Epidermal and Epidermal–Dermal Adhesion: Introduction
|
The cell–cell and cell–basement membrane adhesion in the epidermis provides the skin with its resistance against environmental influences; epidermal integrity is required for protection of the entire organism against mechanical, physical, or microbial insults. The major cellular structures involved are the desmosomes at cell–cell junctions in the epidermis and the hemidesmosome–basement membrane adhesion complexes and related structures at the dermal–epidermal junction. Ultrastructurally, the hemidesmosome closely resembles one-half of the desmosome; however, at the molecular level, these two structures are distinct. Both represent specifically organized assemblies of intracellular and transmembrane molecules. The desmosome anchors cytoskeletal filaments to cell–cell junctions, and the hemidesmosome anchors cytoskeletal filaments of basal epithelial cells to the basement membrane. Our knowledge of the desmosomal, hemidesmosomal, and basement membrane molecules has expanded drastically in recent years due to the great power of both molecular genetics and proteomics. After keratinocyte transmembrane proteins were initially identified as autoantigens in pemphigus and pemphigoid, a multitude of molecules have now been characterized at both protein and gene levels, and their expression, regulation, and functions have been discerned. The antigenic epitopes in different autoimmune blistering skin diseases have been carefully mapped and, to date, mutations in at least 24 different genes have been shown to underlie heritable disorders of epidermal or epidermal–dermal adhesion in humans and mice. Morphologic, molecular, and functional aspects of these adhesion structures are delineated in this chapter.
Epidermal Adhesion
The desmosome (or macula adherens) is the major cell adhesion junction of the epidermis, serving to anchor apposing keratinocyte cell surface membranes to the intracellular keratin intermediate filament network. Desmosomes are present in almost all epithelial tissues, including the oropharynx, gut, liver, heart, lung, bladder, kidney, prostate, thymus, cornea, and central nervous system, although the desmosomal protein isoforms and intermediate filament proteins vary by cell type.1 The primary role of desmosomes in epidermal cell adhesion is evidenced by the histologic findings in epidermal spongiosis, or intercellular edema, in which adjacent keratinocytes remain attached to each other only at desmosomal junctions (Fig. 53-1). These “intercellular bridges” served as the earliest description of desmosomes in tissues, their observation made possible with the advent of light microscopy in the nineteenth century.2 The development of electron microscopy techniques in the mid-twentieth century allowed for higher resolution micrographs that revealed the ultrastructure of these intercellular junctions. Even in the twenty-first century, the desmosome still remains best defined by its electron micrographic appearance, with an electron-dense midline in the intercellular space halfway between apposing plasma membranes, sandwiched by two pairs of electron-dense cytoplasmic plaques (Fig. 53-2A).3 The intercellular space between plasma membranes was called the desmoglea (from the Greek for “desmosomal glue”), because it was presumed to provide the adhesion that kept cells together.4
Figure 53-1
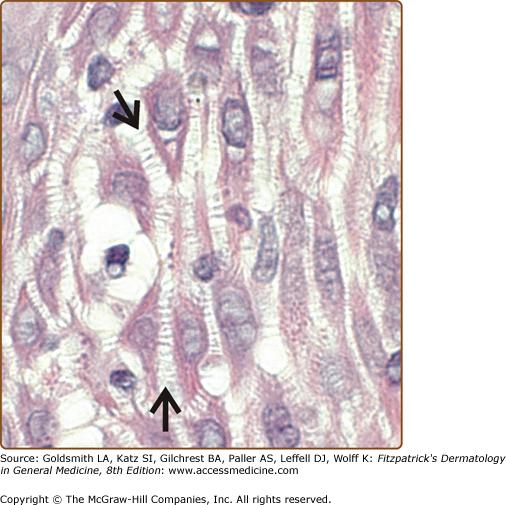
Desmosomes are the primary cell adhesion junction in the epidermis. Epidermal spongiosis, or intercellular edema due to inflammation, causes separation of keratinocytes, which remain attached by intercellular bridges representing desmosomal junctions (arrows). (Photo used with permission from John Seykora, MD, PhD.)
