Disorders of Vascularization
Amy Paller M.D.
Kurt Hirschhorn M.D.
Judith Willner M.D.
Ilona Frieden M.D.
Clinical Pearls
(AP)
(KH)
(JW)
(IF)
Sturge-Weber Syndrome
Synonym
Encephalotrigeminal angiomatosis
Inheritance
Sporadic
Prenatal Diagnosis
None
Incidence
Rare; 2% to 11% of children with facial capillary malformation (0.3% to 0.6% of infants are born with facial capillary malformation) approximately 10% with V1 distribution; M=F
Age at Presentation
Birth; seizures at 1 to 2 years old
Pathogenesis
Defect in morphogenesis within the cephalic neural crest with subsequent abnormal vasculature in the upper facial dermis, choroid, and pia-arachnoid (mesoectodermal tissue); may be an autosomal lethal mutation surviving by mosaicism
Key Features
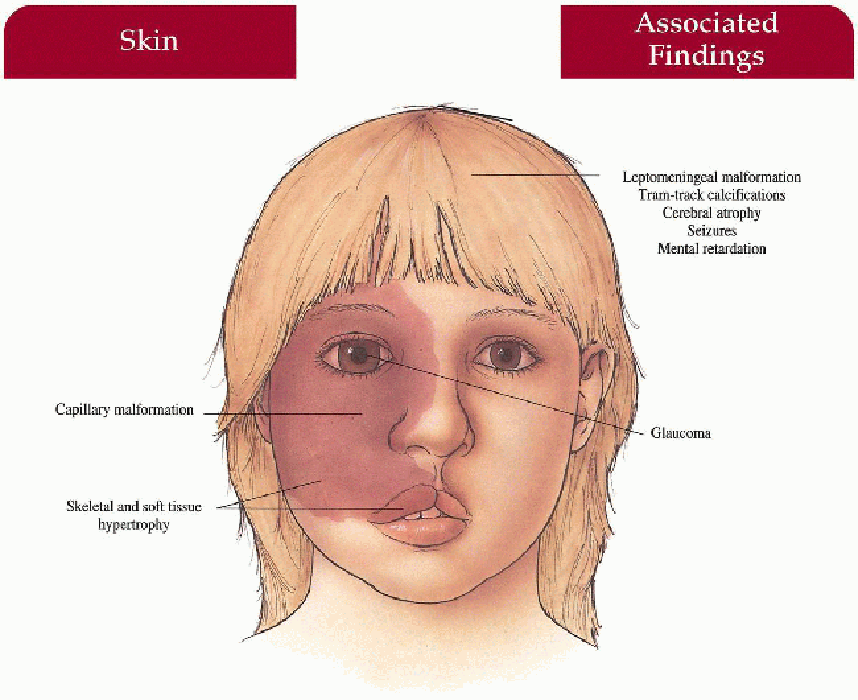
Skin
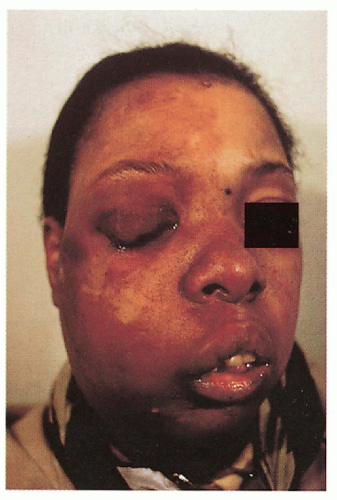
Facial capillary malformation
Trigeminal nerve distribution (V1 ± V2, V3)
Unilateral more common than bilateral
Progressive soft tissue and skeletal hypertrophy beneath malformation
Central Nervous System
Cerebral atrophy
Capillary, venous, and arteriovenous malformations ipsilateral to skin malformation in leptomeninges
Tram-track calcification in temporal and occipital cortex beneath leptomeningeal malformation
Seizures(> 70%), intellectual impairment (50%), hemiparesis, headache
Eyes
Choroid malformation
Ipsilateral glaucoma with secondary buphthalmos, visual loss
Differential Diagnosis
Periorbital hemangioma
Salmon patch
Laboratory Data
Magnetic resonance imaging (MRI) with gadolinium; computed tomography (CT) with contrast
Less than 6 months: positron emission tomography/single-photon emission computed tomography (PET/SPECT) scan may be useful
EEG (if above positive)
Management
Referral to dermatologist—laser treatment of capillary malformation
Referral to ophthalmologist—at presentation to detect and manage glaucoma, preserve vision
Referral to neurologist—seizure control
Referral to orthodontist/oral surgeon-treat complications of maxillary hypertrophy (occlusion deformity, cross-bite), gingival hypertrophy
Prognosis
If seizures are difficult to control, there is an increased frequency of intellectual impairment over time; visual impairment if glaucoma left untreated; normal life span
