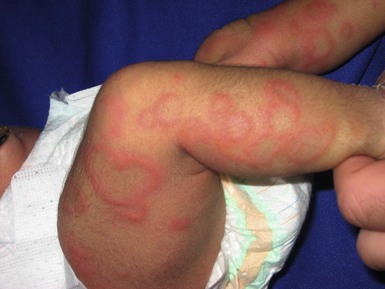
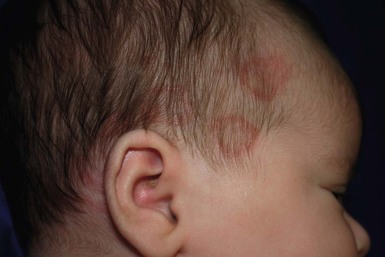
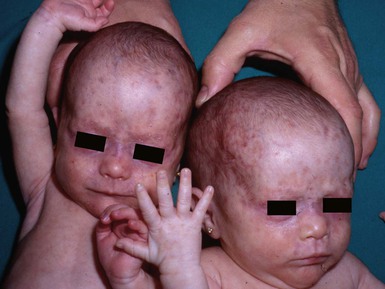
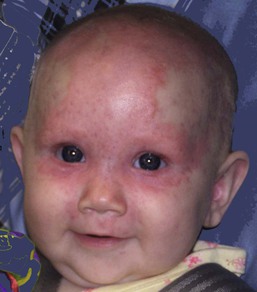
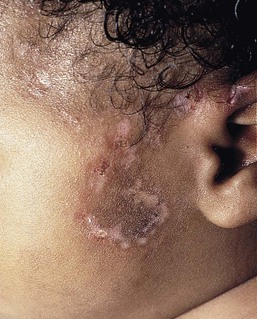
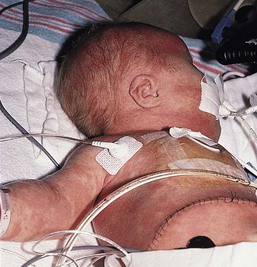
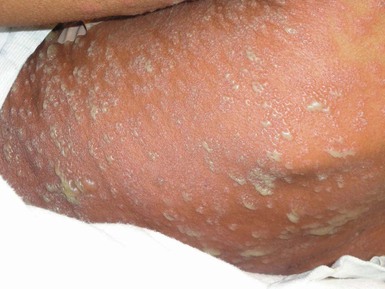
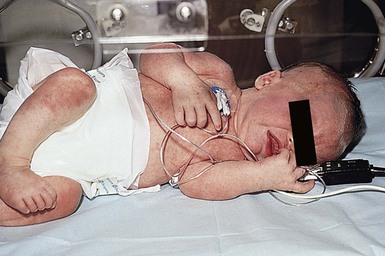
Eulalia Baselga, Angela Hernández-Martín, Antonio Torrelo In this chapter, a number of non-related entities will be discussed. They appear grouped by convenience, and represent a heterogeneous group of genetic and acquired diseases with a common ground of an immunologic pathophysiology. Several disorders appear to represent hypersensitivity reactions. Purpuric eruptions will also be covered in this chapter. The differential diagnosis of purpura is extensive in neonates and young infants, and includes hematological disorders, infections, trauma, metabolic diseases, and iatrogenic disorders. Annular erythema is a descriptive term that encompasses several entities of unknown etiology characterized by circinate polycyclic lesions that extend peripherally from a central focus.1–3 Because of subtle differences in clinical features, age of onset, duration of individual lesions, and total duration of the eruptions, a variety of descriptive terms have been coined for these disorders (Table 20.1). For prognostic reasons, it is useful to subdivide annular erythemas into transient and persistent forms.7 Transient forms include annular erythema of infancy and the less well-established entities erythema gyratum atrophicans transiens neonatale, neutrophilic figurate erythema of infancy and eosinophilic annular erythema. Persistent annular erythemas include erythema annulare centrifugum and familial annular erythema. The most important issue, however, is to exclude entities which require specific evaluation and treatment, such as neonatal lupus, tinea corporis, erythema chronicum migrans, erythema marginatum rheumaticum, erythema gyratum repens, and erythema multiforme. These ‘annular’ erythemas have distinctive clinical or histologic features and are considered below and elsewhere in this book. TABLE 20.1 Annular erythemas Annular erythema of infancy is a benign disease of early infancy characterized by the cyclic appearance of urticarial papules that enlarge peripherally, forming 2–3 cm rings or arcs with firm, raised, cord-like or urticarial borders.4,8 Adjacent lesions become confluent, forming arcuate and polycyclic lesions (Fig. 20.1). Neither vesiculation nor scaling is present at the border. The eruption is asymptomatic. Individual lesions resolve spontaneously without a trace within several days, but new lesions continue to appear in a cyclical fashion until complete resolution within the first year of life. Resolution of the lesions only during febrile episodes has been reported.9 A few cases lasting for years have been described.10,11 The cause of annular erythema is unknown, and there are no associated systemic findings. Histologic studies reveal a superficial and deep, dense, perivascular infiltrate of mononuclear cells and eosinophils. No flame figures are observed. The epidermis is normal or mildly spongiotic. Two variants with a predominantly eosinophilic and neutrophilic infiltrate have been described and renamed as eosinophilic annular erythema and neutrophilic figurate erythema of infancy, respectively.9,12 Laboratory studies are normal. Peripheral eosinophilia does not accompany tissue eosinophilia. Immunoglobulin levels, including IgE levels, are normal. The differential diagnosis should include other annular lesions of infancy (see below). No treatment is warranted because of the self-limited nature of the