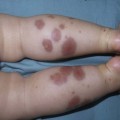
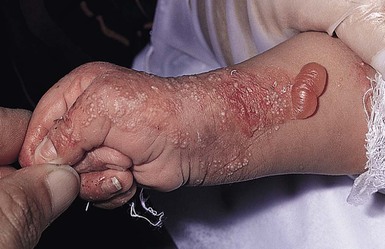
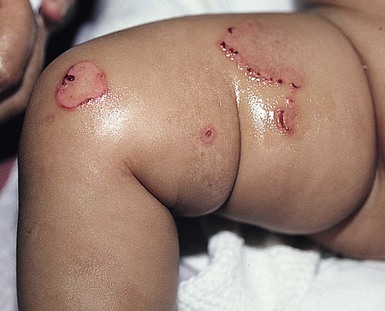
Anna L. Bruckner, Grainne M. O’Regan Epidermolysis bullosa (EB) is a family of rare, inherited disorders characterized by fragility of the skin and sometimes the mucosa in response to minor mechanical trauma. EB is caused by mutations in at least 14 genes1,2 that encode proteins of the basement membrane zone (BMZ) of the skin (Fig. 11.1). BMZ proteins are structural molecules involved in the adhesion of the epidermis and dermis. When an affected protein is absent or abnormal, adhesion strength is diminished and blistering occurs as a response to frictional stress. Data from the National Epidermolysis Bullosa Registry (NEBR) estimates the incidence of all forms of EB in the USA to be 20 cases/million live births.3 The classification of EB was revised in 2008 and utilizes the level of ultrastructural blistering as the primary categorical designation.1 The four major types of EB are: EB simplex (EBS) where cleavage occurs in the epidermis; junctional EB (JEB) with blistering through the BMZ; dystrophic EB (DEB) with cleavage arising in the superficial dermis, and Kindler syndrome where multiple splits can be seen. Each type is further divided into major and minor subtypes based on inheritance pattern, clinical findings, and the implicated molecular and genetic defects (Table 11.1). TABLE 11.1 The classification of epidermolysis bullosa AD, autosomal dominant; AR, autosomal recessive; LOC, laryngo-onycho-cutaneous; *Laminin-332 was formerly called laminin-5. Friction-induced blisters and erosions are the cardinal cutaneous features of EB. The distribution and extent of blistering varies depending on the disease subtype. Some forms of EB, such as EBS and dominant DEB (DDEB), are stereotyped as mild and often localized, whereas blistering in recessive DEB (RDEB) and JEB is often severe and generalized. It is important to remember that these generalizations best apply to older infants and children in whom a ‘mature’ EB phenotype has developed. In contrast, neonates with any type of EB may present with widespread cutaneous blistering and erosions. Mucosal erosions and absent or dystrophic nails can also be seen in neonates with all forms of EB. Thus, diagnosing the specific type and subtype based on clinical findings alone can be difficult in the first weeks of life, and biopsies are indicated (see below). Certain EB subtypes are associated with extracutaneous manifestations and complications that can begin to present in infancy and early childhood. Neonates with EB may present at birth with large ulcers, usually on the lower extremities, called congenital localized absence of skin (CLAS) (Fig. 11.2). The edges of such ulcers are well-demarcated and the base is red and shiny. Bart and colleagues4 originally described the association of CLAS, mucosal blistering, and nail dystrophy and proposed that this triad represented a distinct syndrome, later termed Bart syndrome. Since that description, however, CLAS has been reported as a presenting sign in all types of EB, and Bart’s original kindred was further examined and found to have DDEB.5 CLAS likely results from intrauterine friction, such as the leg rubbing against the uterine wall, and is not specific for any one type of EB. The use of the term Bart syndrome is now discouraged. With or without CLAS, neonates with EB develop friction-induced blistering and erosions after birth. Skin changes may initially correlate with areas traumatized during birthing, such as the scalp and face in cases of vaginal birth. In many instances, blistering may be generalized. After birth, areas that are most subject to friction, such as the hands, diaper area, extensor extremities, and back, are most likely to blister (Fig. 11.3). Intact blisters are filled with serous or hemorrhagic fluid. In JEB and RDEB, the bullae can be quite large, and the pressure of fluid within the blister cavity can cause the lesion to extend (Fig. 11.4). More superficial blisters may rupture easily, leaving open erosions. The blisters in EBS and JEB often heal without scarring, but macular hypo- or hyperpigmentation may be a transient change after blisters heal. DEB blisters heal with scarring that is often atrophic. Milia are most suggestive of DEB but can be seen in all forms of EB (Fig. 11.5). Granulation tissue, which