Clinical Pearls
Having a port wine stain in the distribution of the first branch of the trigeminal nerve is not uncommon; the minority will have Sturge-Weber syndrome … We only perform an MRI scan if the neonate or young infant shows evidence of neurologic disorder, most commonly seizure activity … Atrophic changes may be detected prior to calcifications on MRI … If MRI is initially normal and there is suspicion, I would repeat the scan at 2 to 3 years of age … I get an ophthalmologic examination … Seizures are often controlled at least partially with anticonvulsants, but may require hemispherectomy … Pulsed dye laser can be initiated early for the facial port wine stains. AP
|
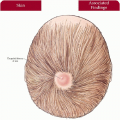
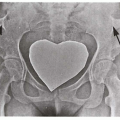
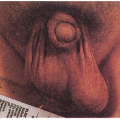
 3.1 Unilateral facial capillary malformation overlying marked soft tissue and bony hypertrophy. (1) |
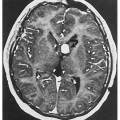
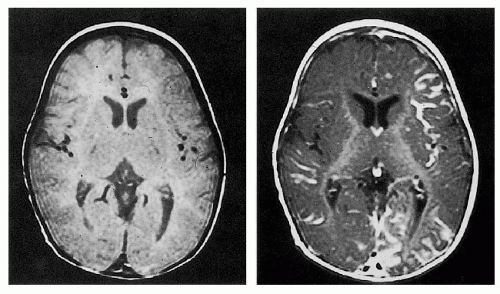 3.2. MRI with gadolinium revealing leptomeningeal malformation. (5) |
Klippel-Trenaunay Syndrome
Synonym
Angio-osteohypertrophy syndrome
Inheritance
Sporadic
Prenatal Diagnosis
None
Incidence
M > F
Age at Presentation
Birth; capillary malformation first sign
Pathogenesis
Increased vascular supply hypothesized as cause of limb hypertrophy
Key Features
Skin
Capillary malformation involving lower extremity (95%), upper extremity (5%), or combined (15%); unilateral in 85%
Musculoskeletal
Soft tissue, muscle and bony hypertrophy below cutaneous malformation with increased limb length and/or girth; rarely hypotrophic limb, polydactyly, syndactyly
Vascular
Superficial venous varicosities, phleboliths, deep venous malformation, arteriovenous fistulas (Parkes-Weber variant), superficial thrombophlebitis, deep vein thrombosis complicated by pulmonary embolism (rare)
Lymphatic
Lymphatic malformation with/without lymphedema
Differential Diagnosis
Proteus syndrome (p. 106)
Neurofibromatosis l (p. 82)
Maffucci syndrome (p. 118)
Capillary malformation
Laboratory Data
Doppler ultrasound in childhood
MRI, magnetic resonance angiography (MRA), venography, lymphography