eruption. Erythema gyratum atrophicans transiens neonatale is a less well-defined entity,13 characterized clinically by annular plaques with an erythematous border and an atrophic center. The lesions appear in the newborn period and resolve within the first year of life. Histologic findings include epidermal atrophy and a mild perivascular mononuclear infiltrate. Immunofluorescence studies reveal granular deposits of IgG, C3, and C4 at the dermoepidermal junction and around capillaries. Erythema gyratum atrophicans transiens neonatale possibly represents a variant of neonatal lupus erythematosus.5 Erythema annulare centrifugum (EAC) is a more persistent type of annular erythema that usually affects adults,14 but may also occur in children and rarely in newborns.7,11,15,16 Two clinicopathologic variants have been identified: a superficial and a deep variety. The lesions consist of annular and polycyclic plaques with an indurated border in the deep variety and a scaly border in the superficial variety. The scales characteristically lag behind the advancing border. Individual lesions resolve spontaneously after a few weeks, but new plaques continue to develop for years, or may be a lifelong condition. There is no associated pruritus. Erythema annulare centrifugum is thought to represent a hypersensitivity reaction to several trigger factors, including infectious agents (Candida,17 Epstein–Barr virus,16 Ascaris,18 Pseudomonas), drugs or foods,6,19 and neoplasia, especially in adults. Intradermal injection of candidin or trichophytin may reproduce the clinical lesions.15 Histologic features for superficial EAC consist of a dense, superficial, perivascular mononuclear infiltrate. There is also parakeratosis or epidermal spongiosis. The deep variant shows a sleeve-like arrangement of the superficial and deep lymphocytic infiltrate, and in some cases of melanophages, subtle vacuolar changes at the dermal–epidermal junction, and individual necrotic keratinocytes, which makes differential diagnosis from tumid lupus erythematosus very difficult.20,21 No therapy has been successful in all cases. Depending on the trigger factor, treatment agents that have been used include oral nystatin, oral amphotericin B, topical antifungals, antihistamines, disodium cromoglycate, and interferon-α.15,17 Familial cases of annular erythema with autosomal dominant inheritance have rarely been described.22–24 The onset is in early infancy. Dermographism and pruritus was marked in the original cases. Lesions resolve with residual hyperpigmentation. Chronicity is the rule and geographic tongue may be associated.23 Differential diagnosis includes other eruptions with ring-like lesions, such as neonatal lupus, erythema multiforme, urticaria, urticarial lesions of pemphigoid, fungal infections, erythema chronicum migrans, and congenital Lyme disease.3,10,25,26 Serum antibody determinations (antinuclear, SS-A, and SS-B) are recommended to exclude neonatal lupus. Scraping any scaly lesion for KOH preparation is also advisable. Neonatal lupus erythematosus (NLE)27–34 is a disease of newborns caused by maternally transmitted autoantibodies. The major manifestations are dermatologic and cardiac. Skin findings are transient. Cardiac disease, which is responsible for the morbidity and mortality of NLE, begins in utero and affects the cardiac conduction system permanently. Other findings include hepatic and hematologic abnormalities. Mothers of infants with neonatal lupus have anti-Ro/SS-A autoantibodies in 95% of cases. Anti-La/SS-B and anti-U1RNP autoantibodies have also been implicated in the pathogenesis of NLE in a minority of patients.35,36 Of infants with NLE, 50% have skin