Figure 53-2
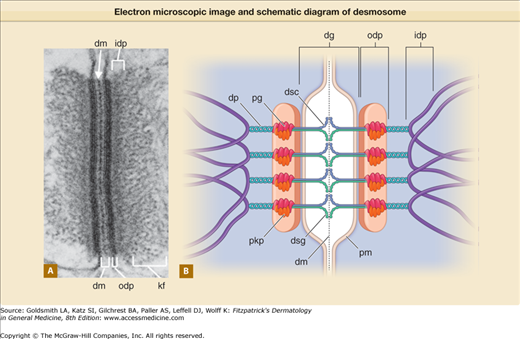
Electron microscopic image (A) and simplified schematic diagram (B) of a desmosome (drawing not to scale). dg = desmoglea; dm = dense midline; dp = desmoplakin; dsc = desmocollin; dsg = desmoglein; idp = inner dense plaque; kf = keratin filaments; odp = outer dense plaque; pg = plakoglobin; pkp = plakophilins; pm = plasma membrane. (Electron micrograph used with permission from Kathleen Green and with permission of Elsevier, adopted from Yin T, Green KJ: Regulation of desmosome assembly and adhesion. Semin Cell Develop Biol 15:665, 2004.)
In anchoring the cell surface to the intermediate filament network, desmosomes create a three-dimensional scaffolding of proteins that extend from the cell surface all the way to the nuclear envelope. This scaffolding is critical to stabilize epithelia in the face of shear stress or external trauma. Early morphologic studies led to the perception of the desmosome as a static structure, a “spot weld” functioning only to maintain intercellular adhesion.5 Over the last three decades, the individual proteins comprising the desmosome have been biochemically characterized and cloned, shedding light on both the dynamic nature of the desmosome structure and the diversity of desmosomal protein function.
Desmosomal proteins fall into three major categories: (1) desmosomal cadherins (desmogleins and desmocollins), (2) armadillo family proteins (plakoglobin and plakophilins), and (3) plakins (desmoplakin, envoplakin, and periplakin). Additional proteins, such as Perp, ninein, kazrin, and corneodesmosin, have also been localized to epidermal desmosomes.6–9 Immunogold electron microscopy labeling studies have further refined our understanding of how these molecular components of desmosomes are ordered within the desmosome ultrastructure10 (Fig. 53-2B). The desmosomal cadherins are transmembrane proteins whose extracellular amino-terminal domains interact to form the trans-adhesive interface between cells, represented by the electron-dense midline of the desmoglea. Intracellularly, approximately 10–20 nm from the plasma membrane, the outer dense plaque contains the desmosomal cadherin cytoplasmic tails, plakoglobin, the desmoplakin amino-terminal domain, and plakophilin. Approximately 40–50 nm from the plasma membrane, the desmoplakin carboxyl-terminus interacts with keratin intermediate filaments, producing the inner dense plaque. The biochemistry, expression pattern, and diseases of each desmosomal component are discussed in further detail below. Although the specific physiologic roles and pathophysiologic mechanisms affecting many of the desmosomal proteins remain under active investigation, current knowledge clearly indicates the importance of desmosomes and their components beyond just cell adhesion.