Management
Compression wraps (young infant), stockings, pump
Routine measurement of limb length and circumference
Referral to orthopedist—correction of limb length discrepancy
Referral to vascular surgeon—treatment of symptomatic varicosities, arteriovenous fistulas
Referral to laser specialist—pulsed dye laser correction of capillary malformation
Prognosis
Progressive limb hypertrophy after birth-degree of hypertrophy dependent on extent of malformation, lymphedema and presence of A-V fistulas
High-output cardiac failure if A-V fistula untreated
Clinical Pearls
Probably the most useful intervention is the use of a compression garment once hypertrophy and varicosities begin to develop … Practically speaking, compression may be very difficult when you are treating a child wearing diapers … If we see a leg length discrepancy > 1 cm, we will consider a leg lift … Doppler imaging is very useful for following patients, particularly if an A-V fistula is present … Distal foot bacterial infections, paronychias, and warts are very common in a lymphedematous leg … May be confused with Proteus syndrome. AP
|
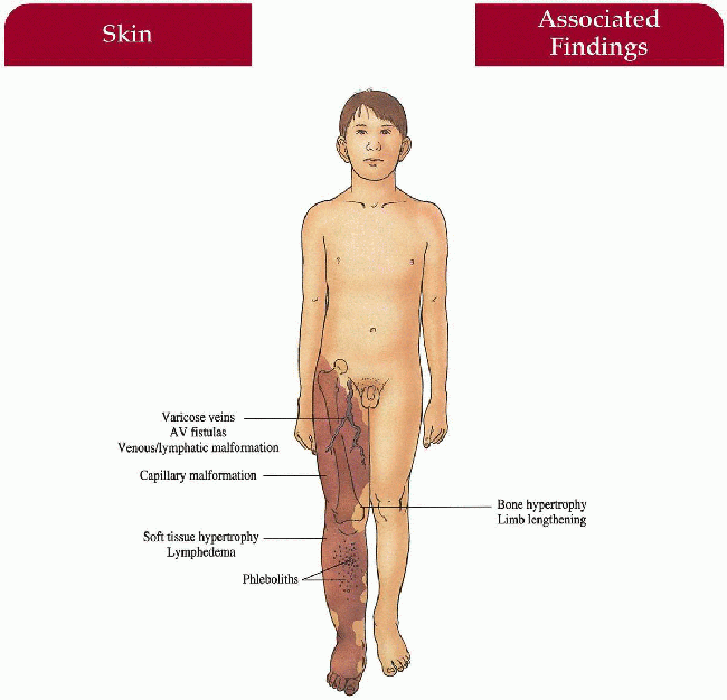
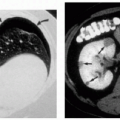
 3.3. Unilateral capillary malformation overlying massive venous-lymphatic malformation in a newborn. (35) |
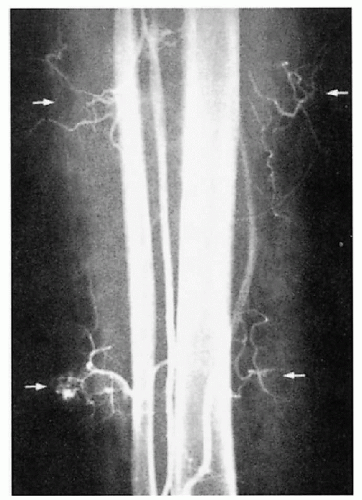 3.4. Angiography reveals discrete areas of arteriovenous malformation (arrows) in the calf of a patient. (40) |
Cobb Syndrome
Synonym
Cutaneomeningospinal angiomatosis
Inheritance
Sporadic
Prenatal Diagnosis
None
Incidence
Rare; spinal arteriovenous malformations (AVMs) more common without skin involvement, M=F
Age at Presentation
Birth; neurologic complications develop in early adulthood
Pathogenesis
Unknown
Key Features
Skin
Posterior thoracic/lumbar/limb vascular lesion in a dermatomal distribution overlying a corresponding segment of spinal cord
Central Nervous System
Fast-flow vascular malformation within the intramedullary (most common) spinal cord with secondary compression/anoxia-secondary pain, weakness, muscle atrophy, and sensation loss below the level of compression; bladder and sphincter dysfunction if malformation extensive; subarachnoid hemorrhage; malformation may involve vertebral body
