Fig. 12.1
Homeostasis of physiological skin barrier and S. aureus-derived virulence factors. KLK kallikrein, LEKTI lymphoepithelial Kazal-type-related inhibitor, NMF natural moisturizing factor, SCCE stratum corneum chymotryptic enzyme, SCTE stratum corneum tryptic enzyme, SKALP skin-derived antileukoprotease, SLPI secretory leukocyte protease inhibitor, SPINK-5 serine protease inhibitor Kazal-type 5, SPRR proteins small proline-rich proteins, TSST-1 toxic shock syndrome toxin-1
12.2.3.1 FLG Mutations
FLG mutations account for 30% of the AD population in European countries, and they are associated with the early onset of AD. FLG mutations in Japan are unique from those found in European-origin populations [17]. A high-throughput FLG mutation detection test revealed that 89 individuals out of the 820 participants were heterozygous for one of the ten FLG mutations, and two individuals were compound heterozygous for two of the ten mutations [18]. A recent study indicates that individuals with bi-allelic FLG mutations do not always have severe AD, and it confirmed that not all individuals with bi-allelic FLG mutations have AD [19].
The FLG mutation-induced disruption of barrier functions in horny layers allows increased epicutaneous penetration of exogenous allergens [20], with subsequent sensitization to the allergens, which results in the development of AD and food allergy [21]. FLG mutations might be involved in the pathogenesis of wheat-dependent exercise-induced anaphylaxis [22]. However, a current study did not identify a close relationship between peanut allergy and loss-of-function variants in the FLG mutations in children with AD [23].
12.2.3.2 SPINK-5 Mutations and Kallikreins (KLKs)
The balance of activity between proteases and their inhibitors is essential to maintain skin barrier functions. As exemplified in Netherton syndrome, a loss-of-function mutation of SPINK-5 attenuates the activity of a protease inhibitor, LEKTI, which in turn results in the enhancement of KLK protease activity. It was demonstrated that the levels of KLKs are increased in the horny layers of AD [24]. The enhancement of protease activity through increased KLK7 expression by T helper cell 2 (Th2) cytokines such as IL-4 and IL-13 might be an important factor for mechanical and chemical epidermal barrier dysfunction in patients with AD [25]. These observations indicate that endogenous KLK activity is increased in the skin lesions of AD, suggesting an association between polymorphism in SPINK-5 and AD [26]. The impairment of skin barrier functions due to endogenous serine proteases such as KLKs increases the risk of S. aureus colonization of the skin, and exogenous proteases released by S. aureus alter the skin barrier functions.
12.3 Staphylococcal Colonization and Virulence
More than 90% of patients with AD are colonized with S. aureus in the lesional skin, whereas most healthy individuals do not harbor this pathogen. Upon colonization of S. aureus in the skin, virulence factors released from S. aureus further damage the skin barrier functions and induce a Th2-skewed inflammatory milieu, which suppresses the innate immune system.
12.3.1 Quorum-Sensing Signals
12.3.1.1 agr Signaling
Cell density-dependent gene regulation known as “quorum sensing” has a crucial role in virulence genes’ expression (Fig. 12.2). The S. aureus agr quorum-sensing cascade includes the RNAIII-dependent and RNAIII-independent pathways, and the RNAII-dependent transcription of agrB, agrD, agrC, and agrA genes [27]. Among these pathways, all of the phenol-soluble modulins (PSMs), except for the hld gene encoding δ-toxin, are upregulated through the RNAIII-independent pathway. The RNAIII-dependent pathway upregulates the expression of proteases, lipase, urease, enterotoxin, leukocidins, α-toxin, and mecA, whereas it downregulates the generation of protein A, SarH1, oligopeptide, and fibronectin-binding proteins.