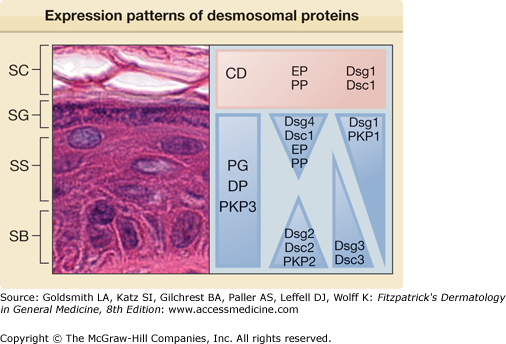
Desmogleins and desmocollins are part of the cadherin superfamily of transmembrane glycoproteins. Members of this superfamily, which includes the adherens junction protein E-cadherin, mediate calcium-dependent adhesion in a variety of epithelial tissues. In humans, there are four desmoglein (Dsg) isoforms and three desmocollin (Dsc) isoforms, each with varying expression patterns within and among epithelia.11–13 Within normal human epidermis, Dsg1, Dsg4, and Dsc1 are expressed predominantly in the differentiated cells of the superficial epidermis, while Dsg2, Dsg3, Dsc2, and Dsc3 are expressed more strongly in the basal and/or suprabasal layers14,15 (Fig. 53-3). Among different epithelial tissues, Dsg1 and Dsg3 expression are largely limited to stratified squamous epithelia in the skin and oropharynx, as well as thymic epithelial cells. Dsg3 is also strongly expressed in squamous cell carcinomas and other head and neck cancers, where it has been proposed as a potential molecular target for therapy.16 Dsg2 is the major desmoglein isoform in most simple and transitional epithelia, as well as cardiac myocytes.17 Dsg4 is prominent in desmosomes of the hair follicle, testis, and prostate.18,19 The expression pattern of the human desmocollins is less well characterized. In normal tissues, Dsc1 expression is largely limited to skin and oral epithelia, while Dsc2 is more widely expressed in most desmosome-containing epithelia and is the only desmocollin isoform in cardiac tissue.11 Dsc3, like Dsg3, is most strongly expressed in the stratified squamous epithelia of the skin and oropharynx,20 although UniGene data suggest weaker expression in a variety of other epithelial tissues. Desmocollin switching has been described in colorectal cancer, with downregulation of Dsc2 and upregulation of Dsc1 and Dsc3.21
The extracellular domains of the desmosomal cadherins consist of four cadherin repeats plus an extracellular anchor domain, each separated by a calcium-binding motif. All cadherins are synthesized as preproproteins, which include an amino-terminal signal sequence and propeptide. The propeptide is thought to prevent intracellular aggregation of newly synthesized cadherins within the secretory pathway of the cell. Proprotein convertases in the late Golgi network cleave the cadherin propeptide, thereby producing the mature adhesive protein.22 Although the crystal structure of the desmosomal cadherins remains unsolved, studies comparing the structure of desmosomes analyzed by cryo-electron tomography of vitreous sections with the solution structure of the classical cadherins suggest common mechanisms of intercellular adhesion, in which a conserved amino-terminal tryptophan residue on one cadherin molecule interacts with a hydrophobic acceptor pocket in the first extracellular domain of another cadherin molecule on a neighboring cell to form the trans-adhesive interface.23–26 Additionally, cadherins may participate in “cis” interactions with cadherin molecules on the same cell through their membrane proximal domains, which may facilitate desmosome assembly. Desmosomal cadherins can engage in both homophilic (i.e., Dsg–Dsg or Dsc–Dsc) as well as heterophilic (i.e., Dsg–Dsc) interactions, although heterophilic interactions are thought to contribute most to strong intercellular adhesion.27–29
The cytoplasmic domains of the desmosomal cadherins are less conserved. Desmocollins have a full length “a” and a shorter “b” splice isoform. The cytoplasmic domains of desmoglein and desmocollin “a” isoforms bind plakoglobin, and some desmoglein and desmocollin isoforms may also directly bind plakophilins30,31 (Fig. 53-2B). Increasing data suggest that desmosomal cadherins are not just adhesive molecules but may also actively regulate intracellular signaling, transcription, and other cellular processes.1 Consistent with this, desmocollin 3-deficiency in mice causes embryonic lethality at day 2.5 before implantation, indicating a central role for desmocollin 3 in early tissue morphogenesis independent of its desmosomal adhesive function.32
In dermatology, the desmosomal cadherins are best known for their role as autoantigens in the immunobullous disease pemphigus (see Chapter 54). The desmoglein 3 gene was originally discovered and cloned because it was the autoantigen in pemphigus vulgaris33 (Fig. 53-4A). Since then, all of the desmosomal cadherins have been associated with human autoimmune, infectious, and/or genetic diseases (summarized below and in Table 53-1).
Figure 53-4
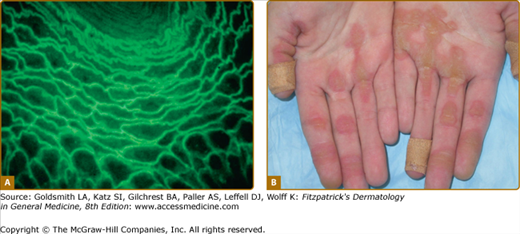
Desmosomal proteins are pathophysiologic targets in human autoimmune and genetic diseases. A. Indirect immunofluorescence on monkey esophagus with pemphigus serum that contains autoantibodies to desmoglein 3. The cell surface–intercellular pattern staining is diagnostic of pemphigus. B. Striate palmoplantar keratoderma is associated with haploinsufficiency of desmoglein 1 or desmoplakin.