Differential Diagnosis
Capillary malformation overlying a meningoencephalocele, spinal dysraphism, tethered spinal cord
Laboratory Data
MRI, MRA
Spinal angiography
Management
Referral to neurologist—complete neurologic examination
Referral to neurosurgeon—extirpation of lesion, embolization
Prognosis
Dependent on degree of symptomatology prior to intervention—may be irreversible
Clinical Pearls
MRI is my imaging technique of choice at time of presentation … Spinal malformations are particularly difficult to approach surgically … Embolization is feasible in certain cases. AP
|
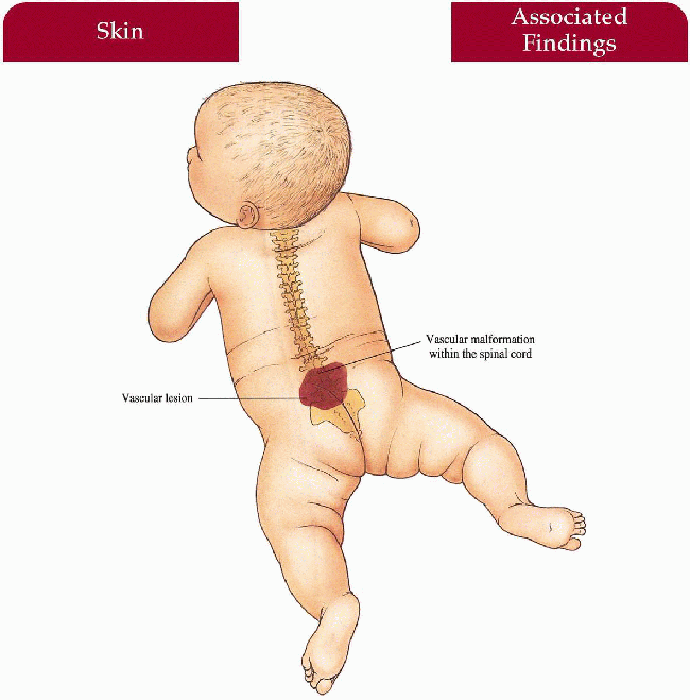
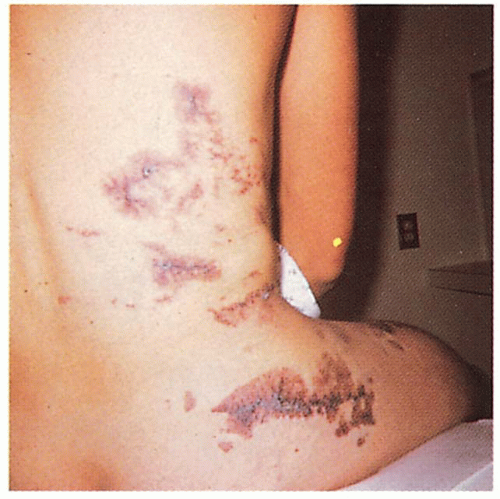 3.5. Extensive capillary malformation extending to involve the lumbosacral spine. (41) |
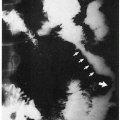
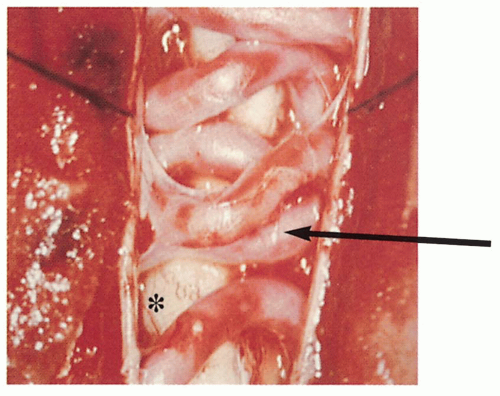 3.6. Arteriovenous malformation (arrow) within the spinal cord (*). (42) |
Proteus Syndrome
Synonym
Most likely includes Riley-Smith and Bannayan syndromes
Inheritance
Sporadic
Prenatal Diagnosis
None
Incidence
Rare; M=F
Age at Presentation
Birth; may develop over time
Pathogenesis
Mosaicism for an autosomal lethal mutation in the PTEN tumor suppressor gene has been reported in some patients
Key Features
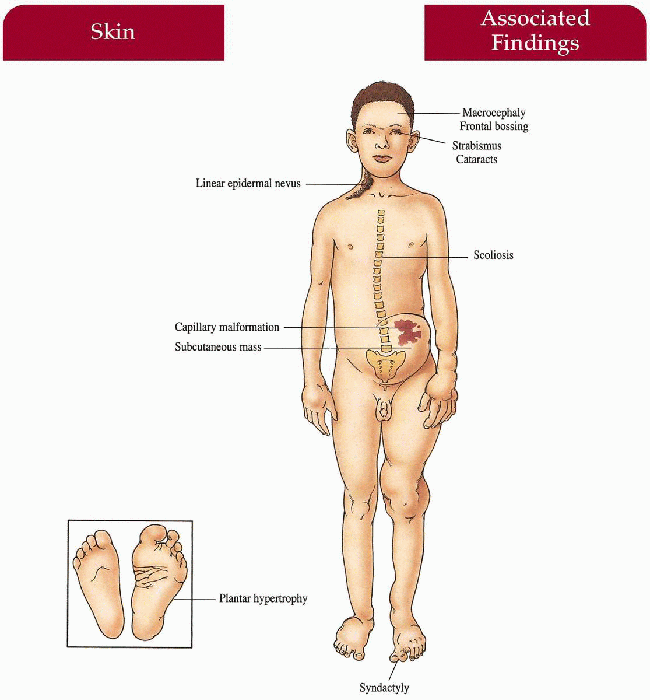
Skin
Soft, subcutaneous masses (likely lymphatic and venous-lymphatic malformations), lipomas, capillary malformations, linear epidermal nevi, plantar/palmar hyperplasia, varicose veins
Musculoskeletal
Macrocephaly, facial asymmetry, skull hyperostoses, frontal bossing, syndactyly, asymmetric soft tissue and bony hypertrophy of hands, feet, limbs; kyphoscoliosis Reports of cataracts, strabismus, microphthalmos, blindness, testicular tumors, penile hypertrophy
Differential Diagnosis
Klippel-Trenaunay-Weber syndrome (p. 102)
Maffucci syndrome (p. 118)
Neurofibromatosis I (p. 82)
Laboratory data
Bone x-rays/MRI
Management
Referral to plastic surgeon, orthopedic surgeon, physiatrist
Referral to symptom-specific subspecialist
Prognosis
Potential for gross deformity and debilitation exists; malignant potential unknown
Clinical Pearls