lesions, and congenital heart block is present in about 10%.27,34 Lesions commonly develop at a few weeks of age but may be apparent at birth, which suggests that ultraviolet (UV) radiation is not essential for the development of skin lesions.37 Ulcerations, bullae, and extensive atrophy may be present, particularly in cases that are present at birth38 and transient bullous lesions from severe vacuolar damage of the basal cell layer39 have been reported as unusual manifestations of NLE. These cases might more accurately be called ‘congenital LE’ rather than NLE (Fig. 20.2). The more common skin manifestations of NLE fall into two main morphologies, papulosquamous and annular. Papulosquamous lesions are more common and are characterized by erythematous, nonindurated scaly plaques (Fig. 20.3), sometimes with an atrophic appearance (Fig. 20.4). In contrast to discoid lupus, scarring and follicular plugging are usually absent. The annular variant consists of well-circumscribed round plaques.40 Lupus profundus and generalized poikiloderma with erosions and patchy alopecia are rare manifestations.41,42 NLE lesions are most common on the face and scalp, predominantly affecting the periorbital and malar areas, often causing the ‘raccoon eyes’ appearance (Figs 20.4, 20.5), but can occur on virtually any body site. The eruption is frequently precipitated or aggravated by sun exposure, but lesions can develop in sun-protected areas (e.g., the diaper region, palms, and soles).37,43,44 Skin lesions are transient and cease to appear around the age of 6 months, after the disappearance of maternal antibodies. Transient or persistent hypopigmentation and epidermal atrophy may result (Fig. 20.6).34 Telangiectases, vascular ectasias resembling petechiae, persistent livedo or cutis marmorata, features of cutis marmorata telangiectatica congenita, and widespread erythema mimicking an extensive capillary malformation have been observed but are much less common manifestations. In some cases, these findings can be an initial sign of NLE, occurring without preceding identifiable inflammatory lesions.33,45–47 A rare report of a case of NLE with a serological profile consistent with drug-induced lupus has been described in a newborn whose mother was treated with α-interferon during pregnancy.48 The most significant manifestation is isolated complete congenital heart block. More than 90% of such cases are due to NLE. Most patients have third-degree block, but progression from a second-degree block has been reported.49 Heart block can often be detected as early as 20 weeks’ gestation. Cardiomyopathy and other types of arrhythmias are associated with NLE.50 Transient liver disease, manifesting as hepatomegaly (with a picture of cholestasis) or elevation of liver enzymes,33,51–53 and thrombocytopenia or other isolated cytopenias, may occur.54 Petechiae and purpura have been described as presenting signs of NLE.55 Less common findings include thrombosis associated with anticardiolipin antibodies, hypocalcemia, spastic paraparesis, pneumonitis, and transient myasthenia gravis.56–58 Central nervous system (CNS) involvement has been emphasized in some reports, and can be asymptomatic, with only ultrasound and CT scan abnormalities, suggesting a transient phenomenon.59 However, hydrocephalus has been reported in 8% of children with NLE.60 NLE is a cause of chondrodysplasia punctata, seen in X-rays as stippling of the epiphyses and the spine.60 Between 30% and 50% of mothers of infants with NLE have a connective tissue disease, most commonly SLE or Sjögren syndrome. Most, however, are asymptomatic. The risk for developing overt connective tissue disease in these mothers is highly