is often described as ‘exuberant’ or ‘hyper-’ in JEB, can be seen in any EB erosion that has been slow to heal but is not often seen in the neonatal period. Oral involvement is most often seen in neonates with JEB or DEB but can be seen in EBS as well. Open erosions or intact vesicles can occur on the lips, gums, and palate. These lesions probably result from the trauma of sucking and may result in pain with feeding. In any form of EB, trauma to the periungual skin can result in nails that are absent, dystrophic, or may be shed. Blisters and erosions are at risk of becoming infected, signaled by the formation of crusts and foul-smelling or purulent drainage. Neonates with EB and extensive erosions are at risk for developing fluid and electrolyte abnormalities as well as sepsis. Neonates with EB and pyloric atresia may present with gastrointestinal obstruction at birth.6 In these cases, polyhydramnios and gastric distension may have been noted on prenatal ultrasound, and genitourinary strictures and obstruction may be present as well. Airway involvement is uncommon in neonates with EB, but can occur. Although it is most often associated with JEB-Herlitz (JEB-H), laryngeal involvement can occur in infants with certain forms of EBS and with RDEB.7 An early sign of blisters and erosions of the larynx is hoarseness, which can progress to stridor as airway obstruction worsens. Lethal acantholytic EB (LAEB) is an extremely rare subtype of EB.8–10 Affected infants present at birth with rapidly progressive erosions with a positive Nikolsky’s sign. Intact vesicles and erosions are not seen. Other common features are complete alopecia, anonychia, and oral erosions. All reported cases have been fatal in the neonatal period. Histology shows suprabasal acantholysis, but immunofluorescence studies will be negative for pemphigus and the typical causes of EB (see below). Ultrastructural studies show that keratin filaments fail to connect properly to desmosomes. Loss of function mutations in DSP that lead to truncation of the desmoplakin protein are causative. Plakophilin deficiency was initially described in 1997 as ectodermal dysplasia/skin fragility syndrome, by McGrath and colleagues.11 Children with this disorder develop spontaneous erosions and fissures, and the perioral area is commonly affected. Painful, fissured keratoderma, absent, sparse or wooly hair, nail dystrophy, and growth failure are other universal features.12 Biopsies of affected skin show acanthosis and hyperkeratosis and widened spaces between keratinocytes in the spinous layer. Desmosomes are small and poorly formed on electron microscopy. This is an autosomal recessive disorder caused by mutations in the PKP1 gene that encodes plakophilin. Basal EBS is the most common and often mildest form of EB. Most forms are transmitted in an autosomal dominant manner. Individuals with localized forms of EBS may not present for medical evaluation, thus precise estimates of the true prevalence of EBS are lacking. For example, the NEBR calculated the prevalence of EBS to be 4.60 cases/million, but also estimated that the registry probably captured only 10% of all individuals affected with EBS.3 Population-based data from Scotland show a prevalence of 28.6 cases/million in 1992.13 EBS, localized (EBS-loc, formerly Weber–Cockayne) is characterized by blisters of the hands and feet. Other than mild oral disease, extracutaneous involvement does not occur. Patients may first develop blisters at any age, including birth, but it is common for the first signs to present in the toddler years, or sometimes as late as adolescence, after a period of marked frictional stress. EBS, other generalized (EBS, gen-nonDM, formerly Koebner) presents at birth and is characterized by intraepidermal blistering in a generalized distribution. Oral involvement can be seen in infancy but improves with age. Atrophic scarring, dyspigmentation, and nail dystrophy may be seen. Extracutaneous involvement in this type of EB is uncommon. For both EBS-loc and EBS, gen-nonDM, life expectancy is normal and the overall prognosis is good. However, affected adults report that acral blistering can be painful and limits daily activities such as walking, thereby affecting quality of life.14,15 EBS Dowling–Meara (EBS-DM), on the other hand, is more severe and can even be fatal in the newborn period.14,16,17 Blistering presents at birth or