It is well documented that Joseph Merrick, the “Elephant Man,” had Proteus syndrome rather than neurofibromatosis … The striking plantar hyperplasia is distinct for this syndrome … The malformations can be so extensive that surgical correction is very difficult. KH, JW
|
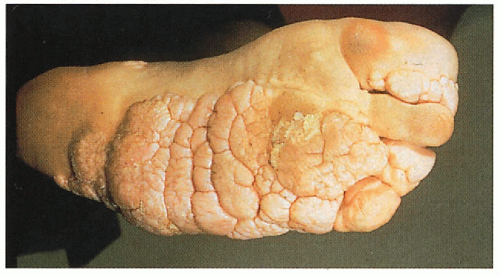 3.7. Characteristic plantar hyperplasia. (43) |
 3.8. Unilateral epidermal nevus with syndactyly and massive soft tissue and bony hypertrophy in a 2-year-old girl. (44) |
Beckwith-Wiedemann Syndrome
Synonym
Exomphalos-macroglossia-gigantism (EMG) syndrome
Inheritance
Most cases are sporadic; variety of transmissions described; p57 (KIP2) gene on 11p15.5
Prenatal Diagnosis
Ultrasound—macrosomia, visceromegaly, omphalocele visualized
DNA analysis in familial cases
Incidence
Unknown; M=F
Age at Presentation
Birth
Pathogenesis
Mutation in the p57 (KIP2) gene, a cyclin-dependent kinase inhibitor gene acting as a negative regulator of cell proliferation, leads to overgrowth of organs and increased susceptibility to malignancies
Key Features
Skin
Capillary malformation on mid-forehead, glabella, and upper eyelids extending to nose and upper lip in some cases
Mouth
Macroglossia
Ears
Linear earlobe crease, circular depressions on rim of posterior helices
Viscera
Hepatomegaly, splenomegaly, nephromegaly, pancreatomegaly, cardiomegaly
Omphalocele
Intestinal malrotation
Endocrine
Neonatal hypoglycemia with secondary neurologic sequelae if unrecognized
Musculoskeletal
Somatic gigantism—birth weight and length greater than 90th percentile
Hemihypertrophy (33%)
Neoplasms (10%)
Wilms’ tumor > hepatoblastoma > adrenal cortical carcinoma, rhabdomyosarcoma; increased in patients with hemihypertrophy
Differential Diagnosis
Down syndrome (p. 346)
Mucopolysaccharidoses (p. 318)
Congenital hypothyroidism
Laboratory Data
Blood glucose level
Abdominal and renal ultrasound at 3-month intervals through early childhood
Serum alpha-fetoprotein levels (screen for hepatoblastoma)
Management
Monitor blood glucose in the neonate
Complete physical examination
Referral to pediatric surgeon
Referral to appropriate subspecialist as necessary
Prognosis