Desmosome Component | Autoimmune Target | Genetic Target | |
|---|---|---|---|
Desmosomal cadherins | Desmoglein 1a | Pemphigus foliaceus, pemphigus vulgaris | Striate PPK (AD) |
Desmoglein 2 | ARVC (AD) | ||
Desmoglein 3 | Pemphigus vulgaris, paraneoplastic pemphigus | ||
Desmoglein 4 | Pemphigus foliaceus,b pemphigus vulgarisb | Hypotrichosis (AR); Monilethrix (AR) | |
Desmocollin 1 | IgA pemphigus (subcorneal pustular dermatosis) | ||
Desmocollin 2 | ARVC (AR and AD) | ||
Desmocollin 3 | Pemphigus vulgaris | Hypotrichosis (AR) | |
Desmosomal plaque proteins | Desmoplakin I/II | Paraneoplastic pemphigus | Striate PPK (AD); Carvajal syndrome (AR): diffuse PPK, wooly hair, left ventricular cardiomyopathy; lethal acantholytic epidermolysis bullosa (AR); skin fragility–wooly hair syndrome (AR): PPK, wooly hair, nail dystrophy |
Other plakinsc | Paraneoplastic pemphigus | ||
Plakoglobin | Naxos disease: diffuse PPK, wooly hair, ARVC (AR) | ||
Plakophilin 1 | Skin fragility and ectodermal dysplasia (AR) | ||
Plakophilin 2 | ARVC (AD) | ||
Stratum corneum desmosome protein | Corneodesmosin | Hypotrichosis simplex of the scalp (AD) |
Desmoglein 1 is the target of pathologic proteolytic cleavage in the infectious disorders bullous impetigo and staphylococcal scalded skin syndrome, as well as the inherited ichthyosis associated with Netherton syndrome (see Chapter 49).34,35 Cleavage of desmoglein 1 by staphylococcal exfoliative toxin occurs between extracellular domains 3 and 4.36 Pathogenic autoantibodies to desmoglein 1 are found in pemphigus foliaceus, mucocutaneous pemphigus vulgaris, and paraneoplastic pemphigus (see Chapters 54 and 55). Most pathogenic pemphigus foliaceus autoantibodies bind the first two extracellular domains of desmoglein 1, overlapping sites that are critical for desmoglein trans-adhesion.37–39 Autosomal dominant mutations causing haploinsufficiency of desmoglein 1 result in palmoplantar keratoderma (PPK; see Chapter 50). The PPK is classically striate (Fig. 53-4B), occurring at sites of greatest trauma or friction, but focal and diffuse forms have also been described.40–42
Desmoglein 2 has been implicated in human cardiovascular disease as a cause of autosomal dominant arrhythmogenic right ventricular cardiomyopathy (ARVC).43 The lack of skin phenotypes in affected patients indicates that desmoglein 2 is not required for epidermal adhesion, likely due to compensatory adhesion from other more highly expressed epidermal desmosomal cadherin isoforms.
Desmoglein 3 is the target of pathogenic autoantibodies in mucosal and mucocutaneous pemphigus vulgaris and paraneoplastic pemphigus (see Chapters 54 and 55.) Most pathogenic autoantibodies in pemphigus vulgaris target the amino-terminal extracellular (EC1–2) domains of desmoglein 3.37,44,45 Because desmoglein 3 deficiency in mice phenotypically resembles autoimmunity to desmoglein 3 in mucosal pemphigus vulgaris with oral suprabasal erosions, pemphigus autoantibodies are thought to cause loss of desmosomal cadherin function.46 More recent research has focused on signaling pathways activated after binding of pemphigus vulgaris autoantibodies to keratinocytes, as several biochemical inhibitors have been shown to prevent blistering in a neonatal mouse passive transfer model47 (discussed in further detail in Chapter 54).