debated, with estimates ranging from 2% to more than 70%.34,61–66 Placentally transmitted maternal IgG autoantibodies are associated with the pathogenesis of NLE.67 The most commonly implicated autoantibodies have been anti-Ro/SS-A and anti-La/SS-B, present in 95% and 60–80%, respectively. A small subset of affected infants have neither Ro or La antibodies, but instead have anti-U1RNP.35,36 Antibodies against the 52/60-kD Ro and 48-kD La ribonucleoproteins are associated with heart block, whereas antibodies against the 50-kD La ribonucleoprotein are associated with cutaneous disease.68 Significantly more symptomatic mothers of children with congenital heart block have anti-La antibodies than do disease-matched mothers with unaffected children.69 Moreover, the mean level of anti-La seems to be higher in mothers of infants with congenital heart block than in mothers of children with cutaneous NLE.70 It is likely that the amount of maternal antibodies, rather than their presence, is associated with fetal injury.68 Why less than 5% of mothers with anti-Ro and anti-La antibodies give birth to affected children is not understood, nor is the fact that mothers of affected infants are often asymptomatic despite having these antibodies. Fraternal twins are often discordant for NLE, and NLE does not occur in every subsequent pregnancy. Genetic factors may be important for the development of NLE in children with maternal lupus antibodies. A link has been suggested between NLE rash and the allele HLA-DRB1*03, as well as a -308A polymorphism in the TNF-α gene.32 Alternatively, maternal and/or sibling microchimerism may play an additional role, as levels of microchimerism have been reported to correlate with NLE disease activity.71 Serologic studies for autoantibodies in the mother and infant demonstrate anti-Ro, anti-La, and/or anti-U1RNP antibodies. Anti-NDNA, anticardiolipin antibodies, antinuclear antibody, and rheumatoid factor may also be present. Anti-Sm antibody, highly specific for systemic lupus erythematosus, is not found in NLE. The maternal antibody titer is usually higher than the infant titer. In apparently seronegative infants, more sensitive studies such as ELISA, immunoblotting, or line immunoassay, should be used instead of immunodiffusion techniques. Skin biopsy, which is usually not necessary for diagnosis, shows changes characteristic of lupus erythematosus, i.e., epidermal atrophy, vacuolization of the basal layer with a sparse lymphohistiocytic infiltrate at the dermoepidermal junction with a periappendageal distribution. In many instances, histopathological features in children with NLE rash are subtle. Direct immunofluorescence is positive in 50% of cases, demonstrating granular deposits of IgG, C3, and IgM at the dermoepidermal junction. The differential diagnosis encompasses congenital infections including rubella, cytomegalovirus and syphilis, congenital graft-versus-host disease, annular erythema of infancy, tinea corporis, and seborrheic dermatitis. False-positive VDRL tests for syphilis may occur in NLE. Telangiectasia and photosensitivity may suggest Bloom syndrome or Rothmund–Thomson syndrome. Serologic studies for autoantibodies in both infant and mother help to confirm the diagnosis. Skin biopsy for histologic and direct immunofluorescence studies is seldom necessary. Neonates with suspected NLE should receive a complete physical examination, electrocardiogram, complete blood count with platelet count, and liver function tests (Box 20.1). Skin lesions are transient. Treatment of skin disease consists of sun protection and the application of topical steroids. Pulsed dye laser therapy may be considered for residual