Normal intellect as long as hypoglycemia well controlled in the neonate; typically large (approximately 2 standard deviations above the mean) adults leading normal lives; may have shortened life span secondary to neoplasm
Clinical Pearls
The phenotype is quite variable … The clinical diagnosis may be difficult in a preemie who lacks macrosomia … Severe neurologic sequelae may result from undetected hypoglycemia … Macroglossia can be surgically reduced if respiratory complications ensue … Ultrasound every 3 months in the first few years, every 6 months thereafter to rule out embryonic tumors linked to mutations in a paternally imprinted region on chromosome 11p. KH, JW
|
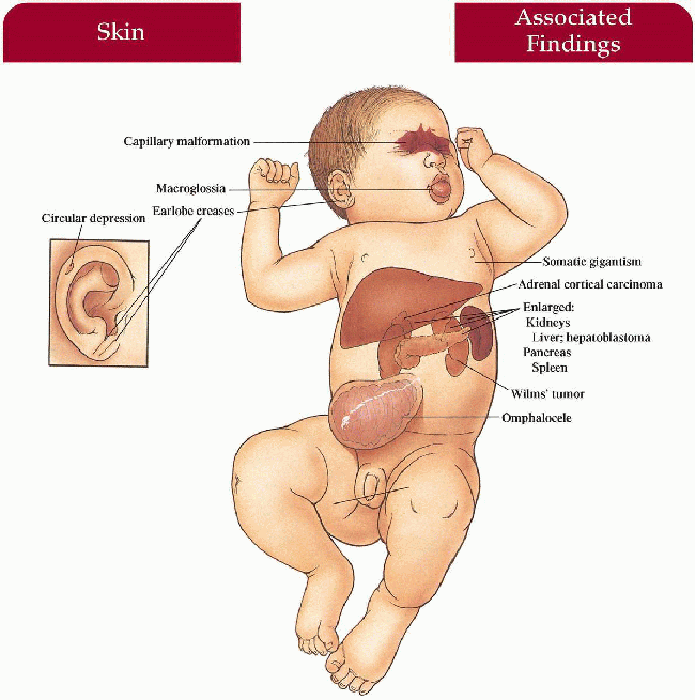
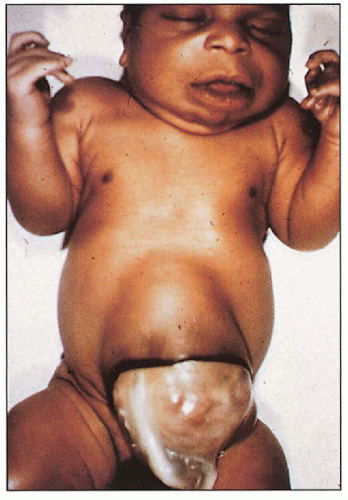 3.9. Newborn with omphalocele, macroglossia. (45) |
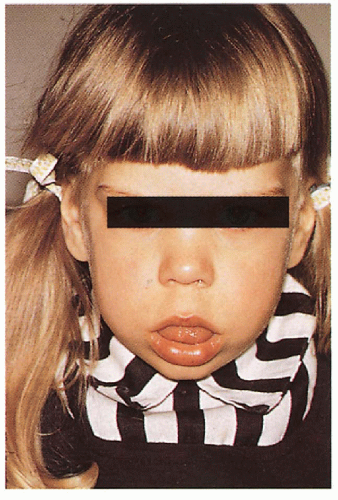 3.10. Open mouth with protruding macroglossia. (46) |
Von Hippel-Lindau Syndrome
Inheritance
Autosomal dominant; VHL gene on 3p26-p25
Prenatal Diagnosis
DNA linkage analysis or mutation detection
Incidence
1:50,000-60,000; M=F
Age at Presentation
Usually by the fourth decade of life
Pathogenesis
Mutation in the VHL tumor suppressor gene leads to phenotype
Key Features
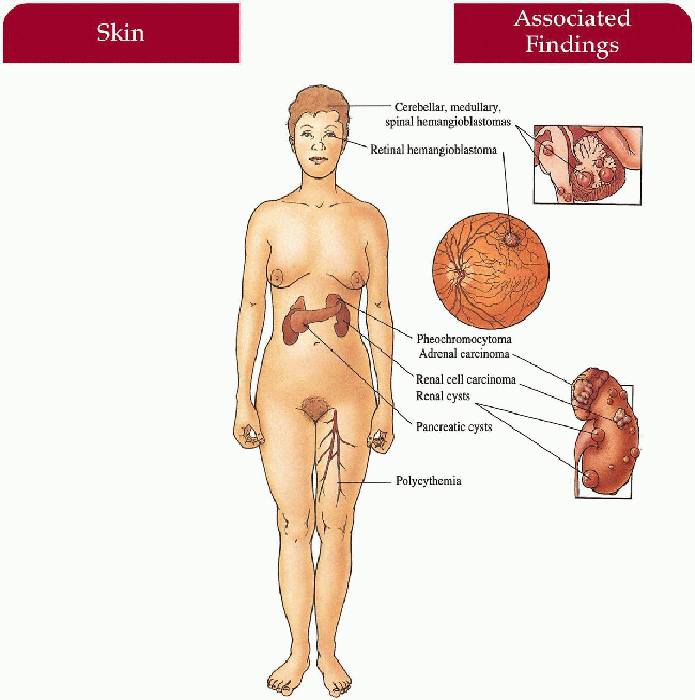
Eyes
Retinal hemangioblastomas with secondary visual impairment, blindness if untreated
Central Nervous System
Cerebellar > medullary, spinal cord hemangioblastomas with secondary signs of increased intracranial pressure (i.e., headache, vomiting, vertigo, ataxia, mental changes) or spinal cord compression (loss of sensation, proprioception, spastic paraparesis)
Kidneys
Renal-cell carcinoma, cysts
Endocrine
Pheochromocytoma, pancreatic cysts, adrenal carcinoma
Skin (< 5%)
Capillary malformation—head and neck
Hematologic
Polycythemia secondary to cerebellar hemangioblastoma and renal cell carcinoma production of erythropoietin
Differential Diagnosis
Cerebellar tumors
Laboratory Data
CT/MRI scan of brain/spinal cord
Abdominal CT/MRI scan
Urinary vanillylmandelic acid (VMA) level screen
Serum catecholamine level screen
Complete blood cell count
Management
Referral to ophthalmologist—photocoagulation or cryocoagulation of tumor
Referral to neurosurgeon/neurologist—surgical removal
Referral to urologist—surgical removal