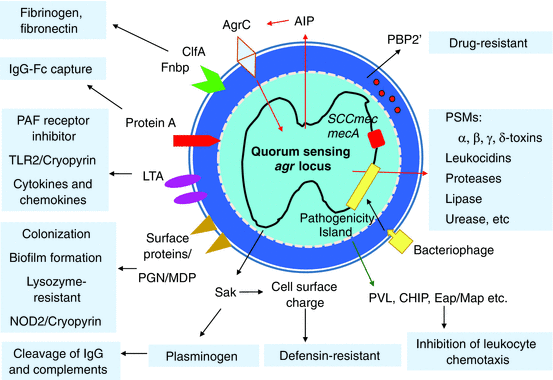
Fig. 12.2
Staphylococcal virulence factors and quorum-sensing circuit. The agr RNAIII-dependent pathway upregulates the expression of virulence factors including proteases, lipase, urease, enterotoxin, leukocidins, α-toxin, and mecA. Agr accessory gene regulator, AIP autoinducing peptide, CHIP chemotaxis inhibitory protein of staphylococci, ClfA clumping factor A, Eap extracellular adherens protein, ET exfoliative toxin, LTA lipoteichoic acid, Map major histocompatibility class II analogue protein, MDP muramyl dipeptide, NOD2 nucleotide-binding oligomerization domain 2, PAF platelet-activating factor, PGN peptidoglycan, PSM phenol-soluble modulin, PVL Panton-Valentine leucocidin, Sak staphylokinase, SCC staphylococcal cassette chromosome, SE staphylococcal enterotoxin, TLR Toll-like receptor
12.3.1.2 agr Mutation
A recent cohort study demonstrated that dysfunctional mutations in the agr locus, especially in agrC gene, were detected only in the strains from the infants without AD [8]. The agr quorum-sensing circuit may thus be involved in the development of AD, and low levels of RNAIII expression caused by the agrC mutations may reduce staphylococcal colonization, thereby blocking the development of AD.
12.3.2 Virulence Factors
Almost all S. aureus strains have the ability to secrete virulence group of enzymes and exotoxins, including four hemolysins (α-, β-, δ-, and γ-toxins), nucleases, proteases, lipases, hyaluronidase, and collagenase (Fig. 12.2). In addition to these microbial products, some strains produce one or more exotoxins targeting the specific molecules that constitute the cell adhesion and host immune systems. Some of these staphylococcal products have biological properties that are harmful to physiological and immunological skin barriers against pathogens.
12.3.2.1 Disruption of Physiological Skin Barriers
The staphylococcal V8 serine protease increases desquamation of the epidermis, and the exfoliative toxins A and B (ETA/B) cleave desmoglein-1, a desmosomal adhesion protein. Excess shedding of corneocytes leads to a disrupted skin barrier and increased bacterial invasion. S. aureus-derived ceramidase increases the permeability of the stratum corneum [28]. Vulnerability to bacterial colonization in the skin of patients with AD is associated with reduced levels of a natural antimicrobial agent, sphingosine, which results from decreased levels of ceramides [29]. In addition to the protease-induced skin barrier defects, possible physiological factors leading to staphylococcal colonization and dysbiosis include a decrease in antimicrobial peptides and sweat containing IgA and dermcidin, and increased skin pH, moisture, lipid content, and more [30–33].
Previous studies have demonstrated that lipoteichoic acid (LTA) and surface proteins of S. aureus are responsible for colonization on the nasal mucosa [34, 35]. Staphylococcal LTA manifests its immunomodulatory effects via the platelet-activating factor receptor (PAF-R), which leads to the production of IL-10 [36]. A toxin-receptor complex, such as the Hla (pore-forming α-hemolysin)-ADAM10 complex, disrupts a cell adhesion molecule, E-cadherin [37]. Furthermore, mice lacking ADAM17 in the epidermis induced AD-like dermatitis [38, 39], and naturally occurring dysbiosis with dominant colonization of S. aureus and Corynebacterium bovis induced eczematous dermatitis [40]. It has been demonstrated that the ADAM17-Notch-c-Fos and/or the ADAM17-EGFR signaling pathways play an essential role to sustain the homeostasis of skin barrier functions [38, 39].
