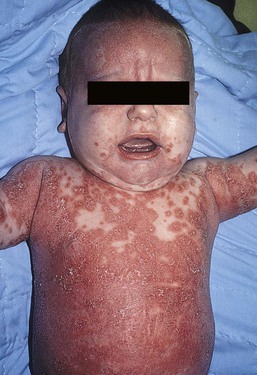
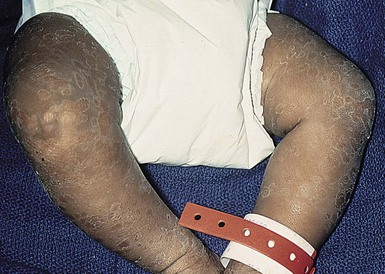
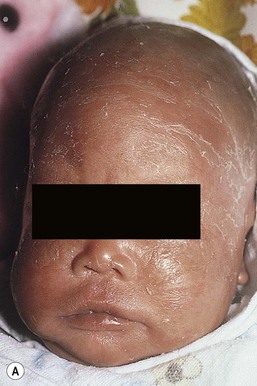
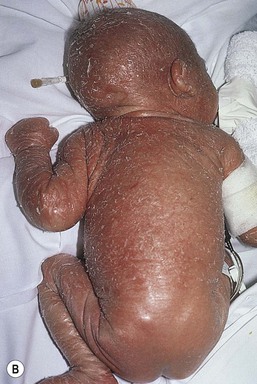
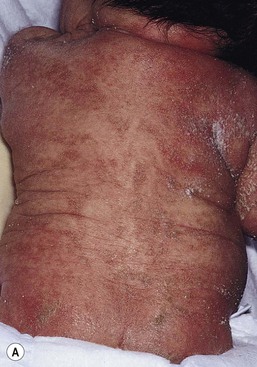
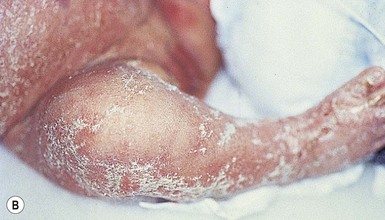
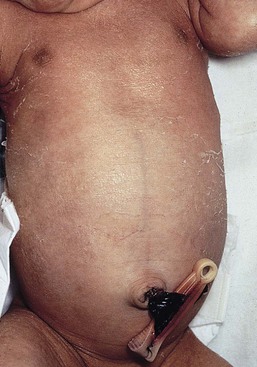
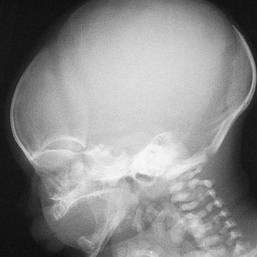
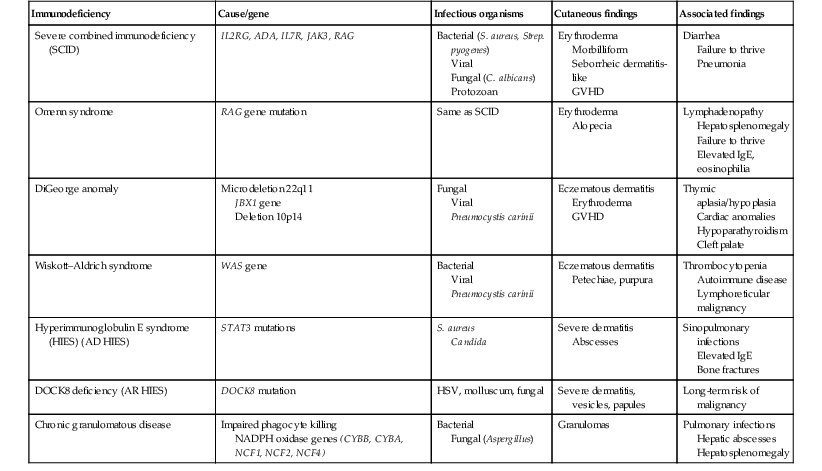
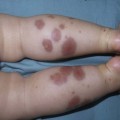
Moise L. Levy The term ‘erythroderma’ is used in dermatology to describe a skin eruption characterized by diffuse erythema, usually in association with scaling. Infantile erythroderma is caused by or associated with a large number of disorders (Box 18.1). The differential diagnosis includes inflammatory, infectious, inherited, and immunologic diseases, many of which have a hereditary basis. Some of these diseases are potentially life-threatening, and erythroderma itself can cause serious medical complications, such as electrolyte imbalance, sepsis, and temperature instability resulting from heat loss. It is therefore important for the physician to accurately diagnose and treat the problem. Severe generalized atopic dermatitis is unusual in neonates, though may often be seen in infancy. Because atopic dermatitis is such a common problem, it is the most common cause of acquired erythroderma in infants (see Chapter 15). Classic infantile atopic dermatitis involves the scalp, cheeks, and extensor surfaces of the extremities and may not appear until the infant is several months of age.1 When the distribution is generalized and the onset is early, diagnosis can be more difficult. The presence of pruritus, an almost invariable feature of this condition, is not always apparent in neonates and young infants. There is often a family history of atopy. Typically, the diaper region is spared, even in cases of widespread atopic dermatitis, as a result of the moist, occlusive environment of diapered skin. In contrast to infants with severe metabolic or immunologic disease, infants with atopic dermatitis usually grow normally and thrive, assuming the disease is recognized and treated promptly. Severe and long-standing disease, however, can be a cause of failure to thrive. Other clinical features such as repeated pneumonia, viral infections such as HSV or molluscum, and skeletal abnormalities may suggest the autosomal dominant hyper-IgE syndrome (HIES) or the autosomal recessive DOCK8 deficiency syndrome.2,3 Atopic dermatitis generally responds rapidly to appropriate therapy with topical anti-inflammatory agents and emollients. Skin biopsy in atopic dermatitis demonstrates acanthosis (thickening of the epidermis) and varying degrees of spongiosis (epidermal edema), as well as lymphohistiocytic inflammatory infiltrates, often with scattered eosinophils and plasma cells.4 Seborrheic dermatitis is a common problem during the neonatal period and is generally easily recognized (see also Chapter 15). Typically, there is scaling and erythema involving seborrheic areas such as the scalp and body folds. The yellow, greasy, scalp scale may encompass the entire forehead, including the eyebrows, and erythema and maceration can involve body folds such as the retroauricular areas, neck, axillae, and groin. Occasionally, a more diffuse pattern of seborrheic dermatitis can occur, which must be distinguished from atopic dermatitis, neonatal candidiasis, psoriasis, and other causes of infantile erythroderma (Box 18.1 and Fig. 18.1). The distribution of the dermatitis is more helpful than any other criterion in differentiating between atopic and seborrheic dermatitis, but it can be difficult and sometimes impossible to differentiate the two conditions accurately early in their course. Although the scalp can be red and scaly in both conditions, seborrheic dermatitis tends to involve the groin and other body folds, which are generally spared in atopic dermatitis. As treatment for both conditions in infancy is similar, from a practical standpoint