All first-degree relatives: annual retinal examination, neurologic examination, regular screening with brain and abdominal scans, VMA and catecholamine screens
Prognosis
Premature death secondary to progressive growth of central nervous system (CNS) hemangioblastomas or metastatic renal cell carcinoma
Clinical Pearls
This is a progressive, universally fatal disease with death by the fourth decade … Hematuria is a common presenting sign … We screen with a sonogram of the belly, MRI and CT of the brain (to pick up both cerebellar tumors and calcifications), and an eye examination … DNA testing for mutations in the VHL gene is available and indicated for appropriate screening in children of affected patients and for confirmation of diagnosis … Angiography usually performed preoperatively … If kidney involvement, these patients may be candidates for renal transplantation. KH, JW
|
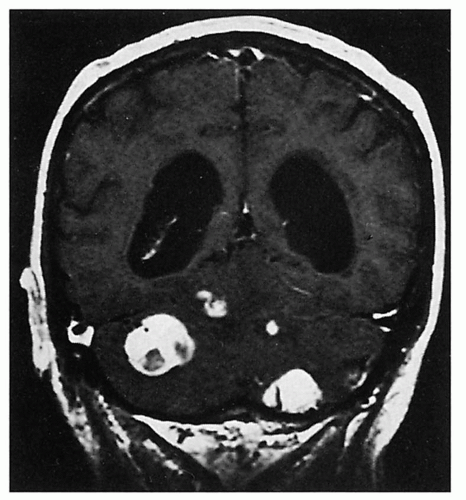 3.11. MRI shows four enhancing cerebellar hemangioblastomas. (36) |
 3.12. Retinal hemangioblastoma. (47) |
Ataxia-Telangiectasia
Synonym
Louis-Bar syndrome
Inheritance
Autosomal recessive; Ataxia-telangiectasia mutated (ATM) gene on 11q22-23
Prenatal Diagnosis
Amniocentesis: chromosomal breaks in amniocytes
Maternal serum: elevated alpha-fetoprotein levels
Molecular DNA analysis
Incidence
1:30,000-100,000 live births; M=F
Age at Presentation
Ataxia presents initially in second or third year of life when child begins to walk; telangiectasias by 3 to 6 years old
Pathogenesis
ATM gene codes for protein important in DNA repair, especially after ionizing radiation exposure; activates (via phosphorylation) repair mechanism utilizing a p53-dependent pathway that regulates apoptosis and cell cycle arrest; defective cellular and humoral immunity
Progressive depletion of Purkinje cells in the cerebellum
Key Features
Skin
Telangiectasias—bulbar conjunctiva first with subsequent ear, eyelid, cheeks, neck, upper chest, and flexor forearms involvement; progeric facies with decreased subcutaneous fat, atrophy, sclerosis; granulomas, café au lait macules
Hair
Canities
Central Nervous System
Cerebellar ataxia, progressive nystagmus, slurred speech, oculomotor apraxia, growth retardation, intellectual impairment