12.3.2.2 Disruption of Immunological Skin Barriers
The subsequent cellular events induced by S. aureus colonization include the activation of phospholipase A2, phosphatidylinositol hydrolysis, the production of nitric oxide, prostaglandin (PG) E2, PGI2 and thromboxane A2, the activation of NF-kB, and the upregulation of various inflammatory cytokines [41, 42]. It has been postulated that bacterial toxins stimulate Rho-family GTPases that activate the downstream signaling pathways affecting the expression of inflammatory mediators and defensins [43].
It was demonstrated that S. aureus-derived δ-toxin is capable of the degranulation of mast cell granules and that S. aureus isolates recovered from AD patients produced large amounts of δ-toxin [44]. In addition, δ-toxin promotes allergic skin inflammation via IgE and IL-4 production. PSMs of S. aureus have various biological properties: antimicrobial function, the recruitment of leukocytes, and the induction of IL-18 and IL-17 cascades [45, 46].
S. aureus induces the expression of the skin-homing receptor cutaneous lymphocyte-associated antigen (CLA) on T cells. Keratinocyte-derived chemokines, thymic stromal lymphopoietin (TSLP), and IL-31 secretion are induced and augmented by S. aureus enterotoxins [47].
12.3.3 Th2 Immunity and Group 2 Innate Lymphoid Cells
Skin barrier dysfunction allows the penetration of immune stimuli which drive dendritic cells to enhance Th2 polarization [48]. The resultant Th2-skewed immune response, characterized by IL-4/IL-13 production, damages the physiological skin barriers by downregulation of filaggrin [49], loricrin, involucrin [50], desmoglein 3 [51], and ceramide [52]. TSLP has also emerged as a key cytokine in the pathogenesis of Th2 cell-associated AD [53]. Early studies have shown that TSLP overexpression in keratinocytes leads to the spontaneous development of AD-like disease in mice [54].
12.3.3.1 Th2 Immunity and the Microbiome
Upon Staphylococcus colonization, a Th2 inflammatory response and persistent IL-18 secretion from keratinocytes are induced by wall LTA and protein A, respectively [36, 55]. Staphylococcus enterotoxins A (SEA) and B (SEB) stimulate the expressions of ICAM-1 and HLA-DR in normal human keratinocytes, and over 50% of AD patients have specific IgE antibodies to SEA and/or SEB in their serum [56]. Epicutaneous sensitization with SEB elicits a local, cutaneous inflammatory response characterized by dermal infiltration with eosinophils and mononuclear cells and an increased mRNA expression of the Th2 cytokine IL-4 but not of the Th1 cytokine IFN-γ.
Cutaneous and systemic bacterial infections are exacerbated by IL-4 signaling because IL-4 hampers neutrophil expansion and migration by antagonizing G-CSF and chemokine receptor-mediated signals via CXCR2-CXCR4 [57]. The level of cytokine IL-22, often coproduced by IL17-secreting cells, is often elevated in AD skin lesions. IL-22 is important for limiting the growth of S. aureus on mechanically injured skin. The blockade of IL-23 and IL-22 may enhance susceptibility to staphylococcal skin infection [5].
12.3.3.2 Superantigens and Regulatory T Cells
Superantigens mediate direct cross-linking of major histocompatibility complex class II (MHCII) molecules on antigen-presenting cells with T-cell receptors, and they induce a strong proliferative response followed by the clonal deletion of a substantial portion of defined Vβ T-cells [58]. Epicutaneous exposure to superantigens skews the immune response toward Th2 cells, leading to the allergic skin inflammation and increased IgE synthesis that are characteristics of AD [59]. Patients with AD have significantly increased numbers of regulatory T (Treg) cells with normal immunosuppressive activity. However, after superantigen stimulation, Treg cells lose their immunosuppressive activity [60].