within the first days of life. Skin involvement ranges from generalized to localization of blisters and erosions to areas of friction. A clinical clue to the diagnosis of EBS-DM is the occurrence of blisters in grouped or annular configurations (Fig. 11.6). This feature, however, is not always reliable and may not be present until after 1 year of age. Milia and atrophic scarring may occur, and nail dystrophy in which nails may be thickened, ridged, or shed, is fairly common. In childhood, a confluent palmoplantar keratoderma develops and becomes more prominent with age, persisting into adulthood. This hyperkeratosis can interfere with ambulation, and joint contractures may be a subsequent complication.14,17 Oral blistering is often present and varies in severity. Laryngeal involvement, presenting as hoarseness, has been reported, but unlike in JEB, does not signal a poor prognosis.18 Gastroesophageal reflux and constipation may also be seen as complications.19 The severity of EBS-DM generally improves over time, with less blistering in adolescence, and rare blistering in adulthood in many cases. Patients may also report reduced blistering during febrile illnesses. EBS with mottled pigmentation (EBS-MP) is a rare subtype.20 It is characterized by acral, nonscarring blistering that presents in infancy. In addition, 2–5 mm hypo- and hyperpigmented macules occur in a reticulate pattern around the neck, axillary, and groin areas. This mottled pigmentation may be congenital or can develop later in infancy. EBS with muscular dystrophy (EBS-MD), EBS with pyloric atresia (EBS-PA), and EBS, Ogna (EBS-Og) are all due to mutations in PLEC1, encoding plectin, although EBS-MD and EBS-PA are inherited recessively and EBS-Og is autosomal dominant.21 EBS-MD is a rare disorder that begins at or shortly after birth with mild generalized blistering and mucous membrane involvement. Other cutaneous findings include milia, atrophic scarring, and nail dystrophy.22 Nonmucosal findings can include respiratory involvement and dental enamel hypoplasia, leading to prominent caries. In most cases, progressive muscle weakness begins in adolescence or adulthood, although onset in infancy has been reported.23 The presentation of neonates with EB with pyloric atresia is discussed below. EBS-DM, EBS-loc, and EBS, gen-nonDM are dominantly inherited, and mutations in KRT5 and KRT14 can be identified in about 75% of cases.24 Keratin-5 and -14 are complementary intermediate filaments expressed in basal keratinocytes. They are crucial components of the cytoskeleton involved in maintaining structural integrity. In addition, they function in the adhesion of these cells to the BMZ by attaching to the hemidesmosome via plectin (see Fig. 11.1).25 Mutations in these keratin genes lead to abnormal keratin intermediate filaments with reduced ability to withstand frictional stress, resulting in basal cell cytolysis histologically and in blistering clinically. Genotype–phenotype correlations for mutations KRT5 and KRT14 suggest that alterations in the highly conserved boundary domains of the α-helical rod domain lead to the Dowling–Meara phenotype, whereas mutations in the less conserved regions produce milder phenotypes.24 Most cases of EBS-MP are due to a specific point mutation in the nonhelical amino-terminal head domain of keratin-5,26 although a case with a mutation in KRT14 has been reported.27 Exactly how these mutations lead to disruption of keratin intermediate filaments or pigmentation is unclear. Plectin, the affected protein in EBS-MD, EBS-PA, and EBS-Og anchors basal keratins to the hemidesmosome and is also expressed in the sarcolemma of muscle. The EBS-MD phenotype tends to correlate with mutations affecting the central rod domain of plectin, while EBS-PA is due to mutations outside this domain.28 The Ogna phenotype is due to a heterozygous missense mutation exerting a dominant negative effect. Junctional epidermolysis bullosa (JEB) is the least common type of EB. Data from the NEBR suggests an incidence of two cases/million live births.3 Inheritance of all forms of JEB is autosomal recessive, and a spectrum of clinical phenotypes can be seen. The JEB–Herlitz (JEB-H) and JEB with pyloric atresia (JEB-PA) subtypes, however, are often incompatible with survival beyond infancy. JEB-H is characterized by generalized skin and mucosal erosions with a propensity for wounds to form