Epidermolysis Bullosa
Juoni Uitto M.D., Ph.D.
Clinical Pearls
(JU)
Epidermolysis Bullosa Simplex
Inheritance
Autosomal dominant; very few autosomal recessive kindreds; keratin 5 and 14 genes on 12q and 17q, respectively
Prenatal Diagnosis
DNA analysis
Incidence
Approximately 10 to 30 cases per million live births; M=F
Age at Presentation
Weber-Cockayne—first to third decade
Generalized (Koebner)—birth to early infancy
Dowling-Meara—birth to first month of life
Pathogenesis
Mutations in keratin 5 and 14 genes produces a weakened basalar cytoskeleton (keratin intermediate filaments) and mechanical fragility with resultant intraepidermal bullae after trauma; plectin gene mutations affect hemidesmosomal protein and play a role in EB with muscular dystrophy and Ogna variant
Key Features
Weber-Cockayne
Skin
Palmoplantar bullae, callouses, hyperhidrosis; with/without pain, superinfection; worsening in summer months, warm temperatures
Generalized (Koebner)
Skin
Generalized bullae with/without superinfection; worsening in summer months, warm temperatures
Mouth
Mucosal erosions (mild)
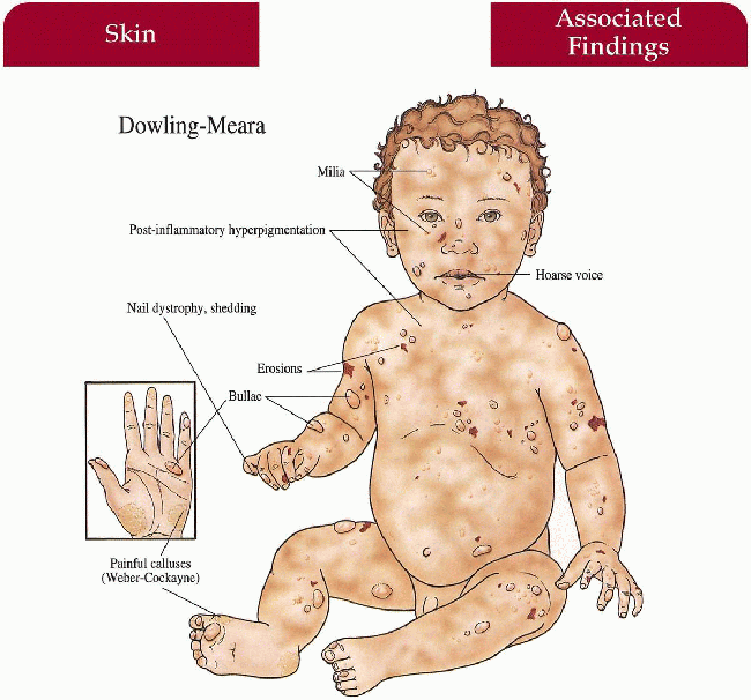
Dowling-Meara
Skin
Widespread bullae with “herpetiform” grouping of lesions—may have marked severity with increased morbidity, mortality in infancy; nonscarring, postinflammatory hyperpigmentation, milia; palmoplantar keratoderma with age
Nails
Dystrophy with shedding
Mucous Membranes
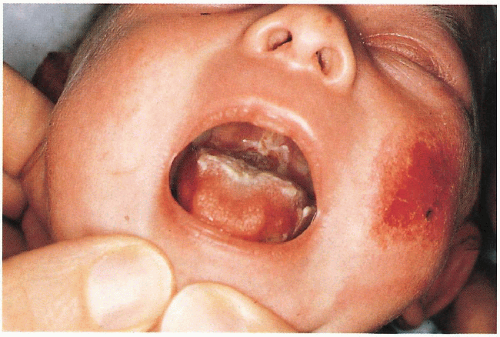
May have blistering, erosions in oral cavity (with/without secondary hoarseness) and esophagus
Differential Diagnosis
Generalized (Koebner) and Dowling-Meara
Neonatal herpes simplex virus (HSV)
Bacterial sepsis
Incontinentia pigmenti (p. 72)
Congenital syphilis
Bullous impetigo
Linear IgA disease
Laboratory Data
Skin biopsy for light microscopy (intraepidermal bullae), electron microscopy (clumped tonofilaments in Dowling-Meara) and immunomapping with monoclonal antibodies (see Junctional and Dystrophic EB, p. 204; 208)
Viral and bacterial cultures
DNA analysis with blood, buccal swabs
Management
Referral to dermatologist—diagnosis, trauma avoidance, wound care with whirlpool, modified Dakin’s solution, topical mupirocin, topical corticosteroids, cool environment with well-ventilated leather shoes; Dowling-Meara patients may improve with increased temperature
Referral to podiatry—silicone, plastizoate orthotics; thin, white cotton socks to decrease friction and sweat
Admit to neonatal intensive care unite (NICU) if severe blistering in neonate—monitor fluids, electrolytes, sepsis