12.3.3.3 Group 2 Innate Lymphoid Cells
Innate lymphoid cells (ILCs) are divided into three different subsets based on their different cytokine expression patterns and transcription factors. ILC1 cells produce type 1 cytokines such as IFN-γ upon stimulation with IL-12, IL-15, and IL-18, and they express a transcription factor, T-bet. ILC2 cells produce Th2 cytokines such as IL-4, IL-5, IL-9, and IL-13 upon stimulation with IL-33, IL-25, TSLP, eicosanoids, and IL-1β, and they express GATA-3. ILC3 produces type 3 cytokines such as IL-17A and IL-22 upon stimulation with IL-1β and IL-23, and they express RORγτ. These cells play a crucial role in defense against extracellular pathogens such as staphylococcal infections [61, 62]. A recent study demonstrated that AD skin contained not only ILC2 cells but also prominent aryl hydrocarbon receptor (AHR)+ ILC3 cells [63].
12.4 Susceptibility to Cutaneous Infections
Keratinocytes serve as the first line of defense against cutaneous pathogens and are able to stimulate an immune response by releasing cytokines and antimicrobial cationic peptides such as defensins and LL-37 to eradicate pathogens. However, despite the upregulation of antimicrobial peptide generation, mice lacking three components of the cornified cell envelope, including envoplakin, periplakin, and involucrin, show an AD phenotype with increased skin bacterial load [64].
12.4.1 Staphylococcal Infections
Human β-defensins (HBD) are present at significantly lower levels in the inflamed skin lesions of AD patients [65, 66] (Fig. 12.3), although a disturbed skin barrier may trigger defensin induction in AD and psoriasis [67]. HBD2 has a less bactericidal activity against Gram-positive cocci such as S. aureus than HBD3 [68]. However, endogenous HBD2 expression was required and sufficient for protection against S. aureus protease SspA/V8-mediated integrity damage, probably via IL-1β-induced protective factors derived from epidermal keratinocytes [69].
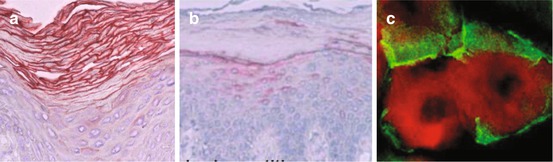
Fig. 12.3
Expression of human β-defensin 2 (HBD2) in psoriasis and atopic dermatitis (Ref. [66]). (a) Increased expression of HBD2 in psoriasis vulgaris, whereas (b) low expression in the atopic dermatitis. (c) HBD2 (green fluorescence) is expressed as surrounding the corneocytes (red fluorescence)
Patients with hyperimmunoglobulin E (IgE) syndrome, type 1 caused by STAT3 mutations, also present with AD-like eczema and recurrent abscess formation because of susceptibility to S. aureus infections [70]. Defects of Th17 cell development due to the defective IL-6 signaling and induced regulatory T (iTreg) cell generation due to defective IL-10 signaling account for the cutaneous pathology at least in part [71].
As ADAM10 cleaves multiple cellular substrates such as E-cadherin, it is of interest to understand the potential role of this protein in staphylococcal disease pathogenesis. Implication of ADAM10 in the development of AD suggests that the Hla (a pore-forming α-hemolysin)-ADAM10 complex may contribute to staphylococcal colonization and invasion [37, 72].
12.4.2 Eczema Herpeticum
Herpes simplex virus type 1 (HSV-1), F35 strains, is clearly associated with eczema herpeticum [73]. A previous study has shown that Th2 predominance and Th1 deficiency are much more pronounced in the subgroup of AD patients at risk for eczema herpeticum [74]. A murine model of eczema herpeticum with an impaired skin barrier demonstrated a critical role of defective NK activities in the development of severe HSV-1-induced skin lesions [75]. Other pathways related to the susceptibility to eczema herpeticum in AD include the inhibition of the IRF2, IRF3, and IRF7 innate immune pathways [76, 77], the expression of indoleamine 2,3-dioxygenease 1 (IDO1) [78], FLG mutations [79], TSLP [80], S. aureus α-toxin [81], and abnormalities in IFN-γ response [82].
Vitamin D levels are significantly lower in children with moderate or severe AD compared to children with mild AD. Children with AD with eczema herpeticum also displayed significantly lower vitamin D levels compared to those with AD alone [83, 84]. Further clinical trials are needed to provide conclusive evidence regarding the involvement of vitamin D in AD.
12.5 Therapeutic Approach Against Microbiota
Antibiotics temporarily reduce S. aureus colonization on the skin. Without signs of infection, oral antibiotics generally have a minimal therapeutic effect on the dermatitis [85]. Apart from their indication for overt secondary bacterial infections, antibiotics (by both systemic and topical applications) should not be recommended for the management of AD because no effect has been observed in clinical improvement or the sparing of steroids [86] and due to the risk of increasing prevalence of antibiotic resistance [87]. Since the production of virulence factors of Staphylococcus species may induce impairment of skin barrier functions and inflammation, there is no doubt that targeting the quorum-sensing signaling pathway and/or anti-toxin therapy is a promising approach for managements of dysbiosis.
12.5.1 Managements of Dysbiosis
Inhibition of bacterial adherence and colonization is the first line of prevention of bacterial infections. Previous studies have indicated that staphylococcal LTA and surface proteins are responsible for the establishment of colonization on nasal mucosa. S. aureus colonizes persistently in approximately 20% of the population and transiently in 60% and never colonizes in the remaining 20% [35]. It is clear that the differences in the individual defense mechanisms on the nasal mucosa are a key to preventing colonization in the host.
Therapeutic procedures to selectively eradicate pathogenic S. aureus, with a simultaneous protection of S. epidermidis, have been used clinically. These procedures include the application of a low pH cream and a gluco-oligosaccharide that inhibits the attachment of S. aureus cells on the epithelial surfaces [88].
In AD patients successfully treated with emollients, the patients’ dysbiosis with dominant staphylococcal colonization is recovered with the overall diversity of the commensal, with the abundance of Stenotrophomonas species [89]. The application of moisturizers significantly reduced the relapse of inflammation in AD patients [90]. These data support the importance of emollients and moisturizers in the management of AD.
12.5.2 Vaccination
A number of investigations are focusing on active or passive immunization against S. aureus infections, using a capsular polysaccharide vaccine [91] and humanized monoclonal antibodies [92]. A prophylactic S. aureus four-antigen vaccine (SA4Ag) is under development for the prevention of invasive S. aureus disease [93].
12.5.3 Anti-exotoxin and Anti-biofilm Therapy
Bacterial glycocalyx is a polysaccharide-containing material produced by bacteria. Bacteria that adhere to implanted medical devices or damaged tissue can become the cause of persistent infections. These bacteria encase themselves in a hydrated matrix of polysaccharide and protein, forming a slimy layer known as a biofilm. Since S. aureus can generally produce a biofilm on damaged skin tissue, antimicrobial agents may not eradicate S. aureus without the help of neutrophils. The S. aureus glycocalyx may play a crucial role in colonization and adherence to damaged skin tissue [94].
Infection with agr or hla (pore-forming toxin α-hemolysin, Hla) deletion mutants, loss of the Hla receptor ADAM10, or neutralization of Hla significantly attenuate virulence in mouse models of skin and soft tissue infections [37]. The blocking of staphylococcal autoinducing peptide 4 (AIP4) in agr-signaling provides a defensive effort against S. aureus virulence [95]. Recent advances in understanding the molecular mechanisms underlying toxin-induced tissue injury and host immune reactions have enabled us to develop a new approach to antitoxin treatments: the direct neutralization of exotoxins and the inhibition of the agr quorum-sensing pathway to produce exotoxins.
References
1.
Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14.Crossref
2.
Grice EA, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324(5931):1190–2.CrossrefPubMedPubMedCentral
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