exuberant granulation tissue. Other associated findings include nail dystrophy (Fig. 11.7), dental enamel hypoplasia, and involvement of the respiratory epithelium. Blistering and erosions are seen at, or shortly after, birth (Fig. 11.8A), although the amount of blistering is not predictive of outcome, as infants with little skin involvement can do poorly. Severe involvement of the back and buttocks is common but not pathognomonic (Fig. 11.8B). In neonates who survive into infancy, the development of exuberant or hypergranulation tissue within erosions is a finding with high specificity for JEB-H. The central face, especially the periorificial areas (Fig. 11.9), periungual skin (paronychial inflammation), and nape of the neck are commonly affected. Absent or dystrophic nails or nail shedding are often present. Ocular erosions can also occur.7 Oral blistering and erosions are often present in the neonatal period and can make feeding difficult. The laryngeal and respiratory epithelia may also be affected, typically first presenting as a weak or hoarse cry.29 Stridor suggests worsening airway obstruction, which can be fatal. Involvement of the gastrointestinal and urinary epithelia can also occur. Anemia, probably due to a combination of iron deficiency and chronic inflammation, is common. Patients with JEB-H typically die in the first few years of life due to failure to thrive, sepsis, pneumonia or respiratory failure. Data from the NEBR reported a cumulative risk of death of ~45% in the first year of life,30 while the Dutch EB registry found that over 23 years, all 22 subjects with JEB-H passed away in the first 3 years, with the average age of death being 5.8 months.31 JEB-non-Herlitz, generalized (JEB-nH gen) has features similar to JEB-H, although it is generally less severe and its overall prognosis tends to be better.32 In the neonatal period, however, clinical signs and pathologic analysis may not fully distinguish between JEB-H and JEB-nH gen, and a final diagnosis may be based on long-term outcome and/or results of genetic mutation analysis. Generalized blistering and mucous membrane involvement are seen in the neonatal period, and both improve as the child ages. Healing with granulation tissue is less common and less pronounced than in JEB-H, but can occur. Cutaneous lesions may heal with atrophy and pigmentary alterations. In hair-bearing areas, progressive alopecia may result, although this finding is most pronounced in adulthood. Likewise, nail dystrophy can begin in infancy and is progressive and marked with increasing age. Laryngeal involvement may occur, sometimes resulting in respiratory failure. Although the overall prognosis for JEB-nH gen is generally better than for JEB-H, the fatalities in the neonatal period and infancy are not uncommon. Data from the NEBR reported a cumulative risk of death of 40% and 49% in the first year and 2 years of life, respectively.30 EB with pyloric atresia (EBS-PA or JEB-PA) presents at birth with upper gastrointestinal obstruction, most often affecting the pylorus. The degree of skin involvement is variable and ranges from extensive CLAS to normal skin, with the onset of blistering at several months of age.33 Prenatal signs of an affected fetus include polyhydramnios and abdominal masses appreciated on ultrasound.34 The ocular, respiratory, and urogenital epithelia are often affected. The prognosis for neonates with EB with pyloric atresia is generally poor, but nonlethal cases are reported.35 Infants with extensive erosions often expire quickly due to fluid and electrolyte imbalances as well as sepsis. If surgical correction of the pyloric atresia is successful, infants may still succumb to sepsis, feeding intolerance, failure to thrive, and respiratory or renal disease before 1 year of age.36 JEB-H and a subset of JEB-nH gen are caused by mutations in three genes, LAMA3, LAMB3, and LAMC2, which encode the constitutive subunits of the basement membrane protein laminin-332.37 As a component of the basal lamina, laminin-332 plays a critical role in the adhesion of keratin intermediate filaments to the basement membrane, and absent or abnormal laminin-332 leads to structural instability, manifesting as blistering through the lamina lucida. In the majority of JEB-H cases, mutations that result in premature termination codons and subsequently, absence of laminin-332 expression, are seen.38,39 In