Desmoglein 4 mutations have been described in rare autosomal recessive forms of hypotrichosis and monilethrix.48–51 One patient demonstrated transient scalp erosions during the first 2 weeks of life. Most of the mutations in Dsg4 are frameshift or nonsense mutations that would be predicted to lead to haploinsufficiency, although missense mutations have also been reported,52 Desmoglein 4 immunoreactivity is observed in pemphigus vulgaris and pemphigus foliaceus sera,51 but subsequent studies have attributed this to cross-reactivity from Dsg1 autoantibodies.53
Desmocollin 1 is the target of autoantibodies in the subcorneal pustular dermatosis of IgA pemphigus (see Chapter 54.)
Desmocollin 2, like desmoglein 2, is mutated in both autosomal dominant and recessive forms of ARVC, with no epidermal phenotype in affected patients.54,55
Desmocollin 3 mutations were found in one Pakistani kindred with autosomal recessive hypotrichosis.56 Autoantibodies to desmocollin 3 have also been found in pemphigus vulgaris patients, particularly those with vegetative lesions.57
Plakoglobin (also known as γ-catenin) directly binds the cytoplasmic tails of the desmogleins and desmocollin “a” isoforms, as well as E-cadherin, the major transmembrane protein of adherens junctions in epidermal keratinocytes.58,59 It is expressed throughout all layers of the epidermis and is ubiquitously expressed in all epithelia. Plakoglobin, like plakophilin, is a member of the armadillo gene family, characterized by a conserved protein structure with head and tail domains that flank multiple homologous arm repeats.60 Various domains of plakoglobin modulate its binding to the desmosomal cadherins.61–63 Other domains bind to desmoplakin, thus linking desmogleins and desmocollins to desmoplakin.64 Plakoglobin can also localize to the nucleus, where it may modulate gene transcription by TCF/LEF family members.65,66 Although most depictions of the desmosome show both plakoglobin and plakophilin binding to desmoplakin, biochemical studies suggest that these interactions are mutually exclusive.67 Likely, the armadillo family proteins play more dynamic roles in recruitment of desmosomal proteins to the plaque, similar to α-catenin in adherens junctions.68,69
Plakoglobin mutations result in Naxos disease, an autosomal recessive syndrome of diffuse PPK, wooly hair, and arrhythmogenic right ventricular cardiomyopathy, the latter of which may present in late childhood to adolescence.70
Desmoplakin exists in two RNA splice variants, desmoplakin I and II.71,72 It is unknown whether different isoforms perform different cellular functions, although human disease mutations suggest that desmoplakin I is required for normal desmosomal function.73 Desmoplakin is part of the plakin gene family,74 which includes the hemidesmosomal proteins bullous pemphigoid antigen 1 and plectin, as well as envoplakin and periplakin. Similar to other desmosomal components, desmoplakin is a modular protein, with different modules fulfilling different functions. The central part of one desmoplakin molecule coils around the central part of another to form a rod-like center. The amino-terminal head domain binds to plakoglobin,64 and the carboxyl-terminal tail binds to keratin.75 Therefore, desmoplakin provides the major link between the keratin filaments and the desmosomal plaque. Desmoplakin also plays a critical role in development independent of its function in desmosomes, as desmoplakin-null mice die early in embryogenesis at E6.5, before desmosomes are formed.76
A broad range of desmoplakin mutations have been associated with human disease, leading to variable phenotypic combinations of PPK (striate or diffuse), dilated cardiomyopathy (left or right), wooly hair, nail abnormalities, and/or skin blisters.52 Haploinsufficiency of desmoplakin leads to autosomal dominant striate PPK.77 Autosomal recessive mutations in desmoplakin were described in three Ecuadorian families with Carvajal syndrome, consisting of diffuse PPK, wooly hair, and arrhythmogenic left ventricular cardiomyopathy.78 A Naxos-like syndrome of PPK, wooly hair, and ARVC occurs with the p.R1267X nonsense mutation, which affects only the desmoplakin I splice isoform, indicating that desmoplakin II is not sufficient to restore normal desmosomal function in epidermis.73 Lethal acantholytic epidermolysis bullosa (widespread epidermolysis, generalized alopecia, anonychia, and neonatal teeth) was attributed to compound heterozygous mutations in desmoplakin that caused loss of the desmoplakin tail.79 Additionally, desmoplakin autoantibodies are observed in paraneoplastic pemphigus sera (see Chapter 55).