Sinopulmonary
Recurrent viral or bacterial infections, progressive respiratory impairment
Endocrine
Ovarian dysgenesis, insulin-resistant diabetes
Neoplasms
Lymphoreticular, breast carcinoma (heterozygotes)
Differential Diagnosis
Hereditary Hemorhagic Telangiectasia Syndrome (p. 114)
Generalized essential telangiectasia
Bloom syndrome (p. 234)
Laboratory Data
Increased T-suppressor cells, decreased T-helper cells
Alpha-fetoprotein (AFP) elevated
Immunoglobulin (Ig) A, IgG2, IgE decreased or absent
Management
Evaluate family members for carriers of ATM mutation-increased incidence of breast cancer and lymphoid malignancies (dominant negative missense mutations most closely associated with carrier morbidity)
Referral to hematologist—oncologist-intravenous γ-globulin, malignancy management
Referral to pulmonologist/infectious disease specialist
Referral to neurologist
Avoid x-rays, radiotherapy, bleomycin
Sun avoidance, sunscreen may help prevent progeric changes
Prognosis
Most patients confined to wheelchair by 10 years of age; premature death secondary to lymphoreticular malignancy or infection in the second decade of life
Clinical Pearls
Major caretaker is the neurologist … While patients usually present to neurology initially, I follow a wheelchair-bound 20-year-old patient who developed telangiectasias in the first years of life and did not develop ataxia until 8 years old … While x-irradiation is a problem, patients seem to be UV sensitive as well … We tend to cover them with sunscreen and protective clothing … If radiotherapy used to treat lymphoma, one must use very small fractionated doses … Family members who are carriers are at increased risk of developing malignancies, and may show chromosomal breaks after irradiation … Risk of breast cancer in female heterozygotes is 5 times that of the normal population … Ultrasound and good physical examination are theoretically safer than mammograms for screening these women. AP
|
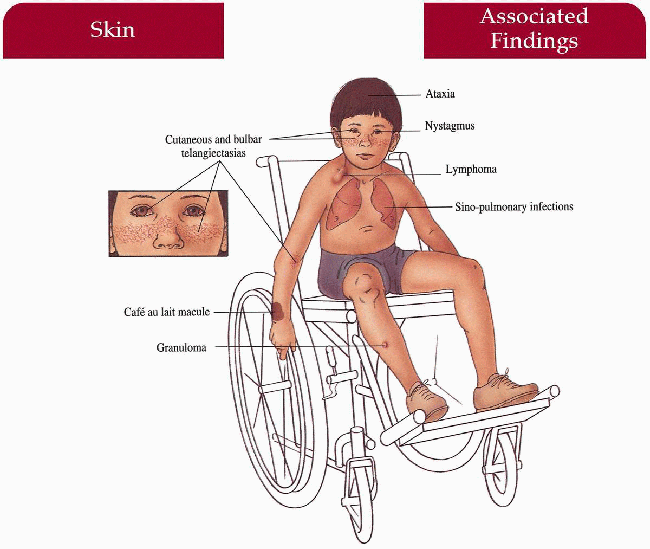
 3.13. Telangiectasias involving bulbar conjunctiva and cheek. (1) |
 3.14. Telangiectasias on external ear. (48)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|