the case of JEB-nH, less deleterious mutations result in an abnormal laminin-332 protein that retains some degree of function.39 Mutations in COL17A1 which encodes type XVII collagen, a component of the hemidesmosome, also produce the JEB-nH gen phenotype, as well as JEB-non-Herlitz, localized.40,41 Mutations in three genes, ITGB4 (~80%), ITGA6 (~5%), and PLEC1 (~15%) produce the EB with pyloric atresia phenotype.42 DDEB may be more common than reported, as individuals with mild phenotypes may not seek medical attention. The NEBR estimated the prevalence of DDEB to be approximately 1/million,3 while Scottish data suggests a point prevalence of 1/14.6 per million.13 Infants with DDEB may present with CLAS and/or with blistering in the newborn period. As the child ages, the tendency for blistering decreases. Blistering most commonly affects areas predisposed to trauma, such as the hands, feet, elbows, and knees, and heals with atrophy and milia. Oral erosions occur but are often mild. Nail dystrophy is common and may be the only clinical sign of disease.43 Extracutaneous complications such as esophageal strictures are uncommon. Transient bullous dermolysis of the newborn is a distinctive variant of DEB that is benign and self-limited. Blistering begins at birth or in the newborn period and dramatically improves or even remits completely, usually within the first year of life. Most reported cases are sporadic, but familial cases are described.44,45 The incidence of RDEB is approximately 1–2/million live births.3,13 The current classification includes RDEB, severe generalized (RDEB-sev gen, formerly Hallopeau–Siemens), RDEB, generalized other (RDEB-O, formerly non-Hallopeau–Siemens), and several rare subtypes with more limited involvement (Table 11.1). In RDEB, blistering and erosions begin at birth and can be extensive. The compromised skin barrier is a risk for infection, and during the neonatal period sepsis is the most worrisome complication. Although death can occur, infants with RDEB generally do fairly well, especially compared with infants with JEB. Affected areas heal with scarring that can lead to the development of joint contractures over time. Recurrent scarring of the hands and feet leads to loss of the interdigital spaces and eventual contracture of the digits, called pseudosyndactyly or ‘mitten’ deformities (Fig. 11.10).46 Joint contractures and pseudosyndactyly may begin in the first year of life. Nail involvement is common with anonychia due to nail shedding and scarring of the nail bed at an early age. Extracutaneous involvement is common in RDEB, and generalized RDEB (and JEB) are effectively multisystem disorders. Mucosal involvement includes the gastrointestinal tract, ocular, and genitourinary system. Oral ulcers are painful and make eating difficult, limiting the ability of the child to take in adequate calories. Oral scarring leads to microstomia, ankyloglossia, and loss of the vestibule.47 Esophageal involvement is extremely common; erosions lead to progressive strictures that produce dysphagia and limit adequate caloric intake. Malabsorption is another gastrointestinal complication that is poorly understood.19 Anal erosions make defecation painful, exacerbating constipation. The cornea and conjunctiva are frequent sites of ocular injury. Recurrent abrasions and ulcers lead to scarring that can affect visual acuity.7 Urinary tract involvement may present with dysuria, hematuria, meatal stenosis, or even sepsis. Ureterovesical obstruction and hydronephrosis can occur.48 Failure to thrive is a common complication of generalized RDEB and results from unmet nutritional needs. Affected neonates and infants demonstrate failure to gain weight adequately before height velocity drops off. Chronic wound healing, blood and protein losses from erosions, and infection increase the body’s need for calories. Oral, esophageal and intestinal involvement hinders adequate intake and absorption of nutrients. In addition to causing growth failure, chronic malnutrition contributes to poor wound healing, the development of anemia, deficiencies of essential minerals and trace elements, and increased susceptibility to infection.49,50 Anemia likely results from both iron deficiency and poor iron utilization due to chronic inflammation.51 Additional long-term complications of RDEB that are most often seen in older children, adolescents or adults