accurate differentiation can be an academic exercise. However, the course of this disease differs: seborrheic dermatitis usually resolves over several months, whereas atopic dermatitis often persists for several years. Skin biopsy findings in seborrheic dermatitis are similar to those in atopic dermatitis. There is mild acanthosis, spongiosis, and a mild lymphohistiocytic inflammatory infiltrate; parakeratotic scale may be present. If clinical features suggest widespread seborrheic dermatitis in an infant who is otherwise well and thriving, and the skin readily clears after the application of low- to mid-potency topical corticosteroids without chronic rebound when therapy is tapered, the diagnosis of seborrheic dermatitis is probably accurate. Otherwise, alternative diagnoses should be considered. Severe seborrheic dermatitis in a child who is not thriving can suggest an immunodeficiency or Netherton syndrome. Less than 1% of all cases of psoriasis are said to occur in children less than 1 year of age. Infantile psoriasis can be difficult to diagnose because of its clinical similarity to both seborrheic dermatitis and atopic dermatitis. Infantile psoriasis can look like that seen in older individuals, with discrete oval erythematous plaques with white scale involving the trunk, extremities, and face. Psoriatic plaques in infants may have less hyperkeratosis than usually seen in adults. Facial involvement is more common in the infant, and the scalp, palms, and soles may have diffuse erythema and scaling. A periumbilical distribution may be helpful in distinguishing psoriasis from either seborrheic or atopic dermatitis. In contrast to atopic dermatitis, psoriasis in young infants often involves the diaper area because it develops in areas of injured skin (the Koebner phenomenon), e.g. after a prior irritant or Candida diaper dermatitis (see Chapter 16).5 Pustular psoriasis, either in a diffuse distribution or limited to the palms and soles, may be seen rarely. A positive family history for psoriasis is helpful. Some neonatal or infantile cases are HLA-B17 positive.6 Rarely, infantile psoriasis is generalized, a presentation that has been reported in young infants, and can even be present at birth. Erythroderma can evolve into and even alternate with pustulosis. Infantile generalized pustular psoriasis can be associated with lytic bone lesions,7 and be complicated by the acute respiratory distress syndrome (pulmonary capillary leak syndrome) that is also described in adults with acute generalized pustular psoriasis (B. Krafchik pers. comm.).8 Skin biopsy can be helpful in differentiating causes of neonatal erythroderma and in some cases, is diagnostic.4 Biopsy usually shows psoriasiform hyperplasia with elongated rete ridges and parakeratotic scale, often containing neutrophils. Occasionally, the diagnostic finding of a spongiform micropustule or microabscess in the upper epidermis is seen. Skin biopsies of erythrodermic psoriasis are often indistinguishable from those of any chronic dermatitis, lacking the classic features, and it may take several biopsies and close observation over time to confirm the diagnosis. Localized psoriasis may be treated with emollients and low-potency topical corticosteroids, but often clears only partially or recurs. Cases of infantile psoriasis may prove to be mild and occasionally even clear completely as the child gets older.9 The prognosis of generalized erythrodermic or pustular psoriasis in infancy is more guarded, and treatment usually requires systemic retinoid therapy, as well as supportive care.10 Erythroderma due to medications is fortunately rare, though cases in pediatric patients, including infants and neonates have been described.13 Severe reactions such as Stevens–Johnson syndrome or the drug reaction with eosinophilia and systemic symptoms (DRESS) represent particular challenges for clinicians.6 The approach to patients, regardless of age, requires a high index of