Abrams ML, Smidt A, Benjamin L, Chen M, Woodley D, Mancini AJ. Arch Dermatol 2011; 147: 337–41.
Epidermolysis bullosa acquisita
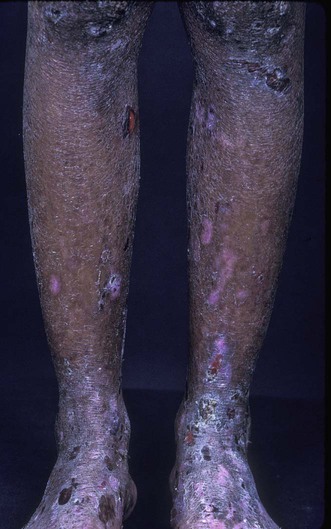
Specific investigations
 Skin biopsy and serum for direct and indirect immunofluorescence, respectively, to detect IgG or IgA class skin basement membrane-specific autoantibodies
Skin biopsy and serum for direct and indirect immunofluorescence, respectively, to detect IgG or IgA class skin basement membrane-specific autoantibodies
 Serum for ELISA to detect IgG or IgA class type VII collagen-specific autoantibodies
Serum for ELISA to detect IgG or IgA class type VII collagen-specific autoantibodies
 Gastrointestinal work-up for possible inflammatory bowel disease
Gastrointestinal work-up for possible inflammatory bowel disease
Congenital epidermolysis bullosa acquisita: vertical transfer of maternal autoantibody from mother to infant.
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