include renal failure, osteopenia and osteoporosis, cardiomyopathy, and aggressive squamous cell carcinomas.7,46 All forms of DEB are caused by mutations in the gene COL7A1, which encodes type VII collagen, the major component of anchoring fibrils.52 Each collagen VII molecule is made of three polypeptide chains that associate and assemble into a triple helix. Two collagen VII molecules align in an antiparallel fashion, and groups of these dimers form the anchoring fibrils that connect the lamina densa to anchoring plaques in the dermis. As with other forms of EB, genotype–phenotype correlations have emerged. In the case of RDEB-sev gen, homozygous or compound heterozygous mutations resulting in premature termination codons and a subsequent lack of collagen VII are most commonly found.52 Less deleterious mutations, such as missense mutations or the combination of a missense mutation and a premature termination codon, are found in other forms of RDEB. Finally, DDEB is typically caused by heterozygous mutations resulting in glycine substitutions in the triple helical region of COL7A1.52 These mutations allow a full-length protein to be formed, but the substitution leads to conformational instability of the mature collagen VII protein (the so-called ‘dominant negative’ effect). Kindler syndrome (KS) is a rare, autosomal recessive disorder in which trauma-induced blistering begins at birth or early in infancy. Based on clinical and laboratory features, it is often diagnosed as a form of EB in the neonate. However, patients with KS develop progressive poikiloderma and cutaneous atrophy in childhood.53 Photosensitivity is variable. Gingivitis and periodontitis are common mucosal findings. In addition, esophageal and urogenital stenosis can occur. KS is caused by mutations in FERMT1 which encodes fermitin family homolog 1 (kindlin-1), involved in cell–matrix interactions.54 When faced with a newborn with blisters and erosions, the most critical immediate task is to exclude infection, especially intrauterine herpes simplex infection. Other potential infectious etiologies include bacterial and fungal infections and neonatal varicella. Other genetic disorders can present with bullous or erosive lesions, although these are not the predominant disease features over time. Incontinentia pigmenti presents with vesicles, and neonates with epidermolytic ichthyosis and ankyloblepharon-ectodermal dysplasia-clefting syndrome55 can present with widespread erosions. Infants born to mothers with immune-mediated blistering disorders may also manifest transient blisters and erosions as neonates. In addition, infants may develop immune-mediated blistering disorders de novo (see below). Diffuse cutaneous mastocytosis may also produce extensive blistering and erosion in newborns and infants. Because an accurate diagnosis of the type and subtype of EB based on clinical findings alone is often impossible in the neonatal period, laboratory evaluation is crucial. Biopsy of an affected area for routine light microscopy is helpful in differentiating EB from other diagnoses. Additional biopsies for immunofluorescence microscopy (IFM) and, in some cavses, transmission electron microscopy (TEM) are needed to rapidly and accurately diagnose the EB type and subtype. This information improves the care provider’s ability to counsel and educate parents on the baby’s prognosis and to guide future therapy. While TEM was formerly considered the ‘gold standard’ for EB diagnosis, IFM testing is preferred now due to broader availability, rapid turnaround time, and lower cost.56 The initial and most common application of IFM involves a limited panel of antibodies to BMZ proteins to map the level of the blister, rapidly diagnosing the EB type. A more specific use of IFM is an expanded panel of monoclonal antibodies to proteins affected in EB, permitting the precise determination of the underlying molecular defect, based on the staining intensity (normal, reduced, or absent) of the targeted proteins (see Fig. 11.11 and Table 11.2). Unfortunately, many dermatopathology services do not maintain the antibodies needed for EB-specific IFM studies, and referral to a laboratory experienced in EB diagnosis may be required. The Stanford Dermatopathology Service (http://dermatopathology.stanford.edu) or Beutner Laboratories (http://www.beutnerlabs.com) are two US laboratories that are recommended.