telangiectasia. Congenital heart block is permanent. Half of newborns with complete congenital heart block require implantation of a pacemaker in the neonatal period.30,64,72 The average mortality rate from complete congenital heart block in the neonatal period is 15%; another 10–20% die of pacemaker complications.25,30 Late-onset cardiomyopathy may develop in a few infants.73–75 Mothers with anti-Ro or anti-La antibodies have a risk of delivering an infant with NLE in the range of 1–20%, depending on whether they have asymptomatic or symptomatic SLE.27,30 The risk of recurrence of congenital heart block in subsequent pregnancies may be as high as 25%.64 Such pregnancies should be closely monitored, ideally by obstetricians familiar with managing high-risk pregnancies. Although NLE is usually self-limited, SLE or other rheumatologic/autoimmune diseases may develop later in life in a small subset of patients.65,76,77 The exact risk is unknown. Collagen vascular disorders seldom appear in newborns and young infants. Both discoid and systemic lupus erythematosus (LE) has been reported in infants below 12 months of age, but skin lesions are very unusual.78–81 A systemic LE-like rash was seen at 12 months of age in an infant with C1q deficiency.82 Patients with familial chilblain lupus have been reported to develop skin lesions in infancy. Familial chilblain lupus is usually due to mutations in the TREX1 gene, and thus is allelic with the Aicardi–Goutières syndrome (AGS).83 In AGS, chilblains are a common symptom, and there is some overlap between AGS and familial chilblain lupus. In one family, a dominant heterozygous mutation in SAMHD1 caused familial chilblain lupus with infantile onset.84 Localized (linear) morphea can be present at birth, but this is very rare.85 It may be initially confused with vascular malformations such as port-wine stains. Cutaneous drug reactions86,87 are very rare in neonates. This is likely due to the relative inability to generate a drug-induced immune response88–90 and the lack of medication exposures together with time lag for sensitization required in many drug hypersensitivities. However, drug eruptions become far more common in older infants. These eruptions may be true hypersensitivity reactions, but many are idiosyncratic reactions, triggered in the setting of concomitant viral illnesses. If a drug eruption is suspected, a detailed history of medications should be obtained. In breast-feeding infants, this should include medications taken by the mother. A history of recent vaccinations can also be relevant. Maculopapular and morbilliform eruptions are the most frequent type of drug reactions in infants and usually have a benign course (Fig. 20.7). These eruptions are characterized by the abrupt onset of multiple small pink-red macules and papules that begin on the head and upper trunk and symmetrically progress downwards (Figs 20.8, 20.9). The lesions appear within the first 2 weeks of starting the offending medication and are often pruriginous. Most benign drug reactions are delayed-type hypersensitivity reactions to antibiotics.91 Distinguishing a drug eruption from a viral exanthem is often difficult. Skin biopsy is not helpful in most cases. It has been proposed that FAS ligand serum concentration might be useful in discriminating between drug rashes and viral exanthems, as it is raised in drug reactions and consistently low in viral rashes,92 but it is not available in most clinical settings. Drug eruptions are usually self-limiting after prompt recognition and discontinuation of the causative drug. Antihistamines in infants older than 6 months of age may help to alleviate the pruritus, but systemic