Plakophilins, like plakoglobin, can localize both to the plasma membrane as well as the nucleus, although their function outside of desmosomal adhesion is not well characterized. Plakophilins directly bind to desmoplakin, and may also directly bind keratins and desmosomal cadherins, which is thought to aid in clustering and lateral stability of the desmosomal plaque.30,31,67 Plakophilin 1 also associates with the eukaryotic translation initiation factor eIF4A1 in the mRNA cap complex, where it functions to regulate translation and cell proliferation.80
Mutations in plakophilin 1 cause ectodermal dysplasia-skin fragility syndrome, suggesting a role for plakophilin 1 in epidermal morphogenesis as well as adhesion.81 Plakophilin 2 mutations are the most common cause of autosomal dominant ARVC.82 Currently, there are no known human diseases associated with plakophilin 3.
Envoplakin and periplakin are desmosomal plaque proteins expressed in the superficial layers of the epidermis. Both proteins incorporate into the corneodesmosomes of the stratum corneum. Mice deficient in envoplakin, periplakin, and involucrin do not demonstrate adhesion defects, but instead show impaired desquamation and epidermal barrier function.83 Envoplakin and periplakin autoantibodies are characteristic of paraneoplastic pemphigus sera (see Chapter 55).
Corneodesmosin is a secreted glycoprotein that incorporates into corneodesmosomes and is also expressed in the inner root sheath of the hair follicle. Heterozygous mutations in corneodesmosin are associated with an autosomal dominant hypotrichosis simplex of the scalp.84 Loss of cohesion in the inner root sheath and aggregates of proteolytically cleaved corneodesmosin around the hair follicle are observed in scalp biopsies of affected patients.
Epidermal–Dermal Adhesion
Basement membranes underlie epithelial and endothelial cells and separate them from each other or from the adjacent stroma. Another form of basement membrane surrounds smooth muscle or nerve cells. The physiologic functions of basement membranes are diverse: in the various organ systems they provide support for differentiated cells, maintain tissue architecture during remodeling and repair, and, in some cases, acquire specialized functions, including the ability to serve as selective permeability barriers (e.g., the glomerular basement membrane or the blood–brain barrier) or acquire strong adhesive properties, like the basement membrane at the dermal–epidermal junction, or that surrounding smooth muscle cells, which provide the tissues resistance against shearing forces. All of these characteristics of basement membranes are also used during development and differentiation of multicellular organisms (Box 53-1).
|
Ultrastructurally, basement membranes most often appear as trilaminar structures, consisting of a central electron-dense region, known as the lamina densa, adjacent on either side to an apparently less-dense area, known as the lamina lucida or lamina rara. The lamina lucida directly abuts the plasma membranes of the adherent cells. The relative size of each of these regions varies in different tissues, among the basement membranes of the same tissue at different ages, and as a consequence of diseases. For example, the trilaminar glomerular basement membrane in humans varies from 240 nm to 340 nm in width, whereas the bilaminar basement membrane of the dermal–epidermal junction measures 50 nm to 90 nm. This ultrastructure demonstrates that basement membranes serve as substrates for the attachment of cells and fix their polarity. Their continuity throughout the various organ systems stabilizes the tissue orientations and provides a template for orderly repair after traumatic injury. Major disruptions in the basement membrane result in the formation of scar tissue and the loss of function in that area.
Different basement membranes contain both common and unique components. All share a basic network structure to which specific macromolecules have been appended. These molecules are responsible for the specialized functions of different basement membranes. The basic constituents of these structures are collagen IV, laminins, nidogens, and proteoglycans of the perlecan type, which all are highly conserved, although the isoforms, the number of the polypeptide subunits, and their individual structures vary among species.84,85 The nearly ubiquitous distribution of heparan sulfate proteoglycans in all basement membranes suggests that these serve as selective permeability barriers in multiple locations, including the kidney and the blood–brain barrier. Ultrafiltration may be especially important during development and morphogenesis of all tissues.