Inherited and Acquired Blistering Diseases
Inherited blistering diseases
Epidermolysis bullosa
Major EB type
Major subtype
Minor subtype
Affected gene(s) (protein)
Typical inheritance
Simplex
Suprabasal
Lethal acantholytic EB
DSP (desmoplakin)
AR
Plakophilin deficiency (ectodermal dysplasia with skin fragility)
PKP1 (plakophilin)
AR
EBS superficialis
?
?
Basal
Localized (formerly Weber–Cockayne)
KRT5 (keratin 5), KRT14 (keratin 14)
AD
Dowling–Meara
KRT5 (keratin 5), KRT14 (keratin 14)
AD
Other generalized (formerly Koebner)
KRT5 (keratin 5), KRT14 (keratin 14)
AD
With mottled pigmentation
KRT5 (keratin 5)
AD
With muscular dystrophy
PLEC1 (plectin)
AR
With pyloric atresia
PLEC1 (plectin), ITGA6, ITGB4 (α6β4 integrin)
AR
Autosomal recessive
KRT14 (keratin 14)
AR
Ogna
PLEC1 (plectin)
AD
Migratory circinate
KRT5 (keratin 5)
AD
Junctional
Herlitz
–
LAMA3, LAMB3, LAMC2 (laminin-332*)
AR
Other
Non-Herlitz, generalized
LAMA3, LAMB3, LAMC2 (laminin-332*), COL17A1 (type XVII collagen)
AR
Non-Herlitz, localized
COL17A1 (type XVII collagen)
AR
With pyloric atresia
ITGA6, ITGB4 (α6β4 integrin)
AR
Inversa
LAMA3, LAMB3, LAMC2 (laminin-332*)
AR
Late onset
COL17A1 (type XVII collagen)
AR
LOC syndrome (Shabbir syndrome)
LAMA3 (laminin-332* α chain)
AR
Dystrophic
Dominant
Generalized
COL7A1 (type VII collagen)
AD
Acral
Pretibial
Pruriginosa
Nails only
Bullous dermolysis of the newborn
Recessive
Severe generalized (former Hallopeau–Siemens)
COL7A1 (type VII collagen)
AR
Generalized other (formerly non-Hallopeau–Siemens)
Inversa
Pretibial
Pruriginosa
Centripetalis