steroids are not indicated unless the reaction is severe, and their efficacy remains controversial. Drug rechallenge to confirm the diagnosis is not recommended,91 and family education about generic and trade drug names is important to avoid recurrences. The primary infection by Epstein–Barr virus (EBV), usually asymptomatic in young children, may display the so-called ‘ampicillin rash’ when patients are given ampicillin, amoxicillin or, much less frequently, other antibiotics such cephalosporins or macrolides. The incidence of ampicillin rash in EBV infection was estimated to appear in 90% of patients, but recent studies have shown that the amoxicillin-induced rash appears only in about 30% of cases.93 The antibiotic-related rash can be macular, petechial, scarlatiniform or urticarial, and it can be differentiated from the spontaneous eruption in EBV infection in that it begins 1–2 days after starting the antibiotic treatment and that it is more severe and generalized, involving the head, neck, trunk, extremities, and even palms and soles. No consistent relationship has been shown for antibiotic dose, duration of treatment, atopic history, or previous exposure to penicillin. The pathogenesis behind the aminopenicillin-associated rash is uncertain. Ampicillin and amoxicillin can be re-administered after viral resolution without any adverse effect, suggesting a toxic etiology and not a true allergy.93,94 Vancomycin, an antibiotic frequently administered to premature newborn infants for Staphylococcus epidermidis nosocomial infections, may produce shock and rash (red-baby syndrome).95,96 This reaction is characterized by the appearance of an intense, macular, erythematous eruption on the neck, face, and upper trunk shortly after the infusion is completed. It may be accompanied by hypotension and shock. The reaction resolves rapidly in a matter of hours. It is frequently associated with rapid infusion; however, lengthening the infusion to more than 1 hour does not completely eliminate the risk. Newborns with AIDS have an increased susceptibility to drug reactions.97,98 Reactions to trimethoprim/sulfamethoxazole in patients with HIV infections can be severe and life-threatening.99 Fixed drug eruptions (FDE) are rare in infants and newborns. The classic cutaneous reaction of FDE consists of one or more round, well-circumscribed, erythematous to violaceous patches of variable size that appear anywhere on the body. Trimethoprim-sulfamethoxasole and non-steroidal anti-inflammatory drugs are well-known causes of fixed drug eruptions. Reactions of the scrotum and penis, with erythema and edema resulting from hydroxyzine hydrochloride, have also been described in early infancy,100 as well as bullous forms.101 Recurrences in the exact same location are common prior to diagnosis. Vegetant bromoderma is a reaction to bromides characterized by coalescing papules and pustules which form vegetant inflammatory or pseudotumoral lesions. It usually affects the scalp, face, and legs. Most cases of vegetant bromoderma have been described in infants after the ingestion of syrups and solutions containing bromide, which has sedative, anticonvulsant and expectorant properties, or the spasmolytic agent scopolamine bromide.102,103 The eruption ceases after withdrawal of bromide. The risk of systemic intoxication, known as bromism, makes it advisable to avoid bromide use in newborns and infants. Other anecdotal reports of toxicoderma in very young infants or newborns have been described, such as a papular eruption from G-CASF for collection of stem cells,104 a lichenoid reaction to ursodeoxycholic acid for neonatal hepatitis,105 and a maculopapular rash from diazoxide