Basement membranes also provide physical separation between epithelia and their underlying extracellular matrices. This barrier is especially important in the containment of tumors. With the exception of certain cells of the immune system, nonmalignant cells seldom cross a basement membrane. In contrast, malignant cells bind the basement membrane, regionally disrupt its structure, and migrate through the rupture. Laminins and integrins mediate the tumor-cell binding, and the basement membrane dissolution is catalyzed at least in part by metalloproteases produced by the tumor cell. The absence of distinguishable basement membranes in tumor biopsies is used as an indicator of malignancy, and there appears to be a high correlation between metastasis and basement membrane disruption. These observations underline the importance of the basal lamina as an obstacle to cell migration.
By binding biologically active signaling molecules, basement membranes regulate a multitude of biologic events. The constituent proteoglycans can bind growth factors that can be released from the complexes. Thus, the basement membranes are potent regulators of cell adhesion and migration, cytoskeleton and cell form, cell division, differentiation and polarization, and apoptosis.85
Ultrastructure of the Dermal–Epidermal Junction
The dermal–epidermal junction is an example of a highly complex form of basement membrane,86,87 which underlies the basal cells and extends into the upper layers of the dermis (Fig. 53-5A). This basement membrane is continuous along the epidermis and skin appendages, including sweat glands, hair follicles, and sebaceous glands. The dermal–epidermal junction can be divided into three distinct zones. The first zone contains the keratin filament–hemidesmosome complex of the basal cells and extends through the lamina lucida to the lamina densa. The plasma membranes of the basal cells in this region contain numerous electron-dense plates known as hemidesmosomes. The intracellular architecture and organization of the basal cells are maintained by keratin intermediate filaments, 7 nm to 10 nm in diameter that course through the basal cells and insert into the desmosomes and hemidesmosomes. External to the plasma membrane is a 25-nm to 50-nm-wide lamina lucida that contains anchoring filaments, 2 nm to 8 nm in diameter, originating in the plasma membrane and inserting into the lamina densa. The anchoring filaments can be seen throughout the lamina lucida but they are concentrated in the regions of the hemidesmosomes. Thus, the anchoring filaments appear to secure the epithelial cells to the lamina densa.
Figure 53-5
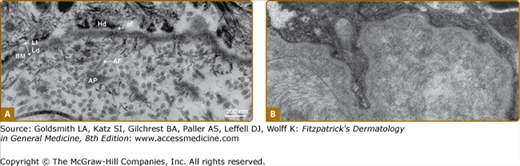
A. Ultrastructure of the human dermal–epidermal junction as visualized by transmission electron microscopy after standard fixation and embedding protocols. af = anchoring filament; AF = anchoring fibril; AP = anchoring plaque; BM = basement membrane; Hd = hemidesmosome; Ld = lamina densa; Ll = lamina lucida. (Bar = 200 nm.) B. Ultrastructure of the human dermal–epidermal junction by transmission electron microscopy following protocols using high-pressure fixation and embedding techniques. Note the dense character of both the basement membrane and the subjacent papillary dermis. Anchoring filaments, anchoring fibrils, and anchoring plaques are not distinguishable. (Both photos used with permission from Douglas R. Keene, MD, Shriners Hospital, Portland, Oregon.)
The existence of the lamina lucida in vivo has been questioned.86,88 When the ultrastructure of the basement membrane is evaluated after high-pressure preservation techniques, the lamina densa appears intimately associated with the epithelial cell surface. When the dermal–epidermal junction is similarly prepared, no distinct lamina lucida is seen (see Fig. 53-5B). This suggests that the lamina lucida may result from shrinkage of the cell surface away from the lamina densa due to dehydration. The appearance of anchoring filaments spanning the lamina lucida may then result from the firm attachment of constituents of the lamina densa at the hemidesmosome that is subsequently pulled from the lamina densa by shrinkage. Other components that are also tightly fixed to the keratinocyte plasma membrane, either at the hemidesmosomes or at other sites along the membrane, may similarly become displaced into the shrinkage space. Regardless of its actual occurrence in vivo, the evaluation of the lamina lucida by standard electron microscopy techniques has allowed identification of specific structures that would otherwise have been difficult to detect. In addition, the morphologic term lamina lucida remains practical in the scientific communication and continues to be used.
