2s are important players in the pathogenesis of AD, where their activity likely depends on the severity of AD, coinfections, and dysfunction of the skin barrier.
Conclusion
With their constitutional plasticity, LCs and ILC2s in the skin play crucial roles in initiating and modulating type 2 immune responses in AD. As many papers have demonstrated, both Th2 cells and ILC2s seem to play crucial roles in the pathogenesis of AD. The magnitude of their effect and/or their interplay, however, remains to be further elucidated.
Recent advances in our knowledge of the skin immune network and pathology in the pathogenesis of AD have opened avenues for the development of therapies that trigger skin DCs and/or ILC2s with specialized properties to control immunity.
Acknowledgments
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology and the Ministry of Health, Labor, and Welfare of Japan.
References
1.
Kaplan DH, Igyarto BZ, Gaspari AA. Early immune events in the induction of allergic contact dermatitis. Nat Rev Immunol. 2012;12(2):114–24. doi:10.1038/nri3150.PubMedPubMedCentral
2.
Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol. 2009;9(10):679–91. doi:10.1038/nri2622.PubMedPubMedCentral
3.
Otsuka A, Kabashima K. Mast cells and basophils in cutaneous immune responses. Allergy. 2015;70(2):131–40. doi:10.1111/all.12526.CrossrefPubMed
4.
Brunner PM, Silverberg JI, Guttman-Yassky E, Paller AS, Kabashima K, Amagai M, et al. Increasing comorbidities suggest that atopic dermatitis is a systemic disorder. J Invest Dermatol. 2016. doi:10.1016/j.jid.2016.08.022.
5.
Dainichi T, Hanakawa S, Kabashima K. Classification of inflammatory skin diseases: a proposal based on the disorders of the three-layered defense systems, barrier, innate immunity and acquired immunity. J Dermatol Sci. 2014;76(2):81–9. doi:10.1016/j.jdermsci.2014.08.010.CrossrefPubMed
6.
Egawa G, Kabashima K. Multifactorial skin barrier deficiency and atopic dermatitis: essential topics to prevent the atopic march. J Allergy Clin Immunol. 2016;138(2):350–8.e1. doi:10.1016/j.jaci.2016.06.002.CrossrefPubMed
7.
Kabashima K. New concept of the pathogenesis of atopic dermatitis: interplay among the barrier, allergy, and pruritus as a trinity. J Dermatol Sci. 2013;70(1):3–11. doi:10.1016/j.jdermsci.2013.02.001.CrossrefPubMed
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