used for neonatal hyperglycemia (Fig. 20.9).106 Serum sickness-like reaction is rare in neonates but has been reported in infants as young as 5 months of age.107 It is characterized by fever, an urticarial eruption, and arthralgias. Lymphadenopathy may be present. In contrast to true serum sickness, there are no immune complexes, vasculitis, or renal impairment. The most commonly implicated drug has been cefaclor,107–109 but this eruption can be seen in infants with an unknown or presumably viral etiology (Fig. 20.10). Acute generalized exanthematous pustulosis (AGEP) is characterized by acute onset of fever and a widespread eruption of less than 5 mm sterile pustules on an erythematous background (Fig. 20.11). It is more common in older children and adults, but a few cases have been reported in infancy.110,111 Known etiologies of AGEP include exposure to systemic medications, mainly antibiotics, recent viral infection, vaccinations,111 and exposure to mercury.112 Histological study shows subcorneal pustules associated with dermal edema, and occasionally vasculitis, eosinophils in the superficial dermis, and focal keratinocyte necrosis. AGEP may be both clinically and histologically difficult to distinguish from pustular psoriasis. In AGEP, however, additional skin lesions such as purpura, vesicles, bullae, and target lesions, may be seen,111,112 there is an antecedent drug exposure in most cases, and the fever and pustules have a shorter duration than in pustular psoriasis. The condition fades away in a few days to weeks after abandoning the offending medication. Drug-induced hypersensitivity syndrome (DISH), also known as ‘drug reaction with eosinophilia and systemic symptoms’ (DRESS) is a serious drug reaction characterized by fever, skin rash, lymphadenopathy, hematological abnormalities and internal organ involvement, especially involving the liver.113–115 It is rare in this age group, although a fatal case in a 3-month-old infant has been reported, as well as a case in a premature infant,116,117 and another in a 22-month-old patient.118 The most commonly implicated drugs are anticonvulsants, particularly phenobarbital, phenytoin, carbamazepine, and lamotrigine, and antibiotics, e.g., trimethoprim-sulfamethoxazole, isoniazid, vancomycin, and amoxicillin.119 It usually occurs within 2 months of the introduction of the offending drug, most often in 2–6 weeks, but cases developing after 3 months of exposure have also been reported.120 However, symptoms can appear earlier and be more severe after re-exposure. The condition may be progressive even after discontinuation of the causative agent. Affected patients are frequently initially misdiagnosed as having less severe conditions such as streptococcal pharyngitis, mononucleosis, or other viral illness, both because the association with medication is not recognized and because early cutaneous manifestations are nonspecific maculopapular rashes. Facial and periorbital swelling are very characteristic and suggestive of DRESS. Diffuse erythroderma, desquamation and occasionally vesicles and bullae evolving into Stevens–Johnson syndrome or toxic epidermal necrolysis may occur (Table 20.2).121 Erythema multiforme-like targetoid lesions and features mimicking Kawasaki disease (KD) have been reported.119,122 In addition to the nearly universal fever, rash and lymphadenopathy, laboratory abnormalities are common, namely eosinophilia, atypical lymphocytosis and elevated transaminases. Lungs, kidneys, muscle, and the gastrointestinal system may also be affected.123 Thyroid disease has been reported in 6–10% of pediatric patients.120,123 Systemic steroids are the most commonly used therapy,113,114,123 and successful treatment with intravenous immunoglobulins in refractory-to-steroid cases has also been reported.124 Although the prognosis is generally good, the condition is potentially life-threatening and early diagnosis and discontinuation of the offending drug are very important. In cases of severe cutaneous or systemic involvement, admission to an ICU or a burn unit for careful monitoring is mandatory (Box 20.2).125 TABLE 20.2 Characteristic findings of severe cutaneous drug reactions