The second zone, the lamina densa, appears as an electron-dense amorphous structure 20 nm to 50 nm in width. At high magnification, it has a granular–fibrous appearance.86 The major molecular components of the lamina densa are collagen IV, nidogens, perlecan, and laminins, which all can polymerize to networks of variable thickness.84,86
The subbasal lamina contains microfibrillar structures. Two of these are readily distinguishable. Anchoring fibrils appear as condensed fibrous aggregates 20 nm to 75 nm in diameter.89 At high resolution, they appear to have a cross-striated banding pattern (see Fig. 53-5A). The length of the anchoring fibril is difficult to measure because of its random orientation in relation to the plane of the section. In toad skin, these structures have lengths of approximately 800 nm. The anchoring fibrils in human skin appear to be somewhat shorter. The ends of the fibrils appear less tightly packed, giving a somewhat frayed appearance. The proximal end inserts into the basal lamina, and the distal end is integrated into the fibrous network of the dermis.90 Many of the anchoring fibrils originating at the lamina densa loop back into the lamina densa in a horseshoe-like manner; others insert their opposite ends into amorphous-appearing structures, termed anchoring plaques.90 These structures are believed to be independent “islands” of electron-dense material, although some controversy exists in the literature.91 Anchoring fibrils are primarily aggregates of collagen VII.
Fibrillin-containing microfibrils, 10 nm to 12 nm in diameter, are also localized in the sublamina densa region. These are elastic-related fibers, because elastic components of the dermis are formed from microfibrillar and amorphous components.92 The microfibrillar component in the presence of abundant amorphous component is known as the elastic fiber. In the papillary dermis, the microfibrils insert into the basal lamina perpendicular to the basement membrane and extend into the dermis, where they gradually merge with the elastic fibers to form a plexus parallel to the dermal–epidermal junction. These two elastic components appear to be continuous with the elastic fibers present deep within the reticular dermis.92
In summary, the ultrastructure of the dermal–epidermal junction strongly suggests that the lamina densa functions as a structural scaffold for the attachment of the epidermal cells at one surface, secured by anchoring filaments extending from the lamina densa to the hemidesmosomes. The latter also serve as insertion points for intracellular keratin filaments that form scaffolding for the basal cells. On the opposite surface, the extracellular matrix suprastructures of the dermis are firmly attached to the lamina densa. The interaction of collagen-containing dermal fibers with the lamina densa appears to be mediated by the anchoring fibrils. The elastic system of the dermis inserts directly into the basal lamina via the microfibrils. Thus, the dermal–epidermal junction provides a continuous series of attachments between the reticular dermis and the cytoskeleton of the basal cells. These observations suggest four major functions for the epidermal basement membrane: (1) a structural foundation for the secure attachment and polarity of the epidermal basal cells; (2) a barrier separating the epidermis and the dermis; (3) firm attachment of the dermis to the epidermis through a continuous system of structural elements; and (4) modification of cellular functions, such as organization of the cytoskeleton, differentiation, or rescue from apoptotic signaling via outside-in signaling mechanisms.
Biochemical Characterization of the Basement Membrane
Basement membranes contain collagenous and noncollagenous glycoproteins and proteoglycans. The content of the collagen-specific amino acids hydroxyproline and hydroxylysine suggests that collagens account for 40% to 65% of the total basement membrane protein. All basement membranes contain certain isoforms of collagen IV, laminin, nidogen, and the heparan sulfate proteoglycan perlecan (Box 53-2). For example, the α3 chain of collagen IV is localized in the basement membrane of the kidney and lung, but not in those of the skin and blood vessels. In contrast, collagens VII and XVII are associated with the squamous epithelia of skin but are not found in glomerular and alveolar basement membranes. In addition, many other tissue-specific components are found in basement membranes, including different collagens, laminins, fibulins, and fibronectin.84,85,92,93 Differences in macromolecular composition are responsible for morphologic and functional variance of basement membranes.
Collagen IV is a heterotrimer of three α chains.84,94 Each of these contains three distinct domains (Fig. 53-6A): (1) the N-terminal cysteine-rich (7-S) domain, (2) a central triple-helical domain, and (3) a C
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