Bullous dermolysis of the newborn
Kindler syndrome
–
–
FERMT1 (kindlin-1)
AR
Clinical features of epidermolysis bullosa
Neonatal features
Specific epidermolysis bullosa subtypes
Suprabasal epidermolysis bullosa simplex
Basal epidermolysis bullosa simplex
Genetics and pathogenesis
Junctional epidermolysis bullosa
Genetics and pathogenesis
Dystrophic epidermolysis bullosa
Genetics and pathogenesis
Kindler syndrome
Differential diagnosis
Evaluating suspected EB
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Inherited and Acquired Blistering Diseases
11

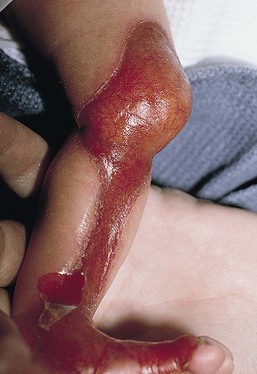
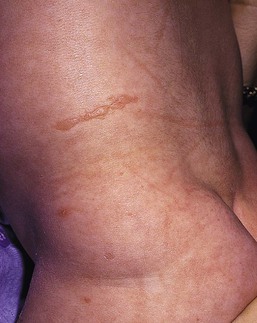
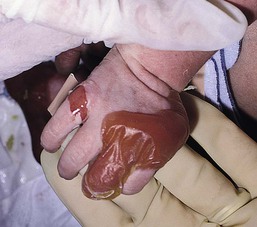
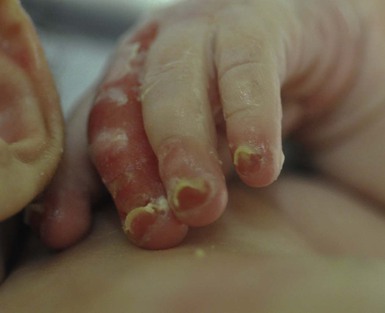
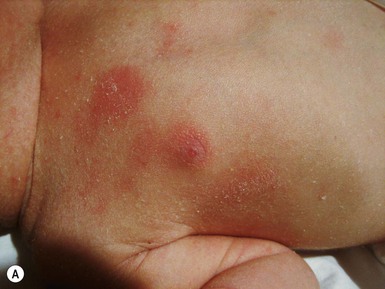
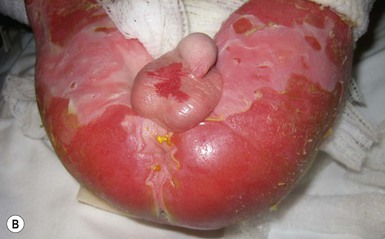
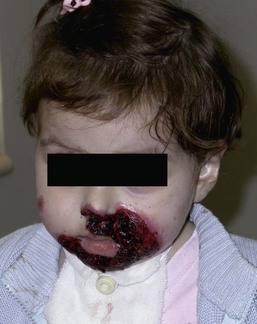
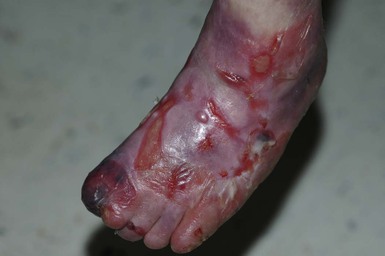
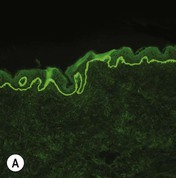
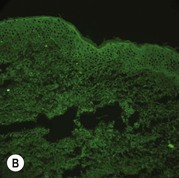
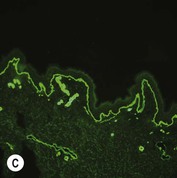
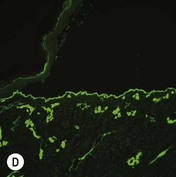
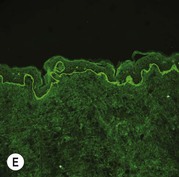
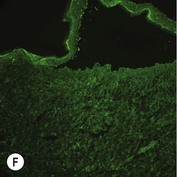
Figure 11.11 Immunofluorescence microscopy for EB. Linear staining with collagen VII antibody in normal skin (A, ×20) is absent in RDEB (B, ×20). Linear staining with collagen IV antibody in intact normal skin (C, ×20) is present in normal intensity on the floor of an induced blister in a patient with JEB (D, ×20). Linear staining with K140 (laminin-332 beta 3 chain antibody) in normal skin (E, ×20) is absent in JEB, Herlitz (F, ×20).