suspicion and a broad understanding of other causes of erythroderma.14 Acute generalized exanthematous pustulosis (AGEP) presents acutely with pustules overlying diffuse erythema after exposure to an offending medication, mercury exposure or viral illness. Antibiotics reported to cause AGEP in infants include amoxicillin and amoxicillin-clavulanic acid. AGEP has also been reported in infants with no antibiotic exposure, suggesting an infectious trigger in these cases.15 Subcorneal pustules are seen by skin biopsy (see Chapter 20).16 Boric acid poisoning is now very rare, but was seen in the past as a result of the frequent use of boric acid-containing powders and lotions for the treatment of diaper dermatitis. It presents with a maculopapular eruption that can evolve into a generalized erythroderma, the appearance of which has been likened to a boiled lobster. A report of this in an adult after ingestion of a boric acid-containing pesticide, has been published.17 A positive Nikolsky sign and desquamation are additional features. Like staphylococcal scalded skin syndrome, the condition may be accentuated in periorificial and intertriginous areas. Alopecia may also develop. Affected infants are usually ill, with fever, irritability, vomiting, and diarrhea, which can progress to shock and even death. The various forms of cutaneous mastocytosis are discussed fully in Chapter 28. Only the rare diffuse cutaneous form of the disease is associated with neonatal erythroderma.18 The affected infant usually has generalized thickening of the skin, which can be subtle. The thickening is due to infiltration of the dermis by mast cells. Because these mast cells release histamine and other vasoactive substances, they cause the skin to be very reactive, with a tendency to develop erythema, flushing, and wheals. Urtication with minor trauma (Darier’s sign), and blisters, which develop either spontaneously or superimposed upon wheals, may be seen. The absence of scale and the presence of the above findings differentiate mastocytosis from other causes of erythroderma. C-kit mutations are seen in most patients with systemic mast cell disease.18 Skin biopsy is diagnostic revealing a dense, band-like infiltrate of mast cells in the upper dermis, which can be confirmed with Giemsa stain. Staphylococcal scalded skin syndrome (SSSS) is an uncommon cause of neonatal erythroderma (see Chapter 12). It is characterized by the abrupt onset of diffuse erythema, which rapidly evolves to erosive desquamation involving most skin surfaces. Although there is often periorificial accentuation, giving a dry, chapped appearance, the staphylococcal toxin requires keratinizing epithelium and therefore spares mucous membrane surfaces. The toxins act at a subcorneal location due to effects on desmoglein 1.6,13 Epidermal sloughing, occurring with minor trauma, helps distinguish SSSS from the other causes of infantile erythroderma, with the exception of toxic epidermal necrolysis (TEN)19 (see Chapter 12) and boric acid poisoning (see above). Rarely, a widespread form of staphylococcal pustulosis has been observed acutely in otherwise healthy infants. The pustules develop on an erythematous macular base and are small and superficial. They subsequently desquamate, thereby mimicking a true erythroderma. Candidal infection can cause neonatal erythroderma in two clinical settings: intrauterine acquisition with the development of congenital candidiasis, and postnatal onset in very premature infants. Congenital candidiasis typically presents either at birth or within the first few days of life with generalized erythema, vesicles, pustules, papules, and scaling. The pustules can be subtle at first, with erythroderma predominating. The palms and soles are often involved, which may be a helpful clue to diagnosis. The condition can occur in either term or preterm infants. Candida albicans can also cause a diffuse burn-like erythema within the first 2 weeks of life in premature infants (see Fig. 14.5A); 20 Also diffuse scaling and erythema, most pronounced over the back may be seen.21 The diagnosis is made by examination of skin scrapings (KOH preparation) and/or surface culture for Candida, or by skin biopsy. The latter will show fungal elements within the epidermis and/or dermis, mixed inflammatory infiltrates, and occasional areas of necrosis and hemorrhage. The risk of extracutaneous disease and the prognosis depend on the gestational age of the infant. In term infants, the prognosis is excellent, and topical anti-yeast therapies are usually curative. In infants weighing less than 1500 g with either congenital or acquired generalized cutaneous candidiasis, there is a significant risk of disseminated disease, and parenteral antifungal agents are recommended. Skin biopsies of affected areas in such infants may be used to predict the ultimate dissemination of disease. In one series, the finding of subcorneal invasion of Candida on skin biopsy was associated with a 69% risk for disseminated disease.21 Both conditions are discussed in more detail in Chapter 14. Although most cases of herpes simplex (HSV) infection have characteristic vesicular lesions localized on the presenting part, the so-called intrauterine variant of HSV can present at birth with either isolated or diffuse erythema, and scaling or crusted erosions on an erythematous base (see Chapter 13). It may be difficult to recognize clinically because vesicles may not be present.22 In fact, 44% of neonates with cutaneous disease do not have vesicles or bullae.22 This type of widespread involvement is generally associated with very severe neurologic disease. Multinucleated giant cells should be demonstrable on Tzanck smears of vesicular lesions. A skin biopsy, scrapings for direct fluorescent antibody staining, and viral cultures will help confirm the diagnosis. Congenital syphilis may cause diffuse erythema and scaling (see Chapter 12). This presentation is most typically seen in infants 6–8 weeks of age, in whom exposure to syphilis occurred either very late in pregnancy or at the time of delivery.23 Superficial erosions or bullae over the hands or feet of a newborn, together with a diffuse scaling dermatitis (Fig. 18.2), should alert the practitioner to the possibility of syphilis. Infiltrated mucosal papules and plaques (condyloma lata) may be seen in a perianal location and are similar to the mucous patches seen on other mucous membrane sites in older patients with secondary syphilis. Periosteal changes of long bones, such as the clavicles, as well as hepatosplenomegaly, are additional features. Appropriate serologies are generally diagnostic, and darkfield examination of mucous membrane lesions should reveal spirochetes. The ichthyoses are a group of genetic disorders characterized by generalized skin scaling; generalized erythroderma is a common presentation for some types of ichthyosis (see Chapter 19). Many infants with this presentation have the autosomal recessive congenital ichthyosis (ARCI) phenotype,24 which is notable for either diffuse erythematous appearance of the skin and overlying fine white scale or thicker plate-like scale (Fig. 18.3). Ectropion can occur. Testing for TGM1, ALOXE3, ALOX12B, or ICHTHYIN genes can be done for individual cases (see Chapter 19). Infants with autosomal dominant bullous ichthyosis, epidermolytic ichthyosis (EI), typically present with diffusely erythematous skin and mild hyperkeratosis, often in association with areas of denuded skin. The lack of mucous membrane involvement in EI helps distinguish it from epidermolysis bullosa. Over subsequent weeks and months, the blistering subsides and is replaced by varying degrees of an ichthyosiform erythroderma. Ultimately, marked hyperkeratosis is evident diffusely, with accentuation on flexural surfaces. The characteristic histopathologic findings of epidermal cytolysis of the upper spinous and granular layers help confirm the diagnosis of EI. Consideration of screening for mutations of keratins 1 and/or 10 is needed for confirmation. The ichthyosis most likely to be confused with other causes of erythroderma is Netherton syndrome, a rare disorder caused by mutations in SPINK5, which encodes a serine protease inhibitor, LEKTI.25 A publication has outlined specific mutations in SPINK5 and the use of such data for prenatal diagnosis.26 Some laboratories are able to stain tissue specimens for this protein.4 Netherton syndrome is marked by severe diffuse erythroderma, scaling, and varying degrees of alopecia, including sparse eyebrows (Fig. 18.4).27,28 Affected infants often fail to thrive as a result of the extreme metabolic demands presented by their skin disease, and they can also develop hypernatremic dehydration. Most have a markedly elevated IgE level. The diagnosis of Netherton syndrome is often delayed because of the late presentation of the diagnostic hair shaft abnormality, trichorrhexis invaginata (bamboo hair). Such hairs, when examined by routine light microscopy, will appear to have telescoped into themselves along the length of the shaft (Fig. 18.5). Small bulbous areas of thickening at the site of the telescoping correspond to the areas of increased fragility and ultimate breakage of affected hairs. Plucking of eyebrow hairs and evaluation of multiple areas of the scalp over time may be required to visualize the characteristic hair changes. Later in the course, patients may continue with generalized, scaling erythroderma or show a distinctive skin finding, ichthyosis linearis circumflexa. This is an erythematous scaling eruption with polycyclic and/or serpiginous morphology and elevated borders. Sjögren–Larsson syndrome is due to a deficiency of fatty aldehyde dehydrogenase (FALDH) and can present with varying degrees of erythroderma.29,30 Most cases are due to mutations in the ALDH3A2 gene, which codes for FALDH, though other factors play a role in some cases.31 A collodion membrane at birth is unusual. Affected infants may have a phenotype consistent with ARCI. Nonprogressive spasticity and mental retardation become apparent during the early years of life. After the first year, many affected patients have distinctive glistening dots seen on careful retinal examination. Chondrodysplasia punctata (Conradi–Hünermann syndrome) presents with either diffuse erythroderma or bands of erythema, and a patterned ichthyosis occurring along Blaschko’s lines (Fig. 18.6).32–34 Alopecia may be seen. These areas of ichthyosis typically resolve and may be replaced by a follicular atrophoderma. This X-linked dominant syndrome is also marked by skeletal defects (dwarfism), cataracts, and other features. Plain radiographs at the time of birth may show stippling of the epiphyseal areas of bones. It is due to a defect in emopamil binding protein (3β-hydroxysteroid-Δ8, Δ7-isomerase). Infantile erythroderma can also be seen in keratosis–ichthyosis–deafness (KID) syndrome,35,36 and neutral lipid storage disease with ichthyosis (Chanarin–Dorfman syndrome).37,38 A report of erythroderma as a presenting sign of Menkes disease has been published.39 Occasionally, males affected with X-linked hypohidrotic ectodermal dysplasia may present at birth with a mild diffuse erythroderma and fine superficial scaling (Fig. 18.7).40 Such infants have the typical facial features of ED and sparse hair, lashes, and eyebrows. Periorbital hyperpigmentation and fine wrinkling can be seen at birth, and lateral plain films of the skull will demonstrate no or few tooth buds (Fig. 18.8). Some of these patients have been reported to have primary immunodeficiency as well.41,42 Several immunologic diseases marked by immunodeficiency may produce similar initial clinical signs, with an eczematous dermatitis, diarrhea and failure to thrive. Children with a persistent eczematous eruption accompanied by failure to thrive may warrant an immunologic evaluation. The following discussion encompasses immunodeficiency syndromes with either erythroderma or other cutaneous manifestations in the neonatal period. These are outlined in Table 18.1. In 1988, Glover and colleagues43 reported a group of five infants with erythroderma, diarrhea, and failure to thrive. None had a yeast opsonization defect (as described in so-called ‘Leiner disease’), however, a variety of other immunologic abnormalities, including elevated IgE levels and hypogammaglobulinemia, were found. Some patients were subsequently diagnosed as having Netherton or Omenn syndromes. This paper established the need to consider the diagnosis of immunodeficiency when evaluating erythrodermic infants, and established the multiple etiologies of what had formerly been called Leiner disease. Infants in this category have a clinical phenotype of (1) noncongenital or acquired erythroderma (Fig. 18.9); (2) diarrhea; and (3) failure to thrive. Infants with these findings need thorough investigations searching for the underlying cause of their disorder. Most of the diseases listed in Box 18.1 need to be considered, especially immunodeficiencies, Netherton syndrome, Omenn syndrome, and eosinophilic gastroenteritis (see below). Baseline immune studies of such infants should include chest radiograph, full blood count, quantitative immunoglobulins, and specific measures of T-cell function. More detailed testing should be pursued as indicated. The prognosis and treatment of this condition are entirely dependent on the specific diagnosis. The associated diarrhea and failure to thrive must be treated aggressively with adequate nutritional support up to and including parenteral hyperalimentation, as indicated. Severe combined immunodeficiency (SCID) is a heterogeneous group of inherited disorders with similar clinical manifestations and immunologic deficiencies, with a profound deficiency of T lymphocytes and defects in both cellular and humoral immunity. Subtypes are classified by the abnormal development of other lymphocyte lineages, predominantly B lymphocytes and natural killer (NK) cells. Most cases are inherited in an autosomal recessive manner, although approximately 40% are X-linked recessive. About 20% of SCID cases are caused by adenosine deaminase (ADA) deficiency.44 Several other genetic defects account for the balance of causes of SCID.45 The immunologic deficiency results in an increased susceptibility to bacterial, viral, fungal and protozoan infections. Recurrent infections, diarrhea, and failure to thrive are evident by 3–6 months of age.46 Among the most common mucocutaneous infections are those due to Candida albicans, Staphylococcus aureus, and Streptococcus pyogenes. In addition to infectious cutaneous manifestations, all reported forms of SCID can present with a diffuse skin involvement, either erythroderma (Fig. 18.10), morbilliform, or seborrheic dermatitis-like eruptions. Patients with SCID may develop graft-versus-host disease from nonirradiated blood products or engraftment of maternal lymphocytes (Fig. 18.11). Cutaneous manifestations vary from a morbilliform rash or exfoliative dermatitis in acute GVHD, to a lichenoid or sclerodermoid rash in chronic GVHD. Early extracutaneous infections may also include viral-induced chronic diarrhea, otitis media, and pneumonia due to bacteria, viruses or Pseudomonas carinii.
Erythrodermas, Immunodeficiency, and Metabolic Disorders
Erythrodermas
Inflammatory diseases
Atopic dermatitis
Seborrheic dermatitis
Psoriasis
Drug exanthem
Acute generalized exanthematous pustulosis
Boric acid poisoning
Diffuse cutaneous mastocytosis
Infectious diseases
Staphylococcal scalded skin syndrome
Candidiasis
Herpes simplex
Syphilis
Genodermatoses
Ichthyosis
Immunologic diseases
Erythroderma and failure to thrive
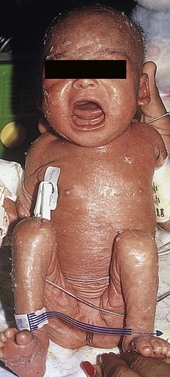
Severe combined immunodeficiency
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Erythrodermas, Immunodeficiency, and Metabolic Disorders
18