Prognosis
Debilitating with normal life span; all types tend to blister less with aging
Dowling-Meara—significant morbidity, mortality in first few months of life
Clinical Pearls
EB is a group of mechano-bullous disorders with one unifying diagnostic feature: fragility of skin. The severity of skin involvement and association of extracutaneous findings produces a broad spectrum of clinical manifestations. This clinical complexity, compounded with a plethora of eponyms, has resulted in the identification of as many as 30 different subtypes (Lamprecht IA, Gedde-Dahl T. Epidermolysis Bullosa. In: Rimoin, Connor M, Pyeritz RE, et al. Principles and Practice of Medical Genetics, 4th edition. New York: Churchill Livingstone, 2002.). More recently, molecularly based classification recognizes four major categories (simplex, hemidesmosomal, junctional, and dystrophic), and distinct mutations in 10 different genes expressed at the cutaneous basement membrane zone have been identified. The level of expression of the mutated genes along the basement membrane zone and in extracutaneous tissues, the types and combinations of mutations, their positions along the mutated genes, and their consequences at the mRNA and protein levels, when superimposed on individuals’ genetic background, explain the tremendous phenotypic variability in this group of diseases. Precise classification of individual cases requires determination of the level of tissue separation by diagnostic immunoepitope mapping and/or transmission electron microscopy. Routine light microscropy may be helpful in identification of the simplex forms, but is often not diagnostic for the junctional and dystrophic subtypes. JU
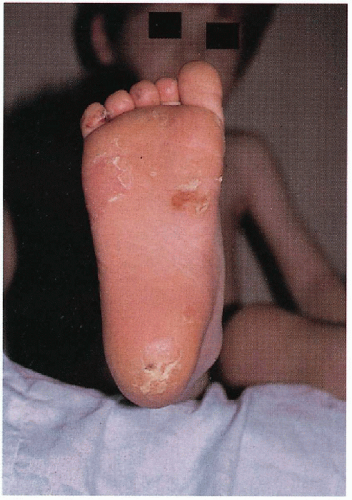 6.1. Bullae, callous, and erosions at points of friction on plantar surface of patient with Weber-Cockayne. (81) |
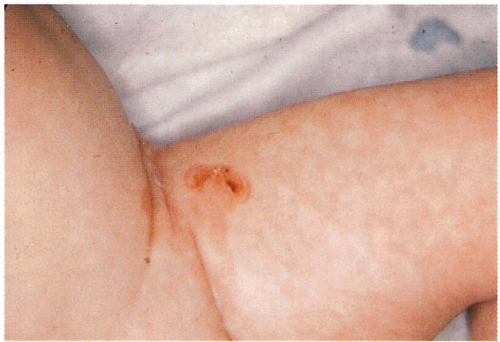
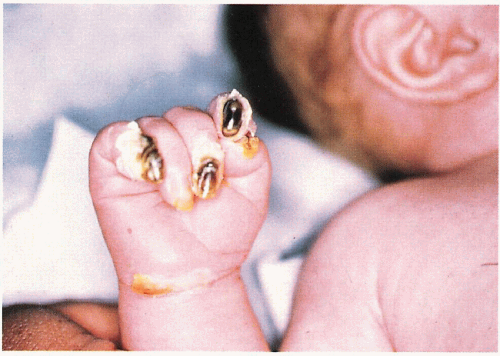 6.2. Severe nail dystrophy prior to shedding in infant with Dowling-Meara. (82) |
|
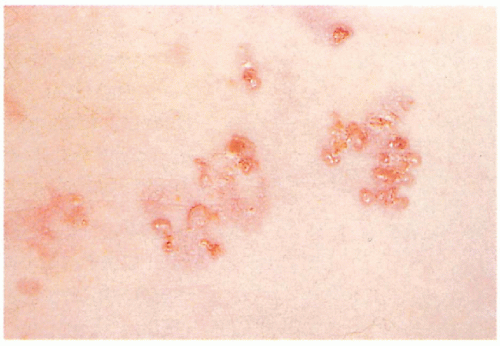 6.5. Close-up of grouped bullae in a patient with Dowling-Meara. (83) |
Junctional Epidermolysis Bullosa (JEB)
Inheritance
Herlitz variant—autosomal recessive; LAMA3, LAMB3 (80% of mutations), and LAMC2 genes encoding laminin 5 polypeptide chains on 1q32
Non-Herlitz variant—autosomal recessive; laminin 5 and COL17A1 on 1q32 and 10q24, respectively (other mutations identified)
JEB-pyloric atresia—autosomal recessive; ITGA6 (integrin α6) and ITGB4 (integrin β4) genes on chromosome 2 and 17q11, respectively
Prenatal Diagnosis
DNA analysis if mutation in family known; preimplantation determination of genotype at eight-cell stage
Incidence
Approximately 2 to 3 cases per million live births; M=F
Age at Presentation
Birth
Pathogenesis
Heterogeneous gene mutations encoding proteins at the dermal-epidermal junction are responsible for phenotype; basal cell adhesion to the basement membrane is altered resulting in a split within the lamina lucida
Herlitz—LAMA3, LAMB3, LAMC2 gene mutations coding for the polypeptide chains within laminin 5 responsible for anchoring filament development in the lamina lucida
Non-Herlitz—laminin 5 and COL17A1 (BP180-180 kDa bullous pemphigoid antigen) gene mutations, the latter encoding type 17 collagen (hemidesmosome protein in the lamina lucida)
JEB with pyloric atresia—ITGB4 and ITGA6 mutations encoding α6, β4 integrin, a hemidesmosome transmembrane protein complex
Key Features
Herlitz Variant
Skin
Generalized bullae without scarring, milia—mild atrophy with healing; nonhealing granulation tissue periorally, scalp, neck, upper trunk, nail folds, buttocks, pinnae of the ears
Nails
Absent (shed)
Ear-Nose-Throat
Dysplastic teeth with enamel defects, oral erosions, laryngeal involvement with hoarseness, croup, edema
Hematologic
Multifactorial anemia
Musculoskeletal
Growth retardation secondary to malnutrition
Non-Herlitz Variant
Skin
Bullae increased on extremities, heal with atrophic scarring; worse in warm environment
Nails
Dystrophy
Hair
Scarring alopecia
Otherwise similar to Herlitz without granulation tissue, anemia, growth retardation and poor prognosis (see Prognosis)
Junctional EBS-Pyloric Atresia
Skin/mucosa
Severe congenital blistering, mucosal erosions
Gastrointestinal
Pyloric atresia
Genitourinary
Hydronephrosis, renail failure secondary to stricture development
Differential Diagnosis
Epidermolytic hyperkeratosis (p.6)
Neonatal HSV
Bullous impetigo
Staphylococcal scalded skin syndrome
Toxic epidermal necrolysis
Laboratory Data
Bacterial, viral cultures
Skin biopsy for light, electron microscopy, immunofluoresence, cell culture
DNA analysis with blood, buccal swabs
Management
Herlitz Variant
JEB-Pyloric Atresia
Referral to surgeon/urologist-surgical release of GI/GU strictures, dilatation and gastrostomy
Prognosis
Herlitz variant—usually fatal by 3 to 4 years of age secondary to profound hypoproteinemia, anemia, and infection
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