Immunologic, Reactive, and Purpuric Disorders
Introduction
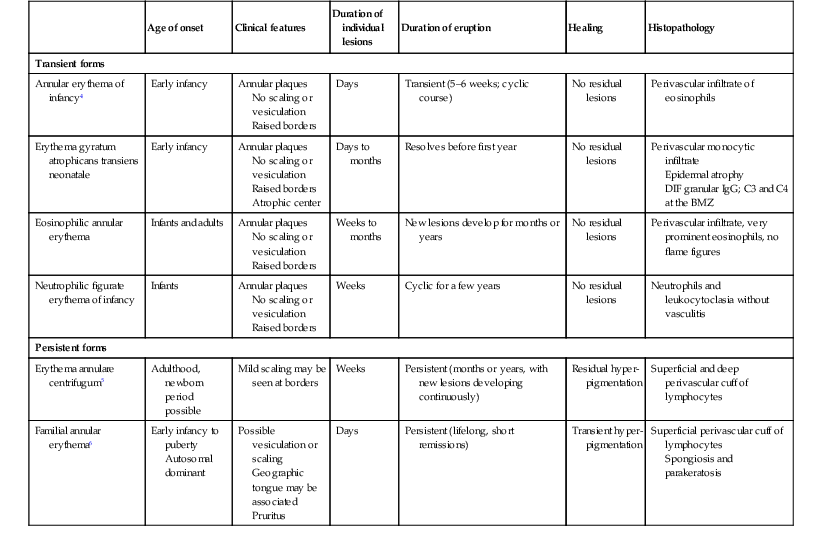
Annular erythemas
Age of onset
Clinical features
Duration of individual lesions
Duration of eruption
Healing
Histopathology
Transient forms
Annular erythema of infancy4
Early infancy
Annular plaques
No scaling or vesiculation
Raised borders
Days
Transient (5–6 weeks; cyclic course)
No residual lesions
Perivascular infiltrate of eosinophils
Erythema gyratum atrophicans transiens neonatale
Early infancy
Annular plaques
No scaling or vesiculation
Raised borders
Atrophic center
Days to months
Resolves before first year
No residual lesions
Perivascular monocytic infiltrate
Epidermal atrophy
DIF granular IgG; C3 and C4 at the BMZ
Eosinophilic annular erythema
Infants and adults
Annular plaques
No scaling or vesiculation
Raised borders
Weeks to months
New lesions develop for months or years
No residual lesions
Perivascular infiltrate, very prominent eosinophils, no flame figures
Neutrophilic figurate erythema of infancy
Infants
Annular plaques
No scaling or vesiculation
Raised borders
Weeks
Cyclic for a few years
No residual lesions
Neutrophils and leukocytoclasia without vasculitis
Persistent forms
Erythema annulare centrifugum5
Adulthood, newborn period possible
Mild scaling may be seen at borders
Weeks
Persistent (months or years, with new lesions developing continuously)
Residual hyper-pigmentation
Superficial and deep perivascular cuff of lymphocytes
Familial annular erythema6
Early infancy to puberty
Autosomal dominant
Possible vesiculation or scaling
Geographic tongue may be associated
Pruritus
Days
Persistent (lifelong, short remissions)
Transient hyper-pigmentation
Superficial perivascular cuff of lymphocytes
Spongiosis and parakeratosis
Annular erythema of infancy
Erythema annulare centrifugum
Familial annular erythema
Differential diagnosis of annular erythemas
Neonatal lupus erythematosus
Cutaneous findings
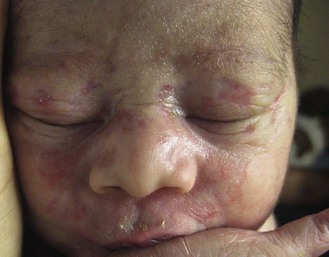
Extracutaneous findings
Etiology and pathogenesis
Laboratory tests and histopathology
Differential diagnosis
Course, management, treatment, and prognosis
Other collagen vascular disorders of the newborn and young infant
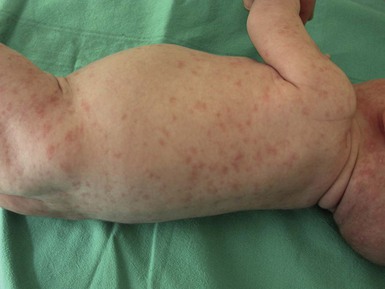
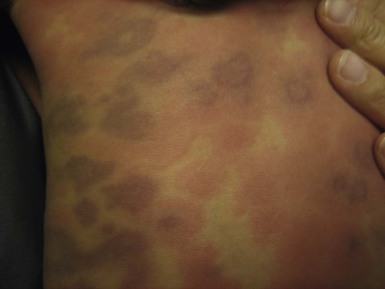
Drug eruptions
Acute generalized exanthematous pustulosis
Drug-induced hypersensitivity syndrome
DRESS
SJS/TEN
AGEP
Onset of eruption
2–6 weeks
1–3 weeks
48 h
Duration of eruption (weeks)
Several
1–3
<1
Fever
+++
+++
+++
Mucocutaneous features
Facial edema, morbilliform eruption, pustules, exfoliative dermatitis, tense bullae, and possible target lesions
Bullae, atypical target lesions, and mucocutaneous erosions
Facial edema, pustules, tense bullae, possible target lesions, and possible mucosal involvement
Histological pattern of skin
Perivascular lymphocytic infiltrate
Epidermal necrosis
Subcorneal pustules
Lymph node enlargement
+++
−
+
Hepatitis
+++
++
++
Other organ involvement
Interstitial nephritis, pneumonitis, myocarditis, and thyroiditis
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree

 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

Immunologic, Reactive